

Abstract
Airway wall remodeling processes are present in the small airways of patients with chronic obstructive pulmonary disease, consisting of tissue repair and epithelial metaplasia that contribute to airway wall thickening and airflow obstruction. With increasing disease severity, there is also increased mucous metaplasia and submucosal gland hypertrophy, peribronchial fibrosis, and an increase in airway smooth muscle mass. Apart from its contractile properties, airway smooth muscle produces inflammatory cytokines, proteases, and growth factors, which may contribute to the remodeling process and induce phenotypic changes of the muscle. Airflow limitation responds minimally to β-agonists and corticosteroid therapy, unlike asthma, perhaps because of alterations in β-receptor or glucocorticoid receptor numbers, alterations in receptor signaling, or the constrictive limitation imposed by peribronchial fibrosis. Better response is observed with the combination of inhaled long-acting β-agonists and corticosteroids. This could result from effects at the level of airway smooth muscle. Airway wall remodeling may involve the release of growth factors from inflammatory or resident cells. The influence of smoking cessation or of current therapies on airway wall remodeling is unknown. Specific therapies for airway wall remodeling may be necessary, together with noninvasive methods of imaging small airway wall remodeling to assess responses.
Keywords: corticosteroids, emphysema, long-acting β-agonists, matrix metalloproteases, transforming growth factor-β
The definition of chronic obstructive pulmonary disease (COPD) provided by the Global Initiative on Obstructive Lung Disease (GOLD) guidelines is: “COPD is a disease state characterized by airflow limitation that is not fully reversible. The airflow limitation is usually both progressive and associated with an abnormal inflammatory response of the lungs to noxious particles or gases” (1). The chronic inflammation of COPD is characterized by an accumulation of neutrophils, macrophages, B cells, lymphoid aggregates, and CD8+ T cells, mainly in the small airways (2), and the degree of inflammation increases with the severity of disease as classified by GOLD (3). Although inflammatory cells contribute to the volume of the tissue wall in the small airways in patients with COPD, structural changes, such as epithelial metaplasia, increase in airway smooth muscle, goblet cell hyperplasia, and submucosal gland hypertrophy are other constituents of this thickening. Hogg and coworkers have indicated that the epithelial thickness is increased by approximately 100%, and the lamina propria, the smooth muscle, and the adventitia are increased together by 50% in GOLD stages 3 and 4, compared with GOLD stage 0, indicating a progressive remodeling of the small airways with increasing disease severity (Figure 1) (3). The degree of airflow limitation as measured by FEV1 is also related to the degree of airway wall thickness in COPD (see Figure 1), providing indirect evidence for the role of airway wall remodeling in airflow obstruction of COPD.
Figure 1.
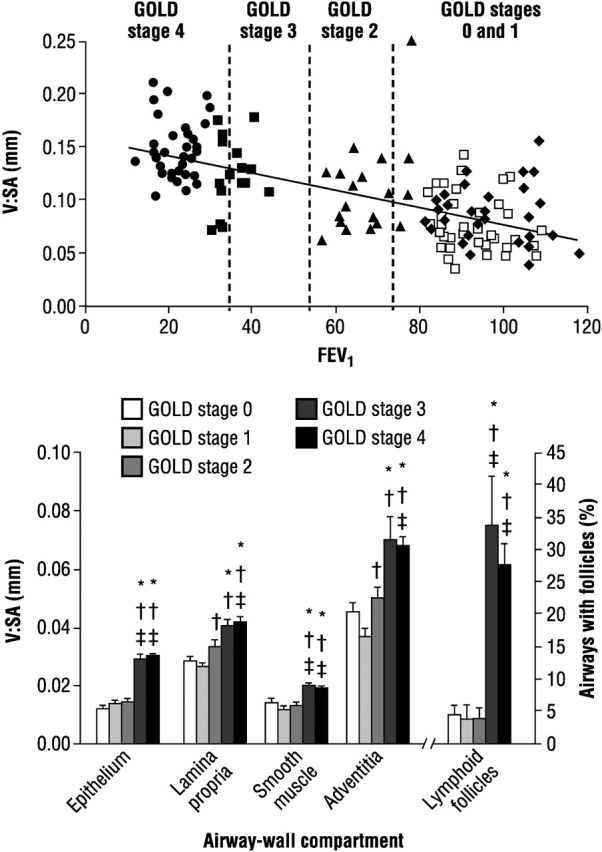
Upper panel: Ratio of volume to surface area (V:SA) as a measure of total wall thickness related to FEV1 in 159 patients with chronic obstructive pulmonary disease (COPD) at different Global Initiative on Obstructive Lung Disease (GOLD) stages. Lower panel: Thickness of epithelium, lamina propria, smooth muscle, and adventitial tissue expressed as total wall thickness (V:SA), and the percentage of airways containing lymphoid follicles in various GOLD stages. *p < 0.001 compared with GOLD stage 0; †p < 0.001 compared with GOLD stage 1; ‡p < 0.001 compared with GOLD stage 2. Reprinted by permission from Reference 3.
The process of remodeling has been better studied in asthma, where changes are present in the large airways, because tissue sampling of the large airways is readily accomplished by the fiberoptic bronchoscope. The process of remodeling in asthma has been described mostly in the large airways from bronchial biopsies, but is present in large and more peripheral airways. The remodeling process includes subepithelial basement membrane fibrosis, epithelial goblet cell hyperplasia, increase in blood vessels, and a proliferative state of the airway smooth muscle, with increased mass comprising hyperplasia and hypertrophy. In COPD, remodeling changes are particularly prominent in the small airways (<2 mm in internal diameter), which are not easily accessible, and most of the information has been available from resection specimens from smokers undergoing lung surgery for tumors or from postmortem specimens. The remodeling changes are also described in the large airways (4). This article examines the components of airway wall remodeling in COPD, their relationship to airflow limitation, the effects of some current treatments, and the potential contribution of airway smooth muscle and matrix changes. The contribution of airway smooth muscle cells is particularly examined in depth.
EPITHELIAL CHANGES
Both shedding of the epithelium and squamous metaplasia have been observed in COPD, but it is likely that the latter, together with goblet cell hyperplasia, is responsible for the increased thickness of the epithelial compartment. The epithelium not only provides a physical barrier between submucosal tissues and the external environment, but also interacts with toxic gases, such as cigarette smoke. Airway wall remodeling may represent the effect of cigarette smoke on the epithelium and attempts by the airway epithelium to protect itself and repair the injury caused by cigarette smoke. The bronchiolar epithelium is altered in smokers, particularly in patients with COPD. Cigarette smoke induces the release of interleukin (IL)-1, IL-8, and granulocyte colony–stimulating factor from bronchial epithelial cells through oxidative pathways, accounting for potential neutrophil and monocytic chemotactic activities released from the epithelium (5, 6). A higher expression of monocyte chemotactic protein-1, transforming growth factor (TGF)-β1, and IL-8 mRNA and protein has been observed in bronchiolar epithelium of smokers with COPD compared with smokers without COPD (7–9). Cultured epithelial cells from smokers and from patients with COPD release more TGF-β in vitro than those from normal patients (8). TGF-β may play an important role in the context of tissue remodeling by stimulation of extracellular matrix production, such as collagen and fibronectin, and reduces matrix degradation by altering the collagenase and collagenase inhibitor balance. Furthermore, TGF-β induces the transformation of fibroblasts to myofibroblasts, which synthesize matrix proteins. Latent TGF-β can be activated through the loss of the integrin, αvβ6, to cause emphysema through alterations of matrix metalloproteinase (MMP)-12 production in macrophages (10). The expression and release of these cytokines and growth factors attest to the role of the epithelial response in submucosal inflammation and fibrosis of COPD.
Goblet Cells, Submucosal Glands, and Mucus Production
The role of chronic sputum production in the development of COPD is uncertain. Although no relationship between the presence of chronic sputum production and the development of COPD was reported in a British cohort (11), a recent Danish study found that chronic sputum production was associated with both the risk of hospitalization because of COPD and excessive yearly decline in FEV1 (12). A postmortem study of lungs from patients dying of COPD showed an increased amount of intraluminal mucus in the bronchioles compared with controls without respiratory disease (13). In surgically resected lung tissues, increasing accumulation of inflammatory exudates with mucus in the small airways was noted with increasing severity of disease (3). Submucosal gland hypertrophy is also seen in the large airways (14, 15). A disproportionate increase in mucous acini and reduction in serous acini has been reported in chronic bronchitis (15). No correlation has been found, however, between mucous gland enlargement and sputum production (16, 17). Goblet cell hyperplasia is a feature of both large and small airways in chronic bronchitis (14). Goblet cells are usually sparse in the small airways, but they are increased in number in the peripheral airways (diameter < 1 mm) of patients with COPD (18). This increase has been associated with an inflammatory process with neutrophil infiltration, supporting the concept that neutrophils may directly cause degranulation of goblet cells through the release of neutrophil elastase and cathepsin G (19). The mechanism of goblet cell hyperplasia itself may involve the activation of the epidermal growth factor receptor, which may be upregulated by oxidants in cigarette smoke and by cytokines, such as tumor necrosis factor (TNF)-α, IL-8, or IL-13 (20, 21). Little is known about the composition of the mucus in COPD. COPD has been specifically associated with increased expression of the mucin MUC5B in the bronchiolar lumen and the mucin MUC5AC in the bronchiolar epithelium (22). Various inducers of MUC5AC, such as neutrophil elastase or cigarette smoke, do so by ligand-dependent activation of an EGFR signaling cascade that could be mediated by ADAM17 and MMP-9 (23–25).
Matrix Changes
In contrast to the thickening of the subepithelial basement membrane in the proximal airways seen in asthma, there is usually no change in this thickness in smokers with chronic bronchitis or COPD. An increase in subepithelial basement membrane associated with tissue eosinophilia was observed, however, in patients with COPD who showed significant reversible airway obstruction that was reversible with corticosteroids (26). In a study of bronchial biopsies of patients with COPD with a mean FEV1 of 56%, a mild increase in subepithelial basement membrane thickness was observed, although not to the same extent as in mild asthma. There were fewer fibroblasts and no changes in collagen III staining in these bronchial biopsies from patients with COPD, whereas in patients with persistent severe asthma, there were more fibroblasts and collagen III staining, indicating the relative lack of fibrotic changes in the proximal large airways in COPD (27).
An increase in matrix deposition in the adventitial compartments of the small airways has been described (3, 28, 29), however, indicating that the fibrotic process may be more important in the small airways. This distribution of matrix proteins in the small airways might contribute to fixed airflow limitation by preventing the airway smooth muscle from relaxing completely, either during hyperinflation or during pharmacologically induced relaxation (Figure 2). The site of matrix deposition is different from that found in asthma, where this is localized predominantly in the subepithelial basement membrane region, whereas in COPD this deposition may be more diffuse throughout the airway wall. The biochemical changes in matrix in COPD, however, have not been intensely investigated. An immunohistochemical study reported reduced expression of the interstitial proteoglycans decorin and biglycan in the peribronchial area of small airways, but without changes in expression of types 1, 3, and 4 collagen, laminin, and fibronectin (30). The significance of this is unclear and further studies are needed.
Figure 2.

Example of extensive small airway remodeling by connective tissue deposition in subepithelial and, particularly, adventitial compartments of the airway wall. Connective tissue deposition seems to encase the airway smooth muscle bundles. Reprinted by permission from Reference 2.
Airway Smooth Muscle Changes
The changes in airway smooth muscle are more prominent in the small airways than in the large airways. In the large airways, alterations in smooth muscle mass were not observed, and the amount of airway smooth muscle did not correlate with airflow limitation (31), although the wall internal to the muscle was significantly thickened and was associated with a reduction in the ratio of FEV1 to forced vital capacity. In biopsies obtained by the fiberoptic bronchoscope, no changes in airway smooth muscle area were observed, and airway smooth muscle size was not increased (27). Furthermore, smooth muscle protein isoforms were not increased, and although myosin light-chain kinase was slightly increased, there was no increase in phosphorylated light-chain myosin (27). These findings are in contrast to those found in patients with severe persistent asthma. In addition, culture of airway smooth muscle cells obtained from biopsies from asthmatic patients showed an increased proliferative rate in serum that persisted over several passages (32), but apparently no such increase was observed in biopsies obtained from patients with emphysema (33).
The airway smooth muscle cells in the small airways have been relatively less well studied, but their properties may be different from those in the proximal airways. A significant increase in airway smooth muscle in small airways of patients with COPD has been reported in several studies (34–36). The amount of airway smooth muscle has also been inversely correlated with lung function (FEV1% predicted) (35). The amount of airway smooth muscle was increased by nearly 50% in patients with more severe COPD, at GOLD stages 3 and 4 (3). In one study (37), the airway smooth muscle mass in the small airways was the only differentiating feature when comparing nonobstructed patients with COPD with patients with asthma. Information on other features of the airway smooth muscle in small airways is not available. Although the airway smooth muscle mass is increased, it is unknown whether this process is caused by an increased number of airway smooth muscle cells, an increase in airway smooth muscle size, or both. Increased proliferative rate of the muscle may be associated with other phenotypic abnormalities. Changes in airway smooth muscle mass seem more prominent in the large airways in asthma, whereas they are more important in the small airways in COPD.
A relationship between force generation in vitro and airway responsiveness measured in vivo has been reported in COPD (38), but other studies reported no such relationship (39–41). Opazo Saez and colleagues examined small airways of average diameter of 2.5 mm in vitro by myography for isometric force generation and isotonic shortening (42). They reported that both force and stress were increased in patients with COPD with airflow obstruction compared with nonobstructed patients, indicating that airway smooth muscle had increased ability to generate force. Maximal isotonic shortening was not significantly different between obstructed and nonobstructed patients. Force and stress correlated well with lung function measurements. Increased ability of the muscle to generate force may contribute to bronchial hyperresponsiveness in COPD, along with loss of lung recoil and fibrosis of the small airways.
Relationship of Changes in Remodeling to Lung Function
The peripheral airways are the major site of obstruction in COPD (43), and airway remodeling and inflammation in these airways are associated with chronic airflow obstruction (29, 34, 44). Furthermore, there is a strong association between disease progression and the increase in epithelium, lamina propria, muscle, and adventitial compartments in COPD (Figure 1) (3). In airway remodeling, mucosal thickening can amplify the effect of mucosal shortening, and muscle thickening can lead to greater muscle shortening against elastic loads (37). In addition, the increased ability of the airway smooth muscle to generate force in chronic obstructive airway disease contributes to the increased airway narrowing when compared with a normal airway. This is a factor in nonspecific bronchial hyperresponsiveness (45), a good predictor of the rapid decline of FEV1 in patients with COPD (46).
The contribution of emphysema to airflow limitation may also be important, but the type of destructive emphysema present may determine lung function changes. Centrilobular emphysema is characterized by focal destruction of lung restricted to respiratory bronchioles and the central portion of the acinus and is usually more severe in the upper portions of the lungs in chronic smokers. Panacinar emphysema involves the uniform destruction of the alveolar walls and is usually found in smokers who develop emphysema early in life and in those with α1-antitrypsin deficiency. Panacinar emphysema is associated with a loss of elastic recoil and higher lung compliance, whereas centrilobular emphysema is associated with a higher degree of airway inflammation and hyperresponsiveness (47–50). In centrilobular emphysema, there is a higher total pathologic score of small airways than in lungs with panacinar emphysema (48). In panacinar emphysema, lung compliance is highest and decreases as the amount of centrilobular emphysema increases. The level of small airway inflammation in smokers correlates with the destruction of alveolar tissue and may facilitate the rupture of the attachment of alveoli with the outer airway wall where mechanical stress is at its maximum (51).
A progressive inflammatory reaction in the small airways has been reported, with connective tissue deposition in the airway wall and a significant degree of emphysema in the most severe cases (29). As the abnormalities in the small airways increased, pulmonary function deteriorated progressively, and tests of small airway function were able to distinguish patients with minimal pathologic changes from those with normal airways (29, 52, 53). The severity of the pathologic abnormalities, including emphysema, correlated with the degree of airflow limitation as measured by FEV1 (54). Direct measurements of peripheral airway resistance show, however, that loss of alveolar support resulting from emphysema is a less important cause of airflow limitation than the changes in airway wall remodeling and in the airway lumen (43).
AIRWAY SMOOTH MUSCLE IN AIRWAY WALL REMODELING
Cytokines and Chemokines
There is a potential for the airway smooth muscle to contribute to the inflammatory and remodeling processes in the small airways (Figure 3). The airway smooth muscle cell not only has contractile properties but also is capable of expressing and releasing cytokines, chemokines, growth factors, and proteases (55, 56), and can participate in the inflammatory and remodeling process (57). Airway smooth muscle cells also produce matrix proteins, and their behavior may depend on interactions with their own matrix (58). Potential cytokines or chemokines of interest in COPD that may be released from airway smooth muscle include IL-6 and -8; monocyte chemotactic protein -1, -2, and -3; growth-related oncogene-α; CXCL10 (IP-10); and granulocyte-macrophage colony–stimulating factor (55, 59–61). Proinflammatory cytokines IL-1β and TNF-α and bradykinin induce the release of IL-8 (60, 62), a potent neutrophil chemoattractant and activator, whereas IFN-γ and TNF-α induce the release of CXCL10, which is also expressed in airway smooth muscle cells of patients with COPD (59). CXCL10 is a potent chemoattractant for human monocytes, neutrophils, natural killer cells, and T cells, preferentially Th1 cells.
Figure 3.

Potential interactions between cytokines, cigarette smoke, and inflammatory cells with airway smooth muscle (ASM) cells that may contribute to small airway wall remodeling in COPD. Interactions of ASM with its own extracellular matrix (ECM) may determine its survival; response to agents, such as β-agonists; and the degree of peribronchial fibrosis. β-AR = β-adrenoceptor; GR = glucocorticoid receptor; GRO-α = growth-related oncogene-α; IL = interleukin; MMP = matrix metalloproteinase; TGF-β = transforming growth factor-β; TNF-α = tumor necrosis factor-α.
Extracellular Matrix Components
The extracellular matrix surrounding the airway smooth muscle can influence airway smooth muscle cell function, such as cytokine production, proliferation, and apoptosis. Fibronectin and collagen I can increase mitogen-induced proliferation in comparison with airway smooth muscle cultured on plastic (63). Airway smooth muscle cells are characterized by comparatively low expression of contractile proteins, such as smooth muscle myosin heavy chain, calponin, and smooth muscle α-actin, but they remain in a proliferative mode. Expression of surface integrins α5 and β2 is involved in survival signaling from collagen IV, collagen V, laminin, and fibronectin, which provide antiapoptotic signals to the airway smooth muscle (64). Disruption of the integrins leads to apoptosis of the airway smooth muscle cell, and this may occur when neutrophils interact with airway smooth muscle. Release of serine proteases from neutrophils, including neutrophil elastase and cathepsin G, induces fibronectin degradation with disruption of integrin binding, leading to apoptosis by detachment of airway smooth muscle cells (65). Neutrophils are prominent cells within airway smooth muscle bundles in COPD (66). This may be a mechanism by which the homeostasis of airway smooth muscle mass can be maintained in the presence of proliferative signals. T cells are also seen in airway smooth muscle bundles in COPD (66), and adhesion of activated T cells can induce airway smooth muscle proliferation through CD44 and vascular cell adhesion molecule (67). The balance of proliferative and apoptotic factors and pathways may determine airway smooth muscle mass.
MMPs
MMPs are zinc-dependent proteolytic enzymes that play a major role in matrix turnover, remodeling, and angiogenesis in COPD, and airway smooth muscle cells may be an important source. In addition to cleaving most of the constituents of the extracellular matrix, MMPs may contribute to smooth muscle hyperplasia by causing the release of immobilized growth factors, such as the release of TGF-β when the extracellular matrix proteoglycan decorin is degraded by MMPs (68). In addition, MMPs may degrade insulin-like growth factor binding proteins, causing the release of insulin-like growth factor (69). Insulin-like growth factor II is released from airway smooth muscle cells and induces airway smooth muscle proliferation (70). Human airway smooth muscle cells constitutively express pro−MMP-2, MMP-3, and membrane type 1 MMP (71). MMP-3 is usually bound to airway smooth muscle−derived matrix, consistent with the staining for MMP-3 in the submucosal matrix of patients with chronic asthma (72). MMP-12, first described from macrophages and thought to be macrophage-specific, is also expressed and released when airway smooth muscle cells are exposed to IL-1β (73), and active MMP-12 is present in airway smooth muscle cells of small airways of smokers and patients with COPD. The role of MMP-12 released from airway smooth muscle cells is still speculative. In addition to acting predominantly as an elastase where extracellular matrix may be degraded, and contributing to emphysema, MMP-12 releases TNF-α from pro−TNF-α (74), ensuring participation in inflammation. The release of MMPs is balanced by the production of tissue inhibitors of metalloproteinase, however, particularly tissue inhibitors of metalloproteinase-1 and -2, which inhibit proteolytic activity (71).
TGF-β
The mediators of airway wall remodeling include the release of growth factors, such as TGF-β and epidermal growth factor. Repair of the epithelial mesenchymal trophic unit, which may result in the production of excessive amounts of these or other growth factors or remodeling mediators, has been postulated in asthma as a major route of airway wall remodeling (75). A similar situation may be present in COPD, with cigarette smoke being the main insult to the epithelium. TGF-β increases the production of extracellular matrix proteins, including collagen and fibronectin, and it is overexpressed in the airway epithelium and airway smooth muscle cells of patients with COPD compared with smokers without COPD (8, 28). Synthesis of procollagen I is regulated by TGF-β in an autocrine fashion (76). TGF-β increases the expression of smooth muscle contractile proteins, such as smooth muscle α-actin and calponin, in airway smooth muscle and fibroblasts, and it increases airway smooth muscle cell size and number (77, 78). TGF-β increases the expression of other growth factors, structural proteins, extracellular matrix proteins, and enzymes from airway smooth muscle cells (55). An interesting effect of TGF-β is the promotion of differentiation of fibroblasts into myofibroblasts secreting interstitial collagen and other growth factors, such as endothelin-1 and vascular endothelial growth factor. There is little work on the fibroblast and the myofibroblast in COPD, and the effects of corticosteroid therapy on the actions of TGF-β are unclear. In vitro studies show, however, that corticosteroids inhibit TGF-β−induced expression of extracellular matrix proteins, including fibronectin, collagen, tissue inhibitors of metalloproteinase, and type 1 plasminogen activator inhibitor (79–81). This action may occur by the direct transrepressive targeting of Smad3, a transcription factor, by the glucocorticoid receptor (79). Whether this occurs in vivo, however, remains to be determined.
Gene expression of the excitatory Smad3 and -4 was unchanged in bronchial biopsies from COPD patients, but the expression of inhibitory Smad7 was decreased, indicating a potential positive enhancement of TGF-β signaling in COPD (82). Although TGF-β may play an important role in remodeling, it has antiinflammatory properties. For example, TGF-β inhibits fractalkine (CX3CL) release induced by TNF-α and IFN-γ from airway smooth muscle cells (83).
AIRWAY SMOOTH MUSCLE AND EFFECTS OF THERAPY
The role of the airway smooth muscle in remodeling is of interest because, as a result of its potential to relax, it is the airway structure that could respond most to the pharmacologic agents used to treat COPD. Response to bronchodilator therapy is small, however, compared with the responses observed in asthma. In addition, corticosteroid therapy during a period of 2 to 4 wk results in little or no improvement in airflow obstruction (26), except in the presence of airway eosinophilia. The mechanisms by which this response is reduced are unknown. Peribronchial adventitial fibrosis may limit the degree of relaxation induced by bronchodilators, such as β-agonists or anticholinergic drugs. In addition, the combination of inhaled corticosteroids and long-acting β-agonists causes a greater degree of bronchodilatation compared with either pharmacologic agent alone (84, 85), indicating possible interaction of these agents at the level of the airway smooth muscle.
The state of the β2-receptor and the glucocorticoid receptor in COPD is unclear. The activity of β-receptors may be decreased together with uncoupling of β2-receptor adenylate cyclase; activity of the phosphodiesterase enzymes that degrade cyclic adenosine monophosphate may be enhanced. IL-1β, which is present in COPD airways, can induce an attenuation of β-receptor−induced airway relaxation (86, 87) through mechanisms involving a reduction in β-receptors, an increase in the Giα subunit, and a defect in adenylyl cyclase activity (87). This effect of IL-1β in impairing β-receptor–mediated relaxation may be reversed partially by corticosteroids through an up-regulation of G-protein–coupled receptor kinases (88), which could underlie the beneficial effect of the combination of corticosteroids and long-acting β-agonists. The presence of TGF-β in COPD may contribute to downregulation of β-receptor function (89, 90). The extracellular matrix may also influence the response of airway smooth muscle to β-receptor stimulation, in that fibronectin increases the response to β-receptor stimulation, whereas collagen V or laminin reduces the accumulation of cyclic adenosine monophosphate through the modulation of Giα (91).
Inhaled corticosteroid therapy does not affect the rate of decline in lung function in patients with COPD (92–94). This may be a reflection of the lack of effect of corticosteroids in reversing the remodeling of the small airways, but there has been no direct study of the effect of corticosteroids on remodeling features. The lack of effect of corticosteroids on the rate of decline in lung function may be secondary to various factors (95) that remain to be determined. Established airway wall remodeling may not be reversed by corticosteroids, but rather may be prevented, as has been demonstrated in remodeling associated with allergic inflammation (96). Alternatively, the lack of response to corticosteroids may be caused by corticosteroid resistance of the mechanisms involved in airway wall remodeling (e.g., the release of cytokines by alveolar macrophages) (97). A defect in recruitment of histone deacetylase activity by corticosteroids in alveolar macrophages of patients with COPD has been proposed (98). A model of corticosteroid resistance in airway smooth muscle related to COPD has not been described. In asthma, the enhanced proliferative phenotype of airway smooth muscle is not inhibited by corticosteroids in vitro because of a specific absence of CCAAT enhancer binding protein-α, a transcription factor (33). No such observation has been made, however, in airway smooth muscle cells from patients with emphysema.
In exacerbations of COPD, systemic administration of corticosteroids is beneficial in increasing the rate of improvement of airflow obstruction and of dyspnea (99). The basis for such improvement may relate to an effect on airway smooth muscle cells through inhibition of proinflammatory cytokine release or through the restoration of β-adrenergic (or cholinergic) receptor function. Prevention of exacerbations in patients with COPD has been shown with inhaled corticosteroid therapy, with combination therapy of corticosteroid and long-acting β-agonists, and with a long-acting anticholinergic drug (94, 100, 101). The mechanisms by which these therapies reduce exacerbations are unknown but could involve an effect on airway smooth muscle cells.
The interaction of corticosteroids and β-agonists may also occur in modulation of cytokine release from airway smooth muscle cells, which could be the basis for the benefit of combination therapy in COPD. Corticosteroids usually inhibit the expression of IL-8 from airway smooth muscle, whereas the addition of β-agonists enhances this effect. Salmeterol potentiated the inhibition of TNF-α−induced IL-8 by fluticasone, whereas on its own it had no effect (102). Salmeterol inhibited TNF-α−induced RANTES (the chemokine regulated on activation, normal T-cell expressed and secreted) and also potentiated the inhibitory effect of dexamethasone on TNF-α–induced RANTES (103). There was potentiation of the release of IL-6 by salmeterol (103), however, indicating that effects of combination treatment on cytokine release are cytokine- or stimulus-specific. Airway smooth muscle cells exposed to cigarette-smoke extracts induced IL-8 release and potentiated TNF-α−induced IL-8 release, but inhibited TNF-α−induced release of RANTES and eotaxin (104). Cigarette smoke–induced IL-8 was inhibited by fluticasone, but there was no potentiation by salmeterol.
Other properties of airway smooth muscle are affected by the combination of β-agonists and corticosteroids. Compared with salmeterol or dexamethasone alone, the combination of the two agents synergistically inhibited migration of human airway smooth muscle cells induced by platelet-derived growth factor (105). Similarly, the combination of formoterol and budesonide synergistically inhibited serum-induced proliferation and induced enhanced activation of p21WAF1/Cip1 stimulated with fetal bovine serum in airway smooth muscle cells (106).
CONCLUSIONS
The small airway wall remodeling process is responsible for a large amount of chronic airflow limitation in COPD, for the decline in lung function, and for the relatively poor responses to available therapies. The process itself remains poorly studied, because access to the small airways is difficult and changes in the small airways are not reflected in the more accessible proximal large airways. Many of the abnormalities underlying these poor responses could be on the airway smooth muscle, which may respond suboptimally to therapies, such as β-agonists and corticosteroids, because of the effect of chronic inflammation or peribronchiolar fibrosis on airway smooth muscle function. To obtain better therapeutic results, more specific targets underlying the airflow limitation need to be aimed at, namely targets of inflammation or airway remodeling processes (107). Noninvasive tools, such as high-resolution CT (108), are urgently needed to delineate the morphologic and physiologic degrees of small airway remodeling (and of emphysema), and to monitor response to treatments.
Supported by the Wellcome Trust (UK), National Institutes of Health, and GlaxoSmithKline (UK).
Conflict of Interest Statement: K.F.C. has received $4,500 in 2004 for attending Advisory Board meetings for GlaxoSmithKline (GSK), Altana, and Novartis. He has also participated in scientific meetings organized by AstraZeneca, GSK, and Altana and has received a travel grant from Boehringer to attend the American Thoracic Society meeting in 2004. He has received an unrestricted research grant from GSK.
References
- 1.Pauwels RA, Buist AS, Calverley PM, Jenkins CR, Hurd SS. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. NHLBI/WHO Global Initiative for Chronic Obstructive Lung Disease (GOLD) Workshop summary. Am J Respir Crit Care Med 2001;163:1256–1276. [DOI] [PubMed] [Google Scholar]
- 2.Hogg JC. Pathophysiology of airflow limitation in chronic obstructive pulmonary disease. Lancet 2004;364:709–721. [DOI] [PubMed] [Google Scholar]
- 3.Hogg JC, Chu F, Utokaparch S, Woods R, Elliott WM, Buzatu L, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med 2004;350:2645–2653. [DOI] [PubMed] [Google Scholar]
- 4.Hamid Q, Cosio M, Lim S. Inflammation and remodeling in chronic obstructive pulmonary disease. J Allergy Clin Immunol 2004;114:1479–1481. [DOI] [PubMed] [Google Scholar]
- 5.Mio T, Romberger DJ, Thompson AB, Robbins RA, Heires A, Rennard SI. Cigarette smoke induces interleukin-8 release from human bronchial epithelial cells. Am J Respir Crit Care Med 1997;155:1770–1776. [DOI] [PubMed] [Google Scholar]
- 6.Masubuchi T, Koyama S, Sato E, Takamizawa A, Kubo K, Sekiguchi M, et al. Smoke extract stimulates lung epithelial cells to release neutrophil and monocyte chemotactic activity. Am J Pathol 1998;153:1903–1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Boer WI, Sont JK, van Schadewijk A, Stolk J, van Krieken JH, Hiemstra PS. Monocyte chemoattractant protein 1, interleukin 8, and chronic airways inflammation in COPD. J Pathol 2000;190:619–626. [DOI] [PubMed] [Google Scholar]
- 8.Takizawa H, Tanaka M, Takami K, Ohtoshi T, Ito K, Satoh M, et al. Increased expression of transforming growth factor-beta1 in small airway epithelium from tobacco smokers and patients with chronic obstructive pulmonary disease (COPD). Am J Respir Crit Care Med 2001;163:1476–1483. [DOI] [PubMed] [Google Scholar]
- 9.Vignola AM, Chanez P, Chiappara G, Merendino A, Pace E, Rizzo A, et al. Transforming growth factor-beta expression in mucosal biopsies in asthma and chronic bronchitis. Am J Respir Crit Care Med 1997;156:591–599. [DOI] [PubMed] [Google Scholar]
- 10.Morris DG, Huang X, Kaminski N, Wang Y, Shapiro SD, Dolganov G, et al. Loss of integrin alpha(v)beta6-mediated TGF-beta activation causes Mmp12-dependent emphysema. Nature 2003;422:169–173. [DOI] [PubMed] [Google Scholar]
- 11.Peto R, Speizer FE, Cochrane AL, Moore F, Fletcher CM, Tinker CM, et al. The relevance in adults of air-flow obstruction, but not of mucus hypersecretion, to mortality from chronic lung disease: results from 20 years of prospective observation. Am Rev Respir Dis 1983;128:491–500. [DOI] [PubMed] [Google Scholar]
- 12.Vestbo J, Prescott E, Lange P. Association of chronic mucus hypersecretion with FEV1 decline and chronic obstructive pulmonary disease morbidity. Copenhagen City Heart Study Group. Am J Respir Crit Care Med 1996;153:1530–1535. [DOI] [PubMed] [Google Scholar]
- 13.Aikawa T, Shimura S, Sasaki H, Takishima T, Yaegashi H, Takahashi T. Morphometric analysis of intraluminal mucus in airways in chronic obstructive pulmonary disease. Am Rev Respir Dis 1989;140:477–482. [DOI] [PubMed] [Google Scholar]
- 14.Reid LM, Melb MB. Pathology of chronic bronchitis. Lancet 1954;i:275–278. [PubMed] [Google Scholar]
- 15.Dunnill MS, Massarella GR, Anderson JA. A comparison of the quantitative anatomy of the bronchi in normal subjects, in status asthmaticus, in chronic bronchitis, and in emphysema. Thorax 1969;24:176–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nagai A, West WW, Thurlbeck WM. The National Institutes of Health Intermittent Positive-Pressure Breathing trial: pathology studies: II. Correlation between morphologic findings, clinical findings, and evidence of expiratory air-flow obstruction. Am Rev Respir Dis 1985;132:946–953. [DOI] [PubMed] [Google Scholar]
- 17.Mullen JB, Wright JL, Wiggs BR, Pare PD, Hogg JC. Reassessment of inflammation of airways in chronic bronchitis. BMJ 1985;291:1235–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saetta M, Turato G, Baraldo S, Zanin A, Braccioni F, Mapp CE et al. Goblet cell hyperplasia and epithelial inflammation in peripheral airways of smokers with both symptoms of chronic bronchitis and chronic airflow limitation. Am J Respir Crit Care Med 2000;161(3 Pt 1):1016–1021. [DOI] [PubMed] [Google Scholar]
- 19.Sommerhoff CP, Nadel JA, Basbaum CB, Caughey GH. Neutrophil elastase and cathepsin G stimulate secretion from cultured bovine airway gland serous cells. J Clin Invest 1990;85:682–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shim JJ, Dabbagh K, Ueki IF, Dao-Pick T, Burgel PR, Takeyama K, et al. IL-13 induces mucin production by stimulating epidermal growth factor receptors and by activating neutrophils. Am J Physiol Lung Cell Mol Physiol 2001;280:L134–L140. [DOI] [PubMed] [Google Scholar]
- 21.Takeyama K, Dabbagh K, Jeong SJ, Dao-Pick T, Ueki IF, Nadel JA. Oxidative stress causes mucin synthesis via transactivation of epidermal growth factor receptor: role of neutrophils. J Immunol 2000;164:1546–1552. [DOI] [PubMed] [Google Scholar]
- 22.Caramori G, Di Gregorio C, Carlstedt I, Casolari P, Guzzinati I, Adcock IM, Barnes PJ, Ciaccia A, Cavllesco G, Chung KF, et al. Mucin expression in peripheral airways of patients with chronic obstructive pulmonary disease. Histopathology 2004;45:477–484. [DOI] [PubMed] [Google Scholar]
- 23.Kohri K, Ueki IF, Nadel JA. Neutrophil elastase induces mucin production by ligand-dependent epidermal growth factor receptor activation. Am J Physiol Lung Cell Mol Physiol 2002;283:L531–L540. [DOI] [PubMed] [Google Scholar]
- 24.Takeyama K, Jung B, Shim JJ, Burgel PR, Dao-Pick T, Ueki IF, et al. Activation of epidermal growth factor receptors is responsible for mucin synthesis induced by cigarette smoke. Am J Physiol Lung Cell Mol Physiol 2001;280:L165–L172. [DOI] [PubMed] [Google Scholar]
- 25.Deshmukh HS, Case LM, Wesselkamper SC, Borchers MT, Martin LD, Shertzer HG, et al. Metalloproteinases mediate mucin 5AC expression by epidermal growth factor receptor activation. Am J Respir Crit Care Med 2005;171:305–314. [DOI] [PubMed] [Google Scholar]
- 26.Chanez P, Vignola AM, O'Shaugnessy T, Enander I, Li D, Jeffery PK, et al. Corticosteroid reversibility in COPD is related to features of asthma. Am J Respir Crit Care Med 1997;155:1529–1534. [DOI] [PubMed] [Google Scholar]
- 27.Benayoun L, Druilhe A, Dombret MC, Aubier M, Pretolani M. Airway structural alterations selectively associated with severe asthma. Am J Respir Crit Care Med 2003;167:1360–1368. [DOI] [PubMed] [Google Scholar]
- 28.de Boer WI, van Schadewijk A, Sont JK, Sharma HS, Stolk J, Hiemstra PS, van Krieken JHJM. Transforming growth factor beta1 and recruitment of macrophages and mast cells in airways in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1998;158:1951–1957. [DOI] [PubMed] [Google Scholar]
- 29.Cosio M, Ghezzo H, Hogg JC, Corbin R, Loveland M, Dosman J, et al. The relations between structural changes in small airways and pulmonary-function tests. N Engl J Med 1978;298:1277–1281. [DOI] [PubMed] [Google Scholar]
- 30.van Straaten JF, Coers W, Noordhoek JA, Huitema S, Flipsen JT, Kauffman HF, et al. Proteoglycan changes in the extracellular matrix of lung tissue from patients with pulmonary emphysema. Mod Pathol 1999;12:697–705. [PubMed] [Google Scholar]
- 31.Tiddens HA, Pare PD, Hogg JC, Hop WC, Lambert R, De Jongste JC. Cartilaginous airway dimensions and airflow obstruction in human lungs. Am J Respir Crit Care Med 1995;152:260–266. [DOI] [PubMed] [Google Scholar]
- 32.Johnson PR, Roth M, Tamm M, Hughes M, Ge Q, King G, et al. Airway smooth muscle cell proliferation is increased in asthma. Am J Respir Crit Care Med 2001;164:474–477. [DOI] [PubMed] [Google Scholar]
- 33.Roth M, Johnson PR, Borger P, Bihl MP, Rudiger JJ, King GG, et al. Dysfunctional interaction of C/EBPalpha and the glucocorticoid receptor in asthmatic bronchial smooth-muscle cells. N Engl J Med 2004;351:560–574. [DOI] [PubMed] [Google Scholar]
- 34.Bosken CH, Wiggs BR, Pare PD, Hogg JC. Small airway dimensions in smokers with obstruction to airflow. Am Rev Respir Dis 1990;142:563–570. [DOI] [PubMed] [Google Scholar]
- 35.Saetta M, Di SA, Turato G, Facchini FM, Corbino L, Mapp CE, et al. CD8+ T-lymphocytes in peripheral airways of smokers with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1998;157:822–826. [DOI] [PubMed] [Google Scholar]
- 36.Cosio MG, Hale KA, Niewoehner DE. Morphologic and morphometric effects of prolonged cigarette smoking on the small airways. Am Rev Respir Dis 1980;122:265–271. [DOI] [PubMed] [Google Scholar]
- 37.Kuwano K, Bosken CH, Pare PD, Bai TR, Wiggs BR, Hogg JC. Small airways dimensions in asthma and in chronic obstructive pulmonary disease. Am Rev Respir Dis 1993;148:1220–1225. [DOI] [PubMed] [Google Scholar]
- 38.De Jongste JC, Sterk PJ, Willems LNA, Mans H, Timmers MC, Kerrebijn KF. Comparison of maximal bronchoconstriction in vivo and airway smooth muscle responses in vitro in nonasthmatic humans. Am Rev Respir Dis 1988;138:321–326. [DOI] [PubMed] [Google Scholar]
- 39.Vincenc CS, Black JL, Yan K, Armour CL, Donnelly PD, Woolcock AJ. Comparison of in vivo and in vitro responses to histamine in human airways. Am Rev Respir Dis 1983;128:875–879. [DOI] [PubMed] [Google Scholar]
- 40.Cerrina J, Ladurie ML, Lebat G, Neffstein B, Bayol A, Brink C. Comparison of human bronchial muscle response to histamine in vivo with histamine and isoproterenol agonists in vitro. Am Rev Respir Dis 1986;134:57–61. [DOI] [PubMed] [Google Scholar]
- 41.Armour CL, Lazar NM, Schellenberg RR, Taylor SM, Chan N, Hogg JC, et al. A comparison of in vivo and in vitro human airway reactivity to histamine. Am Rev Respir Dis 1984;129:907–910. [DOI] [PubMed] [Google Scholar]
- 42.Opazo Saez AM, Seow CY, Pare PD. Peripheral airway smooth muscle mechanics in obstructive airways disease. Am J Respir Crit Care Med 2000;161:910–917. [DOI] [PubMed] [Google Scholar]
- 43.Hogg JC, Macklem PT, Thurlbeck WM. Site and nature of airway obstruction in chronic obstructive lung disease. N Engl J Med 1968;278:1355–1360. [DOI] [PubMed] [Google Scholar]
- 44.Wright JL, Lawson LM, Pare PD, Wiggs BJ, Kennedy S, Hogg JC. Morphology of peripheral airways in current smokers and ex-smokers. Am Rev Respir Dis 1983;127:474–477. [DOI] [PubMed] [Google Scholar]
- 45.Wiggs BR, Bosken C, Pare PD, James A, Hogg JC. A model of airway narrowing in asthma and in chronic obstructive pulmonary disease. Am Rev Respir Dis 1992;145:1251–1258. [DOI] [PubMed] [Google Scholar]
- 46.Tashkin DP, Altose MD, Connett JE, Kanner RE, Lee WW, Wise RA. Methacholine reactivity predicts changes in lung function over time in smokers with early chronic obstructive pulmonary disease. The Lung Health Study Research Group. Am J Respir Crit Care Med 1996;153:1802–1811. [DOI] [PubMed] [Google Scholar]
- 47.Eidelman DH, Ghezzo H, Kim WD, Hyatt RE, Cosio MG. Pressure-volume curves in smokers: comparison with alpha-1-antitrypsin deficiency. Am Rev Respir Dis 1989;139:1452–1458. [DOI] [PubMed] [Google Scholar]
- 48.Kim WD, Eidelman DH, Izquierdo JL, Ghezzo H, Saetta MP, Cosio MG. Centrilobular and panlobular emphysema in smokers: two distinct morphologic and functional entities. Am Rev Respir Dis 1991;144:1385–1390. [DOI] [PubMed] [Google Scholar]
- 49.Finkelstein R, Ma HD, Ghezzo H, Whittaker K, Fraser RS, Cosio MG. Morphometry of small airways in smokers and its relationship to emphysema type and hyperresponsiveness. Am J Respir Crit Care Med 1995;152:267–276. [DOI] [PubMed] [Google Scholar]
- 50.Saetta M, Finkelstein R, Cosio MG. Morphological and cellular basis for airflow limitation in smokers. Eur Respir J 1994;7:1505–1515. [DOI] [PubMed] [Google Scholar]
- 51.Saetta M, Shiner RJ, Angus GE, Kim WD, Wang NS, King M, et al. Destructive index: a measurement of lung parenchymal destruction in smokers. Am Rev Respir Dis 1985;131:764–769. [DOI] [PubMed] [Google Scholar]
- 52.Berend N, Skoog C, Thurlbeck WM. Single-breath nitrogen test in excised human lungs. J Appl Physiol 1981;51:1568–1573. [DOI] [PubMed] [Google Scholar]
- 53.Petty TL, Silvers GW, Stanford RE, Baird MD, Mitchell RS. Small airway pathology is related to increased closing capacity and abnormal slope of phase III in excised human lungs. Am Rev Respir Dis 1980;121:449–456. [DOI] [PubMed] [Google Scholar]
- 54.Hale KA, Ewing SL, Gosnell BA, Niewoehner DE. Lung disease in long-term cigarette smokers with and without chronic air-flow obstruction. Am Rev Respir Dis 1984;130:716–721. [DOI] [PubMed] [Google Scholar]
- 55.Jarai G, Sukkar M, Garrett S, Duroudier N, Westwick J, Adcock I, et al. Effects of interleukin-1beta, interleukin-13 and transforming growth factor-beta on gene expression in human airway smooth muscle using gene microarrays. Eur J Pharmacol 2004;497:255–265. [DOI] [PubMed] [Google Scholar]
- 56.Howarth PH, Knox AJ, Amrani Y, Tliba O, Panettieri RA Jr, Johnson M. Synthetic responses in airway smooth muscle. J Allergy Clin Immunol 2004;114:S32–S50. [DOI] [PubMed] [Google Scholar]
- 57.Chung KF. Airway smooth muscle cells: contributing to and regulating airway mucosal inflammation? Eur Respir J 2000;15:961–968. [DOI] [PubMed] [Google Scholar]
- 58.Johnson PR. Role of human airway smooth muscle in altered extracellular matrix production in asthma. Clin Exp Pharmacol Physiol 2001;28:233–236. [DOI] [PubMed] [Google Scholar]
- 59.Hardaker EL, Bacon AM, Carlson K, Roshak AK, Foley JJ, Schmidt DB, et al. Regulation of TNF-alpha- and IFN-gamma-induced CXCL10 expression: participation of the airway smooth muscle in the pulmonary inflammatory response in chronic obstructive pulmonary disease. FASEB J 2004;18:191–193. [DOI] [PubMed] [Google Scholar]
- 60.John M, Au BT, Jose PJ, Lim S, Saunders M, Barnes PJ et al. Expression and release of interleukin-8 by human airway smooth muscle cells: inhibition by Th-2 cytokines and corticosteroids. Am J Respir Cell Mol Biol 1998;18:84–90. [DOI] [PubMed] [Google Scholar]
- 61.Pype J, Dupont L, Menten J, Van Damme J, Opdenakker G, Chung KF, et al. Expression of monocyte chemoattractant factor (MCP)-1, MCP-2 and MCP-3 by human airway smooth muscle cells: modulation by corticosteroids and Th2 cytokines. Am J Respir Cell Mol Biol 1999;21:528–536. [DOI] [PubMed] [Google Scholar]
- 62.Pang L, Knox AJ. Bradykinin stimulates IL-8 production in cultured human airway smooth muscle cells: role of cyclooxygenase products. J Immunol 1998;161:2509–2515. [PubMed] [Google Scholar]
- 63.Hirst SJ, Twort CH, Lee TH. Differential effects of extracellular matrix proteins on human airway smooth muscle cell proliferation and phenotype. Am J Respir Cell Mol Biol 2000;23:335–344. [DOI] [PubMed] [Google Scholar]
- 64.Freyer AM, Johnson SR, Hall IP. Effects of growth factors and extracellular matrix on survival of human airway smooth muscle cells. Am J Respir Cell Mol Biol 2001;25:569–576. [DOI] [PubMed] [Google Scholar]
- 65.Oltmanns U, Sukkar MB, Xie S, John M, Chung KF. Induction of human airway smooth muscle apoptosis by neutrophils and neutrophil elastase. Am J Respir Cell Mol Biol 2005;32:334–341. [DOI] [PubMed] [Google Scholar]
- 66.Baraldo S, Turato G, Badin C, Bazzan E, Beghe B, Zuin R, et al. Neutrophilic infiltration within the airway smooth muscle in patients with COPD. Thorax 2004;59:308–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lazaar AL, Albelda SM, Pilewski JM, Brennan B, Pure E, Panettieri RA. T lymphocytes adhere to airway smooth muscle cells via integrins and CD44 and induce smooth muscle cell DNA synthesis. J Exp Med 1994;180:807–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Imai K, Hiramatsu A, Fukushima D, Pierschbacher MD, Okada Y. Degradation of decorin by matrix metalloproteinases: identification of the cleavage sites, kinetic analyses and transforming growth factor-beta1 release. Biochem J 1997;322:809–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fowlkes JL, Enghild JJ, Suzuki K, Nagase H. Matrix metalloproteinases degrade insulin-like growth factor-binding protein-3 in dermal fibroblast cultures. J Biol Chem 1994;269:25742–25746. [PubMed] [Google Scholar]
- 70.Noveral JP, Bhala A, Hintz RL, Grunstein MM, Cohen P. Insulin-like growth factor axis in airway smooth muscle cells. Am J Physiol 1994;267:L761–L765. [DOI] [PubMed] [Google Scholar]
- 71.Elshaw SR, Henderson N, Knox AJ, Watson SA, Buttle DJ, Johnson SR. Matrix metalloproteinase expression and activity in human airway smooth muscle cells. Br J Pharmacol 2004;142:1318–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dahlen B, Shute J, Howarth P. Immunohistochemical localisation of the matrix metalloproteinases MMP-3 and MMP-9 within the airways in asthma. Thorax 1999;54:590–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Xie S, Chung KF. Expression and regulation of matrix metalloproteinase-12 in human airway smooth muscle cells. Am J Respir Crit Care Med 2004;169:A193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chandler S, Cossins J, Lury J, Wells G. Macrophage metalloelastase degrades matrix and myelin proteins and processes a tumour necrosis factor-alpha fusion protein. Biochem Biophys Res Commun 1996;228:421–429. [DOI] [PubMed] [Google Scholar]
- 75.Davies DE, Wicks J, Powell RM, Puddicombe SM, Holgate ST. Airway remodeling in asthma: new insights. J Allergy Clin Immunol 2003;111:215–225. [DOI] [PubMed] [Google Scholar]
- 76.Coutts A, Chen G, Stephens N, Hirst S, Douglas D, Eichholtz T, et al. Release of biologically active TGF-beta from airway smooth muscle cells induces autocrine synthesis of collagen. Am J Physiol Lung Cell Mol Physiol 2001;280:L999–1008. [DOI] [PubMed] [Google Scholar]
- 77.Black PN, Young PG, Skinner SJ. Response of airway smooth muscle cells to TGF-beta 1: effects on growth and synthesis of glycosaminoglycans. Am J Physiol 1996;271:L910–L917. [DOI] [PubMed] [Google Scholar]
- 78.Cohen P, Rajah R, Rosenbloom J, Herrick DJ. IGFBP-3 mediates TGF-beta1-induced cell growth in human airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 2000;278:L545–L551. [DOI] [PubMed] [Google Scholar]
- 79.Song CZ, Tian X, Gelehrter TD. Glucocorticoid receptor inhibits transforming growth factor-beta signaling by directly targeting the transcriptional activation function of Smad3. Proc Natl Acad Sci U S A 1999;96:11776–11781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Slavin J, Unemori E, Hunt TK, Amento E. Transforming growth factor beta (TGF-beta) and dexamethasone have direct opposing effects on collagen metabolism in low passage human dermal fibroblasts in vitro. Growth Factors 1994;11:205–213. [DOI] [PubMed] [Google Scholar]
- 81.Su S, Dehnade F, Zafarullah M. Regulation of tissue inhibitor of metalloproteinases-3 gene expression by transforming growth factor-beta and dexamethasone in bovine and human articular chondrocytes. DNA Cell Biol 1996;15:1039–1048. [DOI] [PubMed] [Google Scholar]
- 82.Springer J, Scholz FR, Peiser C, Groneberg DA, Fischer A. SMAD-signaling in chronic obstructive pulmonary disease: transcriptional down-regulation of inhibitory SMAD 6 and 7 by cigarette smoke. Biol Chem 2004;385:649–653. [DOI] [PubMed] [Google Scholar]
- 83.Sukkar MB, Issa R, Xie S, Oltmanns U, Newton R, Chung KF. Fractalkine/CX3CL1 production by human airway smooth muscle cells: induction by IFN-gamma and TNF-alpha and regulation by TGF-beta and corticosteroids. Am J Physiol Lung Cell Mol Physiol 2004;287:L1230–L1240. [DOI] [PubMed] [Google Scholar]
- 84.Calverley PM, Boonsawat W, Cseke Z, Zhong N, Peterson S, Olsson H. Maintenance therapy with budesonide and formoterol in chronic obstructive pulmonary disease. Eur Respir J 2003;22:912–919. [DOI] [PubMed] [Google Scholar]
- 85.Calverley P, Pauwels R, Vestbo J, Jones P, Pride N, Gulsvik A, et al. Combined salmeterol and fluticasone in the treatment of chronic obstructive pulmonary disease: a randomised controlled trial. Lancet 2003;361:449–456. [DOI] [PubMed] [Google Scholar]
- 86.Shore SA, Laporte J, Hall IP, Hardy E, Panettieri RAJ. Effect of IL-1 beta on responses of cultured human airway smooth muscle cells to bronchodilator agonists. Am J Respir Cell Mol Biol 1997;16:702–712. [DOI] [PubMed] [Google Scholar]
- 87.Koto H, Mak JC, Haddad EB, Xu WB, Salmon M, Barnes PJ, et al. Mechanisms of impaired beta-adrenoceptor-induced airway relaxation by interleukin-1beta in vivo in the rat. J Clin Invest 1996;98:1780–1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mak JC, Hisada T, Salmon M, Barnes PJ, Chung KF. Glucocorticoids reverse IL-1beta-induced impairment of beta-adrenoceptor-mediated relaxation and up-regulation of G-protein-coupled receptor kinases. Br J Pharmacol 2002;135:987–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mak JC, Rousell J, Haddad EB, Barnes PJ. Transforming growth factor-beta1 inhibits beta2-adrenoceptor gene transcription. Naunyn Schmiedebergs Arch Pharmacol 2000;362:520–525. [DOI] [PubMed] [Google Scholar]
- 90.Ishikawa T, Kume H, Kondo M, Ito Y, Yamaki K, Shimokata K. Inhibitory effects of interferon-gamma on the heterologous desensitization of beta-adrenoceptors by transforming growth factor-beta 1 in tracheal smooth muscle. Clin Exp Allergy 2003;33:808–815. [DOI] [PubMed] [Google Scholar]
- 91.Freyer AM, Billington CK, Penn RB, Hall IP. Extracellular matrix modulates β2-adrenergic receptor signaling in human airway smooth muscle cells. Am J Respir Cell Mol Biol 2004;31:440–445. [DOI] [PubMed] [Google Scholar]
- 92.Pauwels RA, Lofdahl CG, Laitinen LA, Schouten JP, Postma DS, Pride NB, et al. Long-term treatment with inhaled budesonide in persons with mild chronic obstructive pulmonary disease who continue smoking. European Respiratory Society Study on Chronic Obstructive Pulmonary Disease. N Engl J Med 1999;340:1948–1953. [DOI] [PubMed] [Google Scholar]
- 93.Vestbo J, Sorensen T, Lange P, Brix A, Torre P, Viskum K. Long-term effect of inhaled budesonide in mild and moderate chronic obstructive pulmonary disease: a randomised controlled trial. Lancet 1999;353:1819–1823. [DOI] [PubMed] [Google Scholar]
- 94.Burge PS, Calverley PM, Jones PW, Spencer S, Anderson JA, Maslen TK. Randomised, double blind, placebo controlled study of fluticasone propionate in patients with moderate to severe chronic obstructive pulmonary disease: the ISOLDE trial. BMJ 2000;320:1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Adcock IM, Chung KF. Why are corticosteroids ineffective in COPD? Curr Opin Investig Drugs 2002;3:58–60. [PubMed] [Google Scholar]
- 96.Vanacker NJ, Palmans E, Kips JC, Pauwels RA. Fluticasone inhibits but does not reverse allergen-induced structural airway changes. Am J Respir Crit Care Med 2001;163:674–679. [DOI] [PubMed] [Google Scholar]
- 97.Barnes PJ, Ito K, Adcock IM. Corticosteroid resistance in chronic obstructive pulmonary disease: inactivation of histone deacetylase. Lancet 2004;363:731–733. [DOI] [PubMed] [Google Scholar]
- 98.Ito K, Ito M, Elliott WM, Cosio B, Caramori G, Kon OM, et al. Decreased histone deacetylase activity in chronic obstructive pulmonary disease. N Engl J Med 2005;352:1967–1976. [DOI] [PubMed] [Google Scholar]
- 99.Wood-Baker RR, Gibson PG, Hannay M, Walters EH, Walters JA. Systemic corticosteroids for acute exacerbations of chronic obstructive pulmonary disease. Cochrane Database Syst Rev 2005;CD001288. [DOI] [PubMed]
- 100.Calverley P, Pauwels R, Vestbo J, Jones P, Pride N, Gulsvik A, et al. Combined salmeterol and fluticasone in the treatment of chronic obstructive pulmonary disease: a randomised controlled trial. Lancet 2003;361:449–456. [DOI] [PubMed] [Google Scholar]
- 101.Brusasco V, Hodder R, Miravitlles M, Korducki L, Towse L, Kesten S. Health outcomes following treatment for six months with once daily tiotropium compared with twice daily salmeterol in patients with COPD. Thorax 2003;58:399–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Pang L, Knox AJ. Synergistic inhibition by β2-agonists and corticosteroids on tumor necrosis factor-alpha-induced interleukin-8 release from cultured human airway smooth-muscle cells. Am J Respir Cell Mol Biol 2000;23:79–85. [DOI] [PubMed] [Google Scholar]
- 103.Ammit AJ, Hoffman RK, Amrani Y, Lazaar AL, Hay DW, Torphy TJ, et al. Tumor necrosis factor-alpha-induced secretion of RANTES and interleukin-6 from human airway smooth-muscle cells: modulation by cyclic adenosine monophosphate. Am J Respir Cell Mol Biol 2000;23:794–802. [DOI] [PubMed] [Google Scholar]
- 104.Oltmanns U, Chung KF, Walters M, John M, Mitchell JA. Cigarette smoke induces IL-8, but inhibits eotaxin and RANTES release from airway smooth muscle. Respir Res 2005;6:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Goncharova EA, Billington CK, Irani C, Vorotnikov AV, Tkachuk VA, Penn RB, et al. Cyclic AMP-mobilizing agents and glucocorticoids modulate human smooth muscle cell migration. Am J Respir Cell Mol Biol 2003;29:19–27. [DOI] [PubMed] [Google Scholar]
- 106.Roth M, Johnson PR, Rudiger JJ, King GG, Ge Q, Burgess JK, et al. Interaction between glucocorticoids and beta2 agonists on bronchial airway smooth muscle cells through synchronised cellular signalling. Lancet 2002;360:1293–1299. [DOI] [PubMed] [Google Scholar]
- 107.Barnes PJ, Hansel TT. Prospects for new drugs for chronic obstructive pulmonary disease. Lancet 2004;364:985–996. [DOI] [PubMed] [Google Scholar]
- 108.Nakano Y, Muro S, Sakai H, Hirai T, Chin K, Tsukino M, et al. Computed tomographic measurements of airway dimensions and emphysema in smokers: correlation with lung function. Am J Respir Crit Care Med 2000;162:1102–1108. [DOI] [PubMed] [Google Scholar]