

Abstract
There is now compelling evidence that proteinases and oxidative stress play pathogenetic roles in the following pathologies in chronic obstructive pulmonary disease: airspace enlargement; chronic inflammation in the airways, lung interstitium, and alveolar space; and mucus hypersecretion in the large airways. Proteinases and oxidants may also contribute to remodeling processes in the small airways. In addition, data are emerging that show interactions between classes of proteinases and between proteinases and oxidants, which amplify lung inflammation and injury in chronic obstructive pulmonary disease. This review discusses the biologic roles of proteinases and oxidants, their roles in the pathogenesis of chronic obstructive pulmonary disease, and their potential as targets for therapy.
Keywords: antiinflammatory drugs, antioxidants, oxidative stress, proteinase inhibition, proteinases
PROTEINASES AND OXIDANTS IN THE PATHOGENESIS OF COPD
The proteinase–antiproteinase hypothesis for the pathogenesis of chronic obstructive pulmonary disease (COPD) was formulated almost 40 yr ago in response to two key observations. First, genetic deficiency of α1-antitrypsin (AAT, the major inhibitor of neutrophil elastase [NE] in the lower respiratory tract) is associated with early onset, severe panlobular emphysema (1). Second, instillation of papain (an enzyme with elastase activity) into rat lungs results in progressive airspace enlargement (2). Since then, other elastolytic proteinases have been shown to cause airspace enlargement when instilled into rodent lungs. Consequently, it was thought that an imbalance between proteinases (especially elastases derived from polymorphonuclear neutrophils [PMNs]) and their inhibitors causes pulmonary emphysema. Although excessive activity of PMN-derived elastases is likely to be important in the panlobular pulmonary emphysema associated with AAT deficiency, this is probably an oversimplification of mechanisms underlying common, “garden variety” COPD in cigarette smokers. Other factors have now been implicated in airspace enlargement in COPD, including other classes of proteinases (matrix metalloproteinases [MMPs] and cysteine proteinases), oxidative stress, and apoptosis of lung structural cells. In addition, COPD is clinically and pathologically a heterogeneous disease and includes chronic inflammation in the airways, lung interstitium, and the alveolar space; mucus hypersecretion; and subepithelial fibrosis in the small airways. There is evidence that proteinases and oxidative stress contribute to all of these pathologies in COPD.
CLASSIFICATION, CELL BIOLOGY, AND BIOLOGICAL FUNCTIONS OF PROTEINASES
Classification
Proteinases are enzymes that cleave the internal peptide bonds of polypeptides (3, 4). They can be classified into four groups according to the chemical nature of their active site: serine, metallo-, cysteine, and aspartic proteinases (Table 1). Proteinase inhibitors are generally targeted against proteinases having a specific catalytic mechanism (Table 1). Serine proteinases and MMPs are optimally active at neutral pH (neutral proteinases) and, therefore, play the major role in extracellular proteolysis. Most cysteine and aspartic proteinases are optimally active at acidic pH, and their main role is in intracellular degradation of proteins in the acidic environment of lysosomes. However, some aspartic proteinases (such as renin) function in the extracellular space at neutral pH. Other acidic proteinases degrade extracellular proteins if they retain some of their catalytic activity at neutral pH (as cathepsin S does) or if they are released into an environment having an acidic pH, such as the pericellular environment of activated macrophages, as demonstrated for cathepsins S and L (3). The proteinases implicated in the pathogenesis of COPD are serine, metallo-, and cysteine proteinases. They are expressed by PMNs, macrophages, lymphocytes, and structural cells of the lung (Table 1) (3).
TABLE 1.
Proteinases involved in the pathogenesis of chronic obstructive pulmonary disease
| Class of Protein | Optimal pH | Example | Cellular Source | Inhibitor |
|---|---|---|---|---|
| Serine proteinases | Neutral | Neutrophil elastase | PMNs | α1-Antitrypsin |
| Neutral | Cathepsin G | P monocytes | α1-Antichymotrypsin | |
| Neutral | Proteinase-3 | Secretory leukocyte protease inhibitor | ||
| Elafin | ||||
| Neutral | Tissue kallikrein | PMNs, monocytes, macrophages, submucosal glands |
α1-Antitrypsin | |
| Neutral | Urokinase-type plasminogen activator | PMNs, monocytes, macrophages | Plasminogen activator inhibitors; α2-macroglobulin |
|
| Metalloproteinases | Neutral | MMP-1 to MMP-28; ADAMs | Granulocytes | Tissue inhibitor of metalloproteinase (types 1 to 4) |
| Monocytes | ||||
| Macrophages | ||||
| Epithelial cells | α-Macroglobulin | |||
| Fibroblasts | ||||
| Cysteine proteinases | Acidic | Cathepsins B, S, H, L | Inflammatory cells | Cystatins A, C, S |
| T cells | ||||
| Epithelial cells | α2-Macroglobulin |
Definition of abbreviations: ADAM = cell surface proteinase containing a disintegrin and a metalloproteinase domain; MMP = matrix metalloproteinase; PMN = polymorphonuclear neutrophils; P monocytes = proinflammatory monocytes.
Cell Biology
PMNs do not synthesize proteinases de novo. Serine proteinases and MMPs are produced in PMN and monocyte precursors in the bone marrow. They are stored in various granules of PMNs and are released from degranulating PMNs (3). Unlike PMNs, human monocytes are heterogeneous in their expression of serine proteinases. Only a subpopulation of circulating monocytes (∼ 30%) having a neutrophil-like proinflammatory phenotype (proinflammatory monocytes) contains serine proteinases in their primary granules (5). Cysteine proteinases are stored as processed, active forms within the lysosomes of many cells. MMPs are generally synthesized and secreted by macrophages and structural lung cells in response to cellular activation by various mediators (6). Monocytes have limited capacity to synthesize and secrete MMPs, producing predominantly MMP-7 when they are activated. As monocytes mature into macrophages, they lose their complement of serine proteinases, but develop the capacity to secrete MMP-1, -3, -9, -12, and -14, which increases further when they are activated (3, 6, 7). Unlike serine proteinases, which are stored as active enzymes within cells, MMPs are released as proenzymes and are activated within the extracellular space. Latency is maintained by an interaction between the active site zinc and a conserved cysteine residue in the prodomain. Activation of most pro-MMPs occurs after their secretion into the extracellular space, via disruption of this interaction (the cysteine switch mechanism), which likely is mediated by proteinases and oxidants (8–10). However, MMP-11 and four of the membrane-type subfamily of MMPs (see the next section) are activated inside cells. These pro-MMPs have recognition sequences for proprotein convertase family members, such as furin, which are intracellular serine proteinases (11). Proprotein convertases are thought to cleave and activate these pro-MMPs in the Golgi network before they are secreted from cells.
Membrane-associated Proteinases
A subset of MMPs includes membrane-associated proteinases (membrane-type MMPs). They are anchored to the cell surface by either a transmembrane domain or a glycosylphosphatidylinositol link, and they are expressed on the surface of inflammatory and structural cells (12).
ADAMs are a family of 29 cell surface proteinases anchored to the surface of inflammatory and structural lung cells by a transmembrane domain (13). They are so called because they contain a disintegrin and a metalloproteinase domain. The metalloproteinase domain of ADAMs enables them to shed and activate cell surface molecules involved in inflammation, such as pro−tumor necrosis factor (TNF)-α. TNF-α is produced as an inactive 26-kD transmembrane protein (pro−TNF-α), and ADAM-17 and -10 cleave it, releasing biologically active 17-kD soluble TNF-α (13). ADAMs also shed other membrane-anchored cytokines, growth factors, and ligands for apoptosis, as well as the receptors for these molecules from cells (13). Disintegrins bind to integrins to regulate integrin-mediated cell adhesion (13). Thus, ADAMs may contribute to extracellular proteolysis, and may regulate cell adhesion and migration, inflammation, apoptosis, and cell signaling.
Many of the other MMP family members, and also serine proteinases implicated in the pathogenesis of COPD, lack a transmembrane domain and have been thought to function exclusively as soluble enzymes. However, studies have shown that active forms of NE, cathepsin G, proteinase 3, urokinase-type plasminogen activator, and MMPs, including MMP-8 and -9, are expressed on the surface of inflammatory cells in an inducible manner. Although these surface-bound proteinases are similar in catalytic activity to the soluble forms of the enzymes, they differ from the soluble enzymes because they are substantially resistant to inhibition by naturally occurring proteinase inhibitors (14–18). There is evidence that these membrane-bound forms of proteinases contribute to pathologies in COPD (19–21).
Biological Functions
The main physiologic role of proteinases has been thought to be in degradation of extracellular matrix (ECM) proteins during embryonic development, postnatal growth, and menstruation. During lung inflammation and injury, proteinases may remove damaged ECM and other tissue debris, regulate coagulation and fibrinolysis, and possibly participate in repair processes (3). However, NE and MMP-12 are now known to play critical roles in innate immunity against bacteria and fungi (22, 23). MMP-7 activates antimicrobial defensin polypeptides that lyse bacteria (24). MMPs and serine proteinases also regulate migration of inflammatory cells by cleaving cytokines and chemokines to either generate or remove chemotactic gradients (25–27). In addition, MMPs promote tumor growth and metastasis by clearing paths for tumor cells by locally degrading ECM, activating latent growth factors, and promoting angiogenesis (28). However, MMP-12, -9, and -3 prevent tumor growth and metastasis by generating angiostatin from plasminogen to inhibit angiogenesis (29). MMP-8 also plays an antitumor role in murine skin (25).
CLASSIFICATION AND BIOLOGICAL ROLES OF OXIDANTS AND ANTIOXIDANTS
Classification
Oxidants can be classified as reactive oxygen species (ROS) and reactive nitrogen species (RNS). In health, the main sources of oxidants is inflammatory cells and resident cells, including lung epithelial cells (30). Inflammatory cells generate potent ROS and RNS. They express nicotinamide adenine dinucleotide phosphate (NADPH) oxidase and xanthine oxidase, which convert O2 to the superoxide anion O2−, which can directly cause oxidative injury. This ROS is rapidly dismutated by superoxide dismutase (SOD) to hydrogen peroxide (H2O2; Figure 1). H2O2 is converted to a potent ROS by inflammatory cells using myeloperoxidase (a PMN enzyme), which generates hypochlorous acid (HOCl). Interaction between H2O2 and Fe2+ also generates the potent hydroxyl radical (OH·) by the Fenton and Haber-Weiss reactions (Figure 1). Macrophages also express nitric oxide synthase type 2 (NOS2), which converts l-arginine to nitric oxide (NO) (31). Interaction between O2− and NO generates the potent peroxynitrate (ONOO−).
Figure 1.

Biology of oxidants and antioxidants. Oxidants are shown in red and antioxidants are shown in blue. AP-1 = activator protein-1; ELF = epithelial lining fluid; GCS = glutamylcysteine synthetase; GSH = glutathione; MPO = myeloperoxidase; NADPH = nicotinamide adenine dinucleotide phosphate; NO = nitric oxide; NOS = nitric oxide synthase; SOD = superoxide dismutase.
Antioxidants are present in intracellular and extracellular compartments and protect cells from oxidative injury by rapidly inactivating oxidants (30, 32). Antioxidants can be classified as enzymes, such as catalase, SOD, and glutathione (GSH) peroxidase (GSH peroxidase), or as nonenzymatic scavengers (Figure 1). Scavengers include the low molecular weight reduced GSH, vitamins A and E, and high molecular weight proteins containing many sulfhydryl and/or disulfide groups, such as albumin and mucins (30, 32). Reduced GSH is a major intracellular antioxidant. Important antioxidants in interstitial fluids and airway secretions are GSH, vitamin C, and mucins. Important antioxidants in plasma include albumin and iron-binding proteins, such as ceruloplasmin.
Biological Roles
The main biological role of oxidant is in host defense against infection (33). Bacteria are engulfed by PMNs and macrophages into phagosomes. The lysosomal granules of these phagocytes fuse with the phagosome, releasing proteinases and myeloperoxidase into the phagosome. NADPH oxidase present in the membranes of primary granules of phagocytes generates massive amounts of O2− inside the phagolysosome (34). It has been thought that inflammatory cells use O2− to generate potent ROS and RNS (see the preceding section) that kill bacteria. However, studies show that increased O2− generated inside the phagolysosome causes an anionic load that stimulates a pH-dependent influx of K+ into the phagolysosome, increasing the ionic strength inside the vacuole. This releases serine proteinases bound to sulfated proteoglycans inside the phagolysosome, permitting serine proteinase−mediated bacterial cell wall lysis (35).
Other physiologic roles for oxidants include regulation of growth factor– and cytokine-mediated signaling, and of cell growth and survival by altering the cellular redox state or by modifying oxidative proteins (36, 37). Oxidants also regulate the activity of proteinases. For example, ROS and RNS activate latent pro-MMPs (9, 10). However, HOCl also limits the activity of active MMP-7 in vitro by oxidatively modifying two amino acids within its catalytic domain, preventing its efficient interaction with substrates (38). However, it is unclear whether regulation of MMP activity is an important function of oxidants in vivo.
PROOF OF CONCEPT FOR THE ROLES OF PROTEINASES IN COPD
In addition to the seminal observations by Laurell and Eriksson (1) and Gross and coworkers (2), evidence of roles for proteinases in COPD comes from in vitro studies of the activities of proteinases, correlative studies of proteinase expression in cells and lung samples from human cigarette smokers and patients with COPD, and animal models of COPD. These studies show that proteinases contribute to airspace enlargement, chronic inflammation in the lower respiratory tract, and mucus hypersecretion and other changes in the airways of smokers and patients with COPD (Figure 2).
Figure 2.
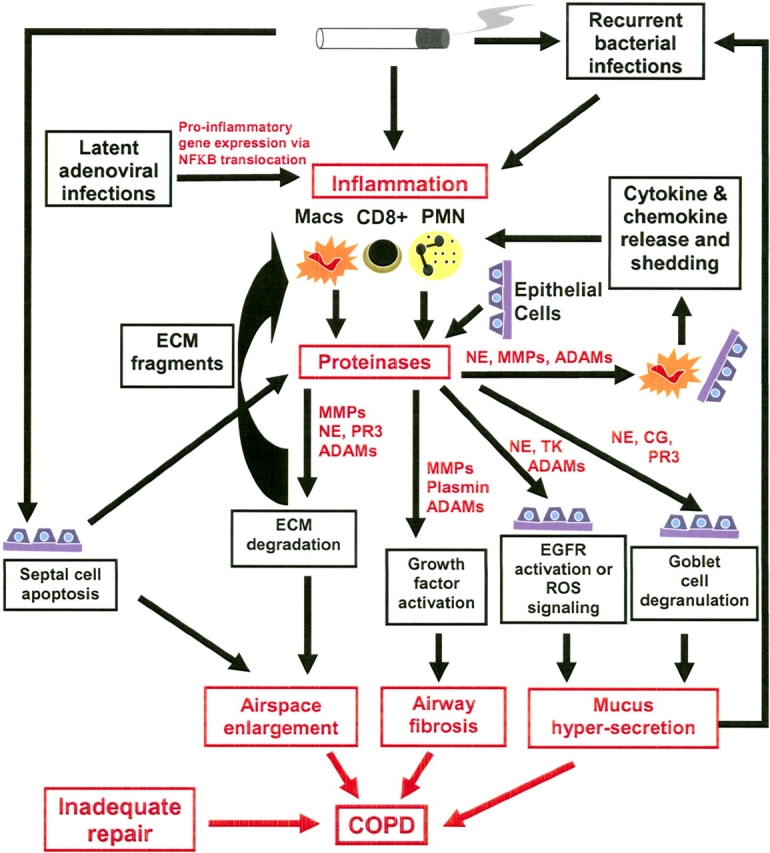
Mechanisms of proteinase-mediated lung injury in COPD. ADAM = cell surface proteinase containing a disintegrin and a metalloproteinase domain; CG = cathepsin G; ECM = extracellular matrix; EGFR = epithelial growth factor receptor; Macs = macrophages; MMP = matrix metalloproteinase; NE = neutrophil elastase; PMN = polymorphonuclear neutrophils; PR3 = proteinase-3; TK = tissue kallikrein.
In Vitro Studies of Proteinases
In vitro studies of the catalytic activities of MMPs, serine proteinases, and cysteine proteinases indicate that they have the capacity to contribute to all the pathologies listed above.
Airspace enlargement.
Proteinases act together to degrade all components of the lung ECM, including structural components (elastin and interstitial collagens) and basement membrane proteins (3), all of which must be degraded when alveolar units are lost to produce airspace enlargement. Proteinases also promote inflammation by several mechanisms (Figure 2). First, MMP and serine proteinase family members cleave and activate cytokines and chemokines. For example, MMP-9 and MMP-8 process interleukin (IL)-8 (39) and lipopolysaccharide-induced CXC chemokine (25), respectively, generating more potent forms of these chemokines. MMP-7 sheds syndecan-1 from epithelial cells, releasing KC (a murine PMN chemokine), which is bound to syndecan-1 (40). Pro−TNF-α is shed and activated by ADAMs and MMPs (13, 41). Serine proteinases activate pro–TNF-α and pro–IL-1β (27). Second, NE stimulates the release of cytokines and chemokines from epithelial cells and mononuclear phagocytes (42, 43). Third, NE and several MMPs cleave elastin (44, 45) and AAT (46, 47), generating fragments that are chemotactic for inflammatory cells. By promoting inflammation, proteinases have the potential to further increase the lung proteinase burden and, thereby, amplify proteinase-mediated inflammation and ECM destruction (Figure 2). It is noteworthy that in human patients with COPD, other mechanisms also amplify and perpetuate inflammation (Figure 2), including recurrent bacterial respiratory tract infections. There is also evidence that in patients with COPD, latent adenoviral infection of epithelial cells amplifies lung inflammation (even when patients have stopped smoking) by increasing synthesis of proinflammatory mediators by activating NF-κB, a proinflammatory gene transcription factor (48).
Airway pathologies.
Proteinases including NE and ADAMs family members contribute to more airway pathologies, including mucus hypersecretion and small airway fibrosis (Figure 2).
Mucus hypersecretion.
Mucus hypersecretion occurs in the large airways of many patients with COPD. Several proteinases have been shown to be involved in increasing mucin synthesis by activating the epithelial growth factor receptor (EGFR) expressed by serous cells in the submucosal glands by shedding inactive, membrane-associated ligands for this receptor. This permits EGFR ligands to bind to, and to activate, this receptor. For example, cigarette smoke activates the EGFR on airway epithelial cells by inducing ADAM-17–mediated shedding and activation of the EGFR ligand amphiregulin (20). NE and ADAM-17 activate the EGFR by shedding another membrane-associated EGFR ligand, pro−transforming growth factor (TGF)-α, leading to an increase in the expression of MUC5AC, a major mucin protein (49, 50). However, NE also increases mucin synthesis by an oxidant-dependent mechanism (51). Tissue kallikrein, a serine proteinase expressed by inflammatory cells and submucosal glands, also stimulates mucin synthesis in COPD airways by shedding and activating pro-EGF, another EGFR ligand (52). Mucus hypersecretion and mucociliary dysfunction in the airways of patients with COPD predispose them to recurrent bacterial infection. The latter may amplify mucus hypersecretion in COPD airways by at least two mechanisms. First, lipoteichoic acid, a product of the cell walls of gram-positive bacteria, stimulates ADAM-10 to shed and activate another EGFR ligand [membrane heparin-binding epithelial growth factor (19, 53)], leading to increased mucin synthesis. Second, PMNs are recruited into the airways of patients with COPD during bacterial infection, and NE, cathepsin G, and proteinase-3 released by activated PMNs or expressed on the surface of PMNs potently stimulate goblet cells to degranulate and release mucus (21).
Small airway fibrosis.
Proteinases also activate latent growth factors in vitro and thus have the potential to induce fibroblasts to deposit interstitial collagens in the walls of the small airways in patients with COPD. For example, plasmin and MMP-9 activate latent TGF-β (54, 55). Insulin-like growth factors circulate in a latent form bound to insulin-like growth factor–binding proteins. Several MMPs and ADAM-9 degrade these binding proteins, releasing active insulin-like growth factors (56, 57). ADAMs may also regulate small airway remodeling by shedding and activating cell surface−bound ligands for the EGFR (see the previous section). However, it is not known whether proteinases contribute to subepithelial fibrosis in the small airways of patients with COPD.
Correlative Studies of Human Patients with COPD
Many studies have shown that the expression of serine proteinases and MMPs (MMP-1, -2, -8, -9, and -14) is increased in bronchoalveolar lavage (BAL) cells (58–61), BAL fluid (62–64), sputum (65–68), and lung tissue (69–71) from smokers and patients with COPD when compared with healthy subjects. In addition, several studies have demonstrated a direct relationship between proteinase levels and disease severity (58, 63, 64, 66–69, 71).
Animal Models of COPD
The strongest support for the roles of proteinases in COPD comes from animal models of COPD. These models include subjecting mice (and other animals) to acute and chronic cigarette smoke. This results in airspace enlargement, inflammation, and subepithelial fibrosis in the small airways, similar to that occurring in human cigarette smokers. Other investigators have studied transgenic mice overexpressing proteinases in the lung, or overexpressing cytokines in an inducible manner in the airway epithelium of adult murine lungs, to induce lung inflammation associated with an increased lung burden of proteinases. Studies of mice genetically deficient in proteinases by gene targeting in these models have provided insights into the pathogenetic roles of individual proteinases. As outlined below, MMP-12, MMP-9, and NE have been shown to contribute significantly to regulating lung inflammation and airspace enlargement in murine models of COPD (72–74). However, little is currently known about the roles of proteinases in airway pathologies in animal models of COPD.
Cigarette smoke exposure models of airspace enlargement.
Mice deficient in MMP-12 (MMP-12−/− mice) exposed to cigarette smoke for 6 mo are 100% protected from developing airspace enlargement, indicating that MMP-12 plays a major role in airspace enlargement in mice (72). MMP-12 also regulates chronic lung inflammation in this model, because wild-type mice exposed to cigarette smoke develop a fivefold increase in macrophage accumulation in the distal airspaces, but this does not occur in MMP-12−/− mice (72). The latter is not due to inability of monocytes from MMP-12−/− mice to migrate into the lung, because delivery of a monocyte chemokine to MMP-12−/− mice fully restores macrophage accumulation in response to cigarette smoke without leading to airspace enlargement. However, BAL fluid from wild-type, but not MMP-12−/−, mice contains chemotactic activity for monocytes. Thus, cigarette smoke likely stimulates constitutive lung macrophages to produce MMP-12, which generates a chemokine gradient, stimulating the recruitment of monocytes from the vasculature into the lung.
Studies of NE−/− mice in the chronic cigarette smoking model showed that NE also plays a major role in airspace enlargement in mice, because NE−/− mice are 60% protected from airspace enlargement (73). Recruitment of PMNs and monocytes is also significantly impaired in NE−/− mice exposed to cigarette smoke, by unknown mechanisms. Thus, NE is required for full PMN and monocyte recruitment during chronic cigarette smoke exposure in mice. However, MMP-9−/− mice are not significantly protected from airspace enlargement in the chronic cigarette smoke exposure model (T. Betsuyaku and S. D. Shapiro, personal communication).
Studies of NE−/− and MMP-12−/− mice exposed to cigarette smoke also demonstrated interactions between these different classes of proteinases. This may explain, in part, the overlapping protection from airspace enlargement that deficiency of either enzyme provides. First, wild-type mice had lower whole-lung levels of AAT than did MMP-12−/− mice, and lower levels of tissue inhibitor of metalloproteinase-1 than did NE−/− mice (73). In vitro, NE cleaves and inactivates tissue inhibitor of metalloproteinase-1 (75), and MMP-12 cleaves and inactivates AAT (76). Thus, it is likely that NE and MMP-12 promote each other's deleterious activities in the lung by inactivating their inhibitors. Second, in mice acutely exposed to cigarette smoke, MMP-12 regulates PMN influx (and the lung NE burden). This is likely mediated by MMP-12 shedding and activating pro−TNF-α from cell surfaces; soluble, active TNF-α upregulates E-selectin expression by endothelial cells, which may promote PMN adhesion and transendothelial migration (41).
Transgenic murine models of airspace enlargement.
Transgenic mice overexpressing MMP-1 in the lung develop enlarged airspaces (77). However, it is unclear whether this represents abnormal alveolar development or destruction of mature interstitial collagens by MMP-1. Inducible overexpression of MMP-1 (and other interstitial collagenases) in the mature murine lung would determine the importance of collagenolytic proteinases in the development of pulmonary emphysema.
Transgenic mice overexpressing cytokines in an inducible manner in the adult lung have been used to assess the roles of immune-mediated inflammation in COPD. Immune cells play important roles in COPD, because CD8+ lymphocytes are present in increased numbers in the lung, along with PMNs and macrophages (78), and immune cells interact with macrophages to stimulate macrophage MMP production (79). Adult transgenic mice overexpressing a helper T cell type 1 cytokine (interferon-γ) or a helper T cell type 2 cytokine (IL-13) in the airway epithelium develop impressive pulmonary inflammation associated with increased production of MMPs and cysteine proteinases, and airspace enlargement (80, 81). In mice overexpressing IL-13, genetic deletion of MMP-12 and -9 confirmed roles for these proteinases in causing airspace enlargement, and MMP-12 was shown to mediate, at least partially, the expression of other MMPs in the lung (74). In addition, genetic deletion of MMP-12 in mice overexpressing IL-13 reduced lung inflammation (74), confirming a role for MMP-12 in promoting pulmonary inflammation in this model.
Proteinases and other mediators in COPD.
Other murine models have shown roles for MMP-12 in airspace enlargement in mice through interactions between MMP-12 and other mediators. TGF-β is one such mediator. TGF-β is known to inhibit macrophage activation and MMP-12 production (82). In addition, αvβ6 integrin expressed by lung epithelial cells is involved in activation of latent TGF-β in the lung (83). In αvβ6−/− mice, the associated loss of TGF-β activation in the lung leads to markedly increased MMP-12 expression by macrophages and airspace enlargement (84). This process is prevented by overexpression of active TGF-β in the lungs of αvβ6−/− mice. Another molecule interacting with MMPs in the lung is surfactant protein D (SP-D). SP-D−/− mice have increased numbers of activated lung macrophages producing increased amounts of MMPs. These mice develop abnormally large airspaces during the first 3 weeks of postnatal lung development (85).
Alveolar septal cell apoptosis models of airspace enlargement.
Apoptosis of the endothelial and epithelial cellular components of the alveolar walls is thought to contribute to airspace enlargement during the development of emphysema. Apoptosis of alveolar septal cells (86–90) and leukocytes (90–92) occurs in patients with COPD. Additional evidence of roles for septal cell apoptosis in emphysema pathogenesis comes from animal studies. For example, pharmacologic blockade of vascular endothelial growth factor receptors (VEGFR) leads to apoptosis of endothelial cells followed by airspace enlargement in the absence of inflammation (93). In addition, transfection of alveolar epithelial cells with the active caspase-3 (a proapoptotic aspartic proteinase) leads to epithelial cell apoptosis followed by airspace enlargement (87). These studies indicate that alveolar septal cell apoptosis is sufficient to cause airspace enlargement in mice. However, increased elastase activity (mediated by proteinases active at acidic pH) is also detected in BAL samples after transfection of alveolar epithelial cells with caspase-3 (87). Thus, proteinases released from dying cells may act synergistically with septal cell apoptosis to cause loss of alveolar units and airspace enlargement by degrading the structural components of the lung ECM. NE hinders clearance of apoptotic PMNs by lung macrophages by cleaving the phosphatidylserine receptor on macrophages, which recognizes phosphatidylserine expressed on the surface of PMNs in the early phase of PMN apoptosis (94). Thus, during acute bacterial exacerbations in patients with COPD, NE released from PMNs recruited into the airways may hinder clearance of PMNs by macrophages, and the prolonged persistence of apoptotic PMN products, including PMN proteinases, may further contribute to proteinase-mediated lung inflammation and injury. Uptake of apoptotic PMNs by macrophages also stimulates macrophage production of antiinflammatory signals such as TGF-β by macrophages, which enhances resolution of inflammation (94). Thus, NE-mediated cleavage of the phosphatidylserine receptor on macrophages may also promote lung inflammation in COPD by inhibiting the generation of antiinflammatory signals by macrophages.
Relevance of murine models of COPD to human disease.
Studies of genetically engineered mice in murine models of COPD indicate that serine proteinases, cysteine proteinases, and metalloproteinases, especially MMP-12, play major roles in airspace enlargement and lung inflammation. However, the exact roles of proteinases in airway pathologies in COPD have yet to be investigated in murine models of COPD.
The relevance of animal models to human COPD depends on the extent to which murine biology mirrors human biology. It is noteworthy that there are important differences in the structure of the respiratory tracts of mice and humans. Mice have fewer branching airways, no respiratory bronchioles, and no submucosal glands. There are also important differences in the profiles of proteinase expressed by murine and human inflammatory and resident lung cells. For example, mice do not express the collagenase MMP-1. MMP-12 is a more prominent MMP in murine macrophages compared with human macrophages. Thus, MMPs other than MMP-12 (e.g., MMP-1, -2, -9, and -14) may be more important in pathologies occurring in human patients with COPD. There are also differences in the pathologies and course of the disease in murine models of COPD versus the human disease. Mice do not develop mucus hypersecretion in the murine cigarette smoke exposure model. Furthermore, mice have fewer circulating PMNs than do humans. Unlike in human patients with COPD, mice housed in barrier facilities, and exposed chronically to cigarette smoke alone, do not develop recurrent respiratory tract bacterial or viral infections. Thus, the PMN and its complement of serine proteinases and MMPs may be relatively more important in human COPD, especially during acute exacerbations.
PROOF OF CONCEPT FOR OXIDATIVE STRESS IN THE PATHOGENESIS OF COPD
Oxidative stress occurs when an imbalance develops between oxidants and antioxidants because of an increased burden of oxidants and/or depletion of antioxidants. The evidence that oxidative stress contributes to the pathogenesis of COPD comes from studies showing oxidant–antioxidant imbalance in smokers and human patients with COPD, and from studies showing that oxidants contribute to pathologies occurring in the lungs of patients with COPD (Figure 3).
Figure 3.
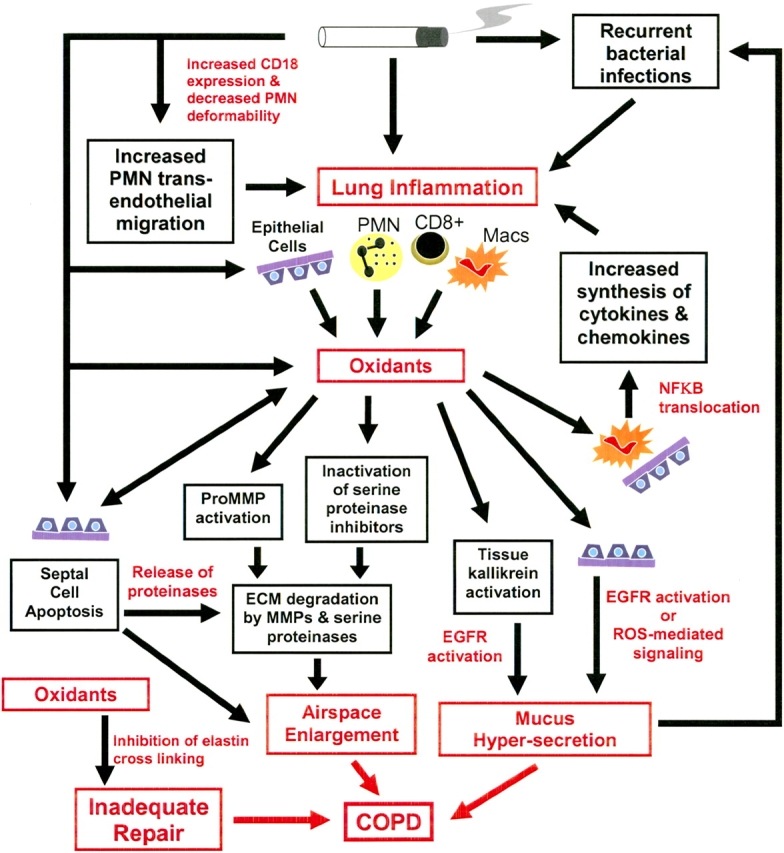
Mechanisms of oxidant-mediated lung injury in COPD. ECM = extracellular matrix; EGFR = epithelial growth factor receptor; Macs = macrophages; MMP = matrix metalloproteinase; NF-κB = nuclear factor-κB; PMN = polymorphonuclear neutrophils; ROS = reactive oxygen species.
Sources of Oxidants in COPD
An important source of the increased burden of oxidants in cigarette smokers and patients with COPD is cigarette smoke itself, which contains large numbers of free radicals and other oxidants (95). The gas phase of cigarette smoke contains predominantly alkyl and peroxyl radicals (1015 radicals per puff). The tar phase of cigarette smoke also contains high concentrations (1017 spins/g, as observed by electron spin resonance) of radicals such OH·, and the semiquinolone radical (which converts O2 to O2−). It also contains H2O2, which is converted to OH· by Fe2+ present in the epithelial lining fluid (30, 96). Modification of the polyunsaturated fatty acids of cell membranes (lipid peroxidation) by free radicals generates peroxides and aldehydes that amplify oxidative stress during smoking (97). Inflammatory cells recruited and activated in the lungs in response to cigarette smoking also contribute to the increased oxidative stress in COPD by generating various ROS and RNS (30, 96). Lipopolysaccharide and TNF-α, which are present in the lungs of patients with COPD, also upregulate NOS2 production of NO by inflammatory cells (31).
Evidence of Increased Oxidative Stress in COPD
Oxidative stress is measured directly by quantifying oxidant production by cells. It is also measured indirectly by quantifying the effects of oxidants on lipids: lipid peroxidation is measured indirectly as thiobarbituric acid–reactive substances or urinary isoprostane F2α-III (98), or more directly as F2-isoprostane (99). Antioxidant capacity or antioxidant responses to oxidative stress are other, indirect measures of oxidative stress (30).
Increased oxidative stress occurs in exhaled breath condensates, BAL fluid, plasma, urine, and leukocytes from cigarette smokers and patients with COPD (98–101). For example, patients with COPD have higher levels of H2O2 in exhaled breath condensates than do healthy smokers, and levels increase further during infective exacerbations (102). Urinary isoprostane F2α-III is increased in patients with COPD and is further increased during exacerbations (98). Cigarette smokers have higher blood levels of F2-isoprostane than do nonsmokers, and healthy smokers (99) and patients with COPD have higher levels of thiobarbituric acid–reactive substances in BAL fluid and plasma than do nonsmokers (101). PMNs from patients with COPD with exacerbations generate more O2− than do cells from patients with COPD in a stable clinical state (103). Alveolar macrophages from cigarette smokers release more ROS (104–106), and also more Fe2+ (107), than do cells from nonsmokers, facilitating the generation of the reactive OH·.
The plasma antioxidant capacity is decreased by cigarette smoking (108, 109), and it decreases further in patients with acute exacerbations of COPD (110). It also correlates negatively with the release of oxidants from circulating PMNs in patients with exacerbations of COPD (110). However, there are adaptive responses in the antioxidant shield to chronic cigarette smoking. Chronic cigarette smokers have increased GSH in BAL fluid (111, 112). This may be due to oxidant-mediated activation of the redox-sensitive transcription factor AP-1 (Figure 1), which upregulates expression of γ-glutamylcysteine synthetase, the rate-limiting enzyme in GSH synthesis (113, 114). However, in human chronic cigarette smokers, 1 h after smoking one to two cigarettes, there is significant depletion of GSH levels in the epithelial lining fluid (30). Acute depletion of intracellular GSH also occurs when epithelial cells are exposed to oxidant in cigarette smoke condensates (112, 115). This is mediated by decreased activity of γ-glutamylcysteine synthetase and leads to increased epithelial permeability (96, 115). These observations suggest that the adaptive increase in antioxidant shield in the epithelial lining fluid of chronic cigarette smokers is not sufficient to protect the lung epithelium during acute smoking.
Mechanisms of Oxidant-mediated Injury in COPD
In vitro and in vivo studies show that oxidants have the potential to contribute to airspace enlargement by inducing septal cell death, lung inflammation, and mucus hypersecretion. Oxidants may also promote proteinase-mediated lung injury (Figure 3).
Airspace enlargement.
As outlined above, there is evidence that apoptosis of the cellular components of the alveolar walls contributes to airspace enlargement during the development of pulmonary emphysema. There is also evidence that oxidative stress in patients with COPD contributes to this process. Oxidants from cigarette smoke injure epithelial cells in vitro (116) and also in vivo, because chronic human cigarette smokers have increased lung epithelial permeability, which increases further during acute smoking (101, 117). Oxidative stress modifies cellular signaling involved in apoptosis. VEGFR blockade leads to septal cell apoptosis in the murine lung associated with increased expression of markers of oxidative stress, leading to airspace enlargement (93, 118). When VEGFR inhibitor–treated mice are given an SOD mimetic, not only are all these processes inhibited, but there is also an increase in septal cell proliferation and enhanced phosphorylation of the antiapoptotic protein Akt (118). In addition, inhibition of caspases in mice treated with the VEGFR inhibitor results in reduced expression of markers of oxidative stress (118). This indicates that oxidative stress and septal cell apoptosis interact to contribute to loss of alveolar units in COPD. Oxidants may also promote proteinase-mediated injury to the ECM components of the alveolar walls (Figure 3) by activating proteinases or inactivating proteinase inhibitors (see below). Together, these oxidant activities may contribute to airspace enlargement in COPD.
Inflammation.
Oxidants increase the lung burden of PMNs during smoking. PMN transit time in the pulmonary microcirculation is delayed after smoking (119). PMNs have a larger diameter (7 μm) than that of the pulmonary capillaries (5 μm) and must deform to transit the pulmonary capillary bed. Oxidants in cigarette smoke decrease PMN deformability by inducing actin polymerization (120). This increases PMN sequestration in the pulmonary microvasculature in human cigarette smokers, allowing more time for PMNs to adhere to the endothelium and migrate through it into the lung. Oxidants in cigarette smoke also increase PMN–endothelial adhesion in rodents by upregulating the expression of CD18 integrins on PMNs (121). In addition, oxidants promote inflammation by regulating the production of proinflammatory mediators in the lung. For example, the levels of TNF-α and IL-8, which promote PMN–endothelial adhesion and PMN chemotaxis, respectively, are increased in sputum and BAL fluids of patients with COPD (122, 123). Oxidants increase the expression of proinflammatory mediators by macrophages and epithelial cells in vitro by increasing nuclear translocation of NF-κB (124, 125), and this process can be inhibited by treating cells with antioxidants (126).
Mucus hypersecretion.
ROS and RNS stimulate the release of mucus by airway epithelial cells (127–129). ROS increase MUC5AC mucin production by airway epithelium by stimulating ligand-independent activation of the EGFR (128).
Oxidant and Proteinase Interactions in COPD
Oxidants may potentiate proteinase-mediated pathologies in COPD by regulating the activities of proteinases. As outlined previously, oxidants increase MMP activity by activating latent pro-MMPs in vitro (Figure 3), but it is unclear whether this occurs in vivo. ROS also activate the serine proteinase, tissue kallikrein. Tissue kallikrein is present in the airways in a latent form that is bound to polymerized hyaluronan. ROS release active tissue kallikrein by inducing hyaluronan depolymerization (52). Thus, ROS promote tissue kallikrein–mediated activities in the airways such as mucus hypersecretion (Figure 3). Oxidants may potentiate serine proteinase activity by oxidatively inactivating two serine proteinase inhibitors, AAT and secretory leukocyte protease inhibitor (130, 131). Both of these inhibitors have a methionine residue at their active sites. Oxidants convert the methionine to methionine sulfoxide, which reduces their capacity to inhibit serine proteinases. Studies in the 1980s yielded conflicting results on whether oxidative inactivation of serine proteinase inhibitors occurs in smokers or patients with COPD (reviewed in Reference 132). However, oxidants are short-lived molecules and are likely to be active only at short distances from the cells that generate them before they are inactivated by antioxidants. Thus, oxidative inactivation of serine proteinase inhibitors could contribute to proteinase-mediated lung injury in cell microenvironments in the lung. Although most of the evidence points to a role for oxidants in increasing proteinase activity, it is noteworthy that oxidants may also limit the activity of MMPs after they have been activated (38).
Repair in COPD
Repair mechanisms are activated in the lung after tissue injury mediated by proteinases and oxidants. However, in patients with COPD, there is inadequate (or abnormal) repair of injured alveolar and airway structures. Our understanding of lung repair mechanisms in COPD has lagged far behind our understanding of the inflammatory and destructive pathways leading to pathologies in COPD. However, the administration of retinoic acid stimulates the formation of new alveoli after the development of airspace enlargement in elastase-exposed adult rats (133), indicating that the injured lung can undergo repair and that exogenous agents can promote repair of injured alveoli. It is likely that structural cells such as epithelial cells, endothelial cells, and fibroblasts play important roles in lung repair processes.
Components of cigarette smoke inhibit fibroblasts and epithelial cell migration and proliferation in vitro (134–136), indicating that cigarette smoke itself interferes with normal repair processes in the lung. However, there is currently little information about the roles of proteinases and oxidants in lung repair in COPD. Proteinases may contribute to scarring around the small airways, which likely represents abnormal repair in patients with COPD (Figure 2). MMPs and ADAMs have the potential to contribute to this process by regulating the activity of growth factors (Figure 2). Oxidants, by activating pro-MMPs could also contribute to this process (Figure 3). Oxidants also inhibit elastin cross-linking and thereby inhibit efficient repair of elastic fibers in the lung interstitium (137). In the bleomycin model of lung injury in mice, MMP-9 promotes migration of epithelial cells in the distal airways into injured alveoli [alveolar bronchiolization (138)], which is probably an aberrant lung repair mechanism. However, it is not clear whether MMP-9 and other proteinases play roles in lung repair processes in COPD.
POTENTIAL OF PROTEINASE INHIBITION/ANTIOXIDANT APPROACHES IN THE TREATMENT OF COPD
Proteinase Inhibition
AAT augmentation therapy is being used in the United States in AAT-deficient patients who have impaired lung function. Although there have been no controlled, randomized clinical trials of AAT augmentation therapy, observational studies of AAT-deficient patients indicate that AAT augmentation slows the rate of decline in lung function, increases quality of life scores, and decreases exacerbation frequency (139).
There have been no long-term clinical trials of proteinase inhibitors in patients with COPD with normal levels of AAT, mainly because of the high cost of funding such trials. However, on the basis of evidence indicating key pathogenetic roles for proteinases in animal models of COPD, it is likely that proteinase inhibition would be an effective new therapeutic strategy. Further support for this approach comes from in vitro studies showing that low molecular weight, synthetic inhibitors of serine proteinases and MMPs effectively inhibit both soluble and membrane-bound proteinases (14, 15, 17, 18), and from studies of proteinase inhibitors in various animal models of COPD showing that proteinase inhibitors effectively block both airspace enlargement and lung inflammation. For example, delivery of synthetic inhibitors of MMPs and cysteine proteinases to mice overexpressing IL-13 significantly prevents airspace enlargement and macrophage accumulation in the lung (74). In animals acutely exposed to cigarette smoke, delivery of synthetic or natural inhibitors of serine proteinases and synthetic inhibitors of MMPs blocks PMN influx into the lung and ECM destruction (140–142). Furthermore, daily oral delivery of synthetic MMP inhibitors to mice prevents airspace enlargement and macrophage accumulation in the lung during 6 mo of exposure to cigarette smoke (143). In further experiments, in which MMP inhibitor therapy was initiated after mice were exposed to cigarette smoke for 3 mo to initiate airspace enlargement, therapy prevented progression of airspace enlargement as smoking continued (143). Thus, proteinase inhibition potentially could prevent disease progression in human patients with COPD. However, it is still unclear which proteinases should be targeted in patients. In addition, proteinases have been shown to have beneficial as well as deleterious roles in the lung (including roles in innate host defense, dampening inflammation, and inhibiting tumor growth and metastasis), which may prove to limit the usefulness of proteinase inhibition.
Antioxidant Therapy
In view of the potential roles of oxidative stress in COPD pathogenesis, approaches to redress the imbalance between oxidants and antioxidants may also prove useful in COPD. There are various options to increase the lung antioxidant defense. The simplest way is to administer antioxidants to patients with COPD. Antioxidants that have been administered to cigarette smokers and patients with COPD include vitamins C and E, β-carotene, and GSH precursors, but the results have been mixed (reviewed in Reference 144). Although vitamin E supplementation in patients with COPD reduces some measurements of lung oxidative stress, vitamin E supplementation for 12 weeks did not augment the depleted levels of vitamin E (145). GSH supplementation has been tried (146), but GSH is not transported well into cells, and an excess of GSH may generate thiyl radicals when oxidative stress is present. Supplementation with cysteine-containing derivatives (cysteine is the rate-limiting amino acid in GSH synthesis) has been assessed in patients with COPD. N-Acetyl cysteine (NAC) and nacystelyn are cysteine-donating molecules that increase cellular GSH synthesis. They also function directly as antioxidants by the interaction of their free thiol groups with electrophilic groups of oxidants. Both compounds are also mucolytic: they split the disulfide bonds in mucin proteins to decrease mucus viscosity and enhance airway clearance of mucus (147). In clinical trials of patients with COPD, oral administration of NAC or nacystelyn increased plasma and BAL fluid GSH levels, increased intracellular levels of GSH in alveolar epithelial cells, and inhibited ROS release from phagocytes (148–151). In open and double-blinded trials of patients with chronic bronchitis and COPD, NAC improved symptoms (decreased sputum viscosity, sputum purulence, and cough severity), decreased bacterial colonization, and reduced the rate of rehospitalization among patients with COPD by 30% (152). In a Swedish study, patients with COPD older than 50 yr were given NAC for 2 yr, which slowed the rate of decline in FEV1 (153). However, there are conflicting reports on the effects of NAC on rates of acute exacerbations in patients with COPD (154–156).
Other potential antioxidant approaches include using more lipophilic methyl esters of GSH, which pass through cell membranes, to increase intracellular GSH, or using thiazolidine, an alternative cysteine donor that protects cells against oxidant injury in vitro (157). However, these have not been tested in clinical trials. Alternative approaches include developing compounds with activity similar to that of catalase or SOD, or upregulating GSH synthesis in vivo by targeting AP-1−mediated activation of γ-glutamylcysteine synthetase. The development and testing of new antioxidant therapies are justified on the basis of evidence indicating the pathogenetic roles of oxidative stress in COPD.
Antiinflammatory Strategies
Approaches to reduce lung inflammatory cell would reduce the lung burden of both proteinases and oxidants in COPD. Inhibitors of phosphodiesterase E4, the major isoenzyme in inflammatory cells, decrease inflammatory cell migration, activation, and release of proteinases. Clinical trials of phosphodiesterase E4 inhibitors in COPD are underway, and the data are promising (see Reference 158). Other antiinflammatory approaches, such as inhibiting NF-κB activation to reduce proinflammatory gene expression, could also potentially inhibit proteinase- and oxidant-mediated lung injury in patients with COPD.
CONCLUSIONS
There is now considerable evidence that proteinases and oxidants can act synergistically to mediate injury to the ECM and lung structural cells and promote inflammation, mucus hypersecretion, and remodeling of the small airways in COPD. Strategies to directly inhibit proteinases and oxidants, alone or in combination with novel antiinflammatory strategies, have the potential to reduce symptoms, disease progression, and mortality in COPD. Determining the safety and efficacy of proteinase inhibitors, antioxidant approaches, and antiinflammatory agents should be a priority for the treatment of COPD.
Acknowledgments
The author thanks the many other investigators who have made important contributions to the fields of proteinases and oxidants in COPD, but whose work was not cited because of space limitations. The author also thanks Steven D. Shapiro, M.D. (Brigham and Women's Hospital and Harvard Medical School, Boston, MA), for critical reading of the manuscript.
Supported by U.S. Public Health Service NHLBI grant RO1 HL63137, by a Career Investigator Award from the Massachusetts Thoracic Society, and by a research grant from the American Thoracic Society/Pulmonary Fibrosis Foundation.
Conflict of Interest Statement: C.A.O. received $1,000 in 2003 for speaking at a conference sponsored by Wyeth and $3,000 for participating as a speaker at a conference sponsored by GlaxoSmithKline. C.A.O. has received a nonclinical research grant of $42,800 in 1999–2003 from GlaxoSmithKline.
References
- 1.Laurell CB, Eriksson S. The electrophoretic α-1-globulin pattern of serum in α-1-antitrypsin deficiency. Scand J Clin Lab Invest 1963;15:132–140. [DOI] [PubMed] [Google Scholar]
- 2.Gross P, Pfitzer EA, Tolker E, Babyak MA, Kaschak M. Experimental emphysema: its production with papain in normal and silicotic rats. Arch Environ Health 1965;11:50–58. [DOI] [PubMed] [Google Scholar]
- 3.Owen CA, Campbell EJ. The cell biology of leukocyte-mediated proteolysis. J Leukoc Biol 1999;65:137–150. [DOI] [PubMed] [Google Scholar]
- 4.Barrett AJ. The many forms and functions of cellular proteinases. Fed Proc 1980;39:9–14. [PubMed] [Google Scholar]
- 5.Owen CA, Campbell MA, Boukedes SS, Stockley RA, Campbell EJ. A discrete subpopulation of human monocytes expresses a neutrophil-like pro-inflammatory (P) phenotype. Am J Physiol Lung Cell Mol Physiol 1994;267:L775–L785. [DOI] [PubMed] [Google Scholar]
- 6.Shapiro SD, Campbell EJ, Senior RM, Welgus HG. Proteinases secreted by human mononuclear phagocytes. J Rheumatol 1991;27:95–98. [PubMed] [Google Scholar]
- 7.Rajavashisth TB, Xu XP, Jovinge S, Meisel S, Xu XO, Chai NN, Fishbein MC, Kaul S, Cercek B, Sharifi B, et al. Membrane type 1 matrix metalloproteinase expression in human atherosclerotic plaques: evidence for activation by proinflammatory mediators. Circulation 1999;99:3103–3109. [DOI] [PubMed] [Google Scholar]
- 8.Shapiro SD, Fliszar C, Broeckelman T, Mecham RP, Senior RM, Welgus HG. Activation of the 92 kD gelatinase by stromelysin and APMA: differential processing and stabilization of the C-terminal domain by TIMP. J Biol Chem 1995;270:6351–6356. [DOI] [PubMed] [Google Scholar]
- 9.Capodici C, Berg RA. Oxidant activation of neutrophil collagenase by cathepsin G. Fed Proc 1987;46:1963. [DOI] [PubMed] [Google Scholar]
- 10.Okamoto T, Akaike T, Nagano T, Miyajima S, Suga M, Ando M, Ichimori K, Maeda H. Activation of human neutrophil procollagenase by nitrogen dioxide and peroxynitrite: a novel mechanism for procollagenase activation involving nitric oxide. Arch Biochem Biophys 1997;342:261–274. [DOI] [PubMed] [Google Scholar]
- 11.Bassi DE, Mahloogi H, Klein-Szanto AJ. The proprotein convertases furin and PACE4 play a significant role in tumor progression. Mol Carcinog 2000;28:63–69. [PubMed] [Google Scholar]
- 12.Knauper V, Murphy G. Membrane-type metalloproteinases and cell surface-associated activation cascades for matrix metalloproteinases. In: Parks W, Mecham R, editors. Matrix metalloproteinases. San Diego, CA: Academic Press; 1998. pp. 199–217.
- 13.Primakoff P, Myles DG. The ADAM gene family: surface proteins with adhesion and protease activity. Trends Genet 2000;16:83–87. [DOI] [PubMed] [Google Scholar]
- 14.Owen CA, Campbell MA, Sannes PL, Boukedes SS, Campbell EJ. Cell-surface-bound elastase and cathepsin G on human neutrophils: a novel, non-oxidative mechanism by which neutrophils focus and preserve catalytic activity of serine proteinases. J Cell Biol 1995;131:775–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Campbell EJ, Campbell MA, Owen CA. Bioactive proteinase 3 on the cell surface of human neutrophils: quantification, catalytic activity, and susceptibility to inhibition. J Immunol 2000;165:3366–3374. [DOI] [PubMed] [Google Scholar]
- 16.Vassalli J-D, Sappino AP, Belin D. The plasminogen activator/plasmin system. J Clin Invest 1991;88:1067–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Owen CA, Hu Z, Barrick B, Shapiro SD. Inducible expression of tissue inhibitor of metalloproteinase-resistant matrix metalloproteinase-9 on the cell surface of neutrophils. Am J Respir Cell Mol Biol 2003;29:283–294. [DOI] [PubMed] [Google Scholar]
- 18.Owen CA, Hu Z, Lopez-Otin C, Shapiro SD. Membrane-bound matrix metalloproteinase-8 on activated polymorphonuclear cells is a potent, tissue inhibitor of metalloproteinase-resistant collagenase and serpinase. J Immunol 2004;172:7791–7803. [DOI] [PubMed] [Google Scholar]
- 19.Basbaum C, Li D, Gensch E, Gallup M, Lemjabbar H. Mechanisms by which gram-positive bacteria and tobacco smoke stimulate mucin induction through the epidermal growth factor receptor (EGFR). Novartis Found Symp 2002;248:171–176. [PubMed] [Google Scholar]
- 20.Lemjabbar H, Li D, Gallup M, Sidhu S, Drori E, Basbaum C. Tobacco smoke-induced lung cell proliferation mediated by tumor necrosis factor α-converting enzyme and amphiregulin. J Biol Chem 2003;278:26202–26207. [DOI] [PubMed] [Google Scholar]
- 21.Takeyama K, Agusti C, Ueki I, Lausier J, Cardell LO, Nadel JA. Neutrophil-dependent goblet cell degranulation: role of membrane-bound elastase and adhesion molecules. Am J Physiol 1998;275:L294–L302. [DOI] [PubMed] [Google Scholar]
- 22.Belaaouaj A, Kim KS, Shapiro SD. Degradation of outer membrane protein A in Escherichia coli killing by neutrophil elastase. Science 2000;289:1185–1188. [DOI] [PubMed] [Google Scholar]
- 23.Hartzell W, Shapiro SD. Macrophage elastase prevents Gemella morbillorum infection and improves outcome following murine bone marrow transplantation. Chest 1999;116:31S–32S. [DOI] [PubMed] [Google Scholar]
- 24.Wilson CL, Ouellette AJ, Satchell DP, Ayabe T, Lopez-Boado YS, Stratman JL, Hultgren SJ, Matrisian LM, Parks WC. Regulation of intestinal α-defensin activation by the metalloproteinase matrilysin in innate host defense. Science 1999;286:113–117. [DOI] [PubMed] [Google Scholar]
- 25.Balbin M, Fueyo A, Tester AM, Pendas AM, Pitiot AS, Astudillo A, Overall CM, Shapiro SD, Lopez-Otin C. Loss of collagenase-2 confers increased skin tumor susceptibility to male mice. Nat Genet 2003;35:252–257. [DOI] [PubMed] [Google Scholar]
- 26.McQuibban GA, Gong JH, Tam EM, McCulloch CA, Clark-Lewis I, Overall CM. Inflammation dampened by gelatinase A cleavage of monocyte chemoattractant protein-3. Science 2000;289:1202–1206. [DOI] [PubMed] [Google Scholar]
- 27.Coeshott C, Ohnemus C, Pilyavskaya A, Ross S, Wieczorek M, Kroona H, Leimer AH, Cheronis J. Converting enzyme-independent release of tumor necrosis factor α and IL-1β from a stimulated human monocytic cell line in the presence of activated neutrophils or purified proteinase 3. Proc Natl Acad Sci USA 1999;96:6261–6266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stamenkovic I. Matrix metalloproteinases in tumor invasion and metastasis. Semin Cancer Biol 2000;10:415–433. [DOI] [PubMed] [Google Scholar]
- 29.Cornelius LA, Nehring LC, Harding E, Bolanowski M, Welgus HG, Kobayashi DK, Pierce RA, Shapiro SD. Matrix metalloproteinases generate angiostatin: effects on neovascularization. J Immunol 1998;161:6845–6852. [PubMed] [Google Scholar]
- 30.MacNee W. Oxidants/antioxidants and chronic obstructive pulmonary disease: pathogenesis to therapy. Novartis Found Symp 2001;234:169–185. [PubMed] [Google Scholar]
- 31.Pechkovsky DV, Zissel G, Stamme C, Goldmann T, Ari JH, Einhaus M, Taube C, Magnussen H, Schlaak M, Muller-Quernheim J. Human alveolar epithelial cells induce nitric oxide synthase-2 expression in alveolar macrophages. Eur Respir J 2002;19:672–683. [DOI] [PubMed] [Google Scholar]
- 32.Repine JE, Bast A, Lankhorst I; Oxidative Stress Study Group. Oxidative stress in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1997;156:341–357. [DOI] [PubMed] [Google Scholar]
- 33.Babior BM. Oxygen-dependent microbial killing by phagocytes. N Engl J Med 1978;298:659–668. [DOI] [PubMed] [Google Scholar]
- 34.Babior BM. NADPH oxidase: an update. Blood 1999;93:1464–1476. [PubMed] [Google Scholar]
- 35.Reeves EP, Lu H, Jacobs HL, Messina CG, Bolsover S, Gabella G, Potma EO, Warley A, Roes J, Segal AW. Killing activity of neutrophils is mediated through activation of proteases by K+ flux. Nature 2002;416:291–297. [DOI] [PubMed] [Google Scholar]
- 36.Rahman I. Oxidative stress, transcription factors and chromatin remodelling in lung inflammation. Biochem Pharmacol 2002;64:935–942. [DOI] [PubMed] [Google Scholar]
- 37.Thannickal VJ, Fanburg BL. Reactive oxygen species in cell signaling. Am J Physiol Lung Cell Mol Physiol 2000;279:L1005–L1028. [DOI] [PubMed] [Google Scholar]
- 38.Fu X, Kassim SY, Parks WC, Heinecke JW. Hypochlorous acid generated by myeloperoxidase modifies adjacent tryptophan and glycine residues in the catalytic domain of matrix metalloproteinase-7 (matrilysin): an oxidative mechanism for restraining proteolytic activity during inflammation. J Biol Chem 2003;278:28403–28409. [DOI] [PubMed] [Google Scholar]
- 39.Van Den Steen PE, Proost P, Wuyts A, Van Damme J, Opdenakker G. Neutrophil gelatinase B potentiates interleukin-8 tenfold by aminoterminal processing, whereas it degrades CTAP-III, PF-4, and GRO-α and leaves RANTES and MCP-2 intact. Blood 2000;96:2673–2681. [PubMed] [Google Scholar]
- 40.Li Q, Park PW, Wilson CL, Parks WC. Matrilysin shedding of syndecan-1 regulates chemokine mobilization and transepithelial efflux of neutrophils in acute lung injury. Cell 2002;111:635–646. [DOI] [PubMed] [Google Scholar]
- 41.Churg A, Wang RD, Tai H, Wang X, Xie C, Dai J, Shapiro SD, Wright JL. Macrophage metalloelastase mediates acute cigarette smoke-induced inflammation via tumor necrosis factor-α release. Am J Respir Crit Care Med 2003;167:1083–1089. [DOI] [PubMed] [Google Scholar]
- 42.Hubbard RC, Fells G, Gadek J, Pacholok S, Humes J, Crystal RG. Neutrophil accumulation in the lung in α1-antitrypsin deficiency: spontaneous release of leukotriene B4 by alveolar macrophages. J Clin Invest 1991;88:891–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bedard M, McClure CD, Schiller NL, Francoeur C, Cantin A, Denis M. Release of interleukin-8, interleukin-6, and colony-stimulating factors by upper airway epithelial cells: implications for cystic fibrosis. Am J Respir Cell Mol Biol 1993;9:455–462. [DOI] [PubMed] [Google Scholar]
- 44.Senior RM, Griffin GL, Mecham RP. Chemotactic activity of elastin-derived peptides. J Clin Invest 1980;66:859–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hunninghake GW, Davidson JM, Rennard S, Szapiel S, Gadek JE, Crystal RG. Elastin fragments attract macrophage precursors to diseased sites in pulmonary emphysema. Science 1981;212:925–927. [DOI] [PubMed] [Google Scholar]
- 46.Banda MJ, Rice AG, Griffin GL, Senior RM. α1-Proteinase inhibitor is a neutrophil chemoattractant after proteolytic inactivation by macrophage elastase. J Biol Chem 1988;263:4481–4484. [PubMed] [Google Scholar]
- 47.Stockley RA, Shaw J, Afford SC, Morrison HM, Burnett D. Effect of α-1-proteinase inhibitor on neutrophil chemotaxis. Am J Respir Cell Mol Biol 1990;2:163–170. [DOI] [PubMed] [Google Scholar]
- 48.Retamales I, Elliott WM, Meshi B, Coxson HO, Pare PD, Sciurba FC, Rogers RM, Hayashi S, Hogg JC. Ampification of inflammation in emphysema and its association with latent adenoviral infection. Am J Respir Crit Care Med 2001;164:469–473. [DOI] [PubMed] [Google Scholar]
- 49.Shao MX, Nakanaga T, Nadel JA. Cigarette smoke induces MUC5AC mucin overproduction via tumor necrosis factor-α-converting enzyme in human airway epithelial (NCI-H292) cells. Am J Physiol Lung Cell Mol Physiol 2004;287:L420–L427. [DOI] [PubMed] [Google Scholar]
- 50.Kohri K, Ueki IF, Nadel JA. Neutrophil elastase induces mucin production by ligand-dependent epidermal growth factor receptor activation. Am J Physiol Lung Cell Mol Physiol 2002;283:L531–L540. [DOI] [PubMed] [Google Scholar]
- 51.Fischer BM, Voynow JA. Neutrophil elastase induces MUC5AC gene expression in airway epithelium via a pathway involving reactive oxygen species. Am J Respir Cell Mol Biol 2002;26:447–452. [DOI] [PubMed] [Google Scholar]
- 52.Casalino-Matsuda SM, Monzon ME, Conner GE, Salathe M, Forteza RM. Role of hyaluronan and reactive oxygen species in tissue kallikrein-mediated epidermal growth factor receptor activation in human airways. J Biol Chem 2004;279:21606–21616. [DOI] [PubMed] [Google Scholar]
- 53.Lemjabbar H, Basbaum C. Platelet-activating factor receptor and ADAM10 mediate responses to Staphylococcus aureus in epithelial cells. Nat Med 2002;8:41–46. [DOI] [PubMed] [Google Scholar]
- 54.Yu Q, Stamenkovic I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-β and promotes tumor invasion and angiogenesis. Genes Dev 2000;14:163–176. [PMC free article] [PubMed] [Google Scholar]
- 55.Taipale J, Koli K, Keski-Oja J. Release of transforming growth factor-β1 from the pericellular matrix of cultured fibroblasts and fibrosarcoma cells by plasmin and thrombin. J Biol Chem 1992;267:25378–25384. [PubMed] [Google Scholar]
- 56.Mohan S, Thompson GR, Amaar YG, Hathaway G, Tschesche H, Baylink DJ. ADAM-9 is an insulin-like growth factor binding protein-5 protease produced and secreted by human osteoblasts. Biochemistry 2002;41:15394–15403. [DOI] [PubMed] [Google Scholar]
- 57.Fowlkes JL, Serra DM, Nagase H, Thrailkill KM. MMPs are IGFBP-degrading proteinases: implications for cell proliferation and tissue growth. Ann N Y Acad Sci 1999;878:696–699. [DOI] [PubMed] [Google Scholar]
- 58.Reilly JJ, Chapman HA Jr. Association between alveolar macrophage plasminogen activator activity and indices of lung function in young cigarette smokers. Am Rev Respir Dis 1988;138:1422–1428. [DOI] [PubMed] [Google Scholar]
- 59.Finlay GA, O'Driscoll L, Russell KJ, D'Arcy EM, Masterson JBL, Fitzgerald MX, O'Connor CM. Matrix metalloproteinase expression and production by alveolar macrophages in emphysema. Am J Respir Crit Care Med 1997;156:240–247. [DOI] [PubMed] [Google Scholar]
- 60.Russell RE, Culpitt SV, DeMatos C, Donnelly L, Smith M, Wiggins J, Barnes PJ. Release and activity of matrix metalloproteinase-9 and tissue inhibitor of metalloproteinase-1 by alveolar macrophages from patients with chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol 2002;26:602–609. [DOI] [PubMed] [Google Scholar]
- 61.Lim S, Roche N, Oliver BG, Mattos W, Barnes PJ, Chung KF. Balance of matrix metalloprotease-9 and tissue inhibitor of metalloprotease-1 from alveolar macrophages in cigarette smokers: regulation by interleukin-10. Am J Respir Crit Care Med 2000;162:1355–1360. [DOI] [PubMed] [Google Scholar]
- 62.Betsuyaku T, Nishimura M, Takeyabu K, Tanino M, Venge P, Xu S, Kawakami Y. Neutrophil granule proteins in bronchoalveolar lavage fluid from subjects with subclinical emphysema. Am J Respir Crit Care Med 1999;159:1985–1991. [DOI] [PubMed] [Google Scholar]
- 63.Betsuyaku T, Nishimura M, Takeyabu K, Tanino M, Miyamoto K, Kawakami Y. Decline in FEV1 in community-based older volunteers with higher levels of neutrophil elastase in bronchoalveolar lavage fluid. Respiration (Herrlisheim) 2000;67:261–267. [DOI] [PubMed] [Google Scholar]
- 64.Betsuyaku T, Nishimura M, Yoshioka A, Takeyabu K, Miyamoto K, Kawakami Y. [Neutrophil elastase and elastin-derived peptides in BAL fluid and emphysematous changes on CT scans]. Nihon Kyobu Shikkan Gakkai Zasshi 1996;34:69–74. [PubMed]
- 65.Cataldo D, Munaut C, Noel A, Frankenne F, Bartsch P, Foidart JM, Louis R. MMP-2- and MMP-9-linked gelatinolytic activity in the sputum from patients with asthma and chronic obstructive pulmonary disease. Int Arch Allergy Immunol 2000;123:259–267. [DOI] [PubMed] [Google Scholar]
- 66.Hill AT, Bayley D, Stockley RA. The interrelationship of sputum inflammatory markers in patients with chronic bronchitis. Am J Respir Crit Care Med 1999;160:893–898. [DOI] [PubMed] [Google Scholar]
- 67.Beeh KM, Beier J, Kornmann O, Buhl R. Sputum matrix metalloproteinase-9, tissue inhibitor of metalloprotinease-1, and their molar ratio in patients with chronic obstructive pulmonary disease, idiopathic pulmonary fibrosis and healthy subjects. Respir Med 2003;97:634–639. [DOI] [PubMed] [Google Scholar]
- 68.Culpitt SV, Rogers DF, Traves SL, Barnes PJ, Donnelly LE. Sputum matrix metalloproteases: comparison between chronic obstructive pulmonary disease and asthma. Respir Med 2005;99:703–710. [DOI] [PubMed] [Google Scholar]
- 69.Damiano VV, Tsang A, Kucich U, Abrams WR, Rosenbloom J, Kimbel P, Weinbaum G. Immunolocalization of elastase in human emphysematous lungs. J Clin Invest 1986;78:482–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Imai K, Dalal SS, Chen ES, Downey R, Schulman LL, Ginsburg M, D'Armiento J. Human collagenase (matrix metalloproteinase-1) expression in the lungs of patients with emphysema. Am J Respir Crit Care Med 2001;163:786–791. [DOI] [PubMed] [Google Scholar]
- 71.Kang MJ, Oh YM, Lee JC, Kim DG, Park MJ, Lee MG, Hyun IG, Han SK, Shim YS, Jung KS. Lung matrix metalloproteinase-9 correlates with cigarette smoking and obstruction of airflow. J Korean Med Sci 2003;18:821–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hautamaki RD, Kobayashi DK, Senior RM, Shapiro SD. Requirement for macrophage elastase for cigarette smoke-induced emphysema in mice. Science 1997;277:2002–2004. [DOI] [PubMed] [Google Scholar]
- 73.Shapiro SD, Goldstein NM, Houghton AM, Kobayashi DK, Kelley D, Belaaouaj A. Neutrophil elastase contributes to cigarette smoke-induced emphysema in mice. Am J Pathol 2003;163:2329–2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lanone S, Zheng T, Zhu Z, Liu W, Lee CG, Ma B, Chen Q, Homer RJ, Wang J, Rabach LA, et al. Overlapping and enzyme-specific contributions of matrix metalloproteinases-9 and -12 in IL-13-induced inflammation and remodeling. J Clin Invest 2002;110:463–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Okada Y, Watanabe S, Nakanishi I, Kishi J, Hayakawa T, Watorek W, Travis J, Nagase H. Inactivation of tissue inhibitor of metalloproteinases by neutrophil elastase and other serine proteinases. FEBS Lett 1988;229:157–160. [DOI] [PubMed] [Google Scholar]
- 76.Gronski TJ Jr, Martin RL, Kobayashi DK, Walsh BC, Holman MC, Huber M, Van Wart HE, Shapiro SD. Hydrolysis of a broad spectrum of extracellular matrix proteins by human macrophage elastase. J Biol Chem 1997;272:12189–12194. [DOI] [PubMed] [Google Scholar]
- 77.D'Armiento J, Dalal SS, Okada Y, Berg RA, Chada K. Collagenase expression in the lungs of transgenic mice causes pulmonary emphysema. Cell 1992;71:955–961. [DOI] [PubMed] [Google Scholar]
- 78.Saetta M, Baraldo S, Corbino L, Turato G, Braccioni F, Rea F, Cavallesco G, Tropeano G, Mapp CE, Maestrelli P, et al. CD8+ve cells in the lungs of smokers with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1999;160:711–717. [DOI] [PubMed] [Google Scholar]
- 79.Lacraz S, Isler P, Vey E, Welgus HG, Dayer JM. Direct contact between T lymphocytes and monocytes is a major pathway for induction of metalloproteinase expression. J Biol Chem 1994;269:22027–22033. [PubMed] [Google Scholar]
- 80.Wang Z, Zheng T, Zhu Z, Homer RJ, Riese RJ, Chapman HA Jr, Shapiro SD, Elias JA. Interferon γ induction of pulmonary emphysema in the adult murine lung. J Exp Med 2000;192:1587–1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zheng T, Zhu Z, Wang Z, Homer RJ, Ma B, Riese RJ Jr, Chapman HA Jr, Shapiro SD, Elias JA. Inducible targeting of IL-13 to the adult lung causes matrix metalloproteinase- and cathepsin-dependent emphysema. J Clin Invest 2000;106:1081–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Feinberg MW, Jain MK, Werner F, Sibinga NE, Wiesel P, Wang H, Topper JN, Perrella MA, Lee ME. Transforming growth factor-β1 inhibits cytokine-mediated induction of human metalloelastase in macrophages. J Biol Chem 2000;275:25766–25773. [DOI] [PubMed] [Google Scholar]
- 83.Munger JS, Huang X, Kawakatsu H, Griffiths MJ, Dalton SL, Wu J, Pittet JF, Kaminski N, Garat C, Matthay MA, et al. The integrin αvβ6 binds and activates latent TGF-β1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell 1999;96:319–328. [DOI] [PubMed] [Google Scholar]
- 84.Morris DG, Huang X, Kaminski N, Wang Y, Shapiro SD, Dolganov G, Glick A, Sheppard D. Loss of integrin αvβ6-mediated TGF-β activation causes MMP12-dependent emphysema. Nature 2003;422:169–173. [DOI] [PubMed] [Google Scholar]
- 85.Wert SE, Yoshida M, LeVine AM, Ikegami M, Jones T, Ross GF, Fisher JH, Korfhagen TR, Whitsett JA. Increased metalloproteinase activity, oxidant production, and emphysema in surfactant protein D gene-inactivated mice. Proc Natl Acad Sci USA 2000;97:5972–5977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kasahara Y, Tuder RM, Cool CD, Lynch DA, Flores SC, Voelkel NF. Endothelial cell death and decreased expression of vascular endothelial growth factor and vascular endothelial growth factor receptor 2 in emphysema. Am J Respir Crit Care Med 2001;163:737–744. [DOI] [PubMed] [Google Scholar]
- 87.Aoshiba K, Yokohori N, Nagai A. Alveolar wall apoptosis causes lung destruction and emphysematous changes. Am J Respir Cell Mol Biol 2003;28:555–562. [DOI] [PubMed] [Google Scholar]
- 88.Yokohori N, Aoshiba K, Nagai A. Increased levels of cell death and proliferation in alveolar wall cells in patients with pulmonary emphysema. Chest 2004;125:626–632. [DOI] [PubMed] [Google Scholar]
- 89.Segura-Valdez L, Pardo A, Gaxiola M, Uhal BD, Becerril C, Selman M. Upregulation of gelatinases A and B, collagenases 1 and 2, and increased parenchymal cell death in COPD. Chest 2000;117:684–694. [DOI] [PubMed] [Google Scholar]
- 90.Hodge S, Hodge G, Holmes M, Reynolds PN. Increased airway epithelial and T-cell apoptosis in COPD remains despite smoking cessation. Eur Respir J 2005;25:447–454. [DOI] [PubMed] [Google Scholar]
- 91.Aoshiba K, Tamaoki J, Nagai A. Acute cigarette smoke exposure induces apoptosis of alveolar macrophages. Am J Physiol Lung Cell Mol Physiol 2001;281:L1392–L1401. [DOI] [PubMed] [Google Scholar]
- 92.Noguera A, Sala E, Pons AR, Iglesias J, MacNee W, Agusti AG. Expression of adhesion molecules during apoptosis of circulating neutrophils in COPD. Chest 2004;125:1837–1842. [DOI] [PubMed] [Google Scholar]
- 93.Kasahara Y, Tuder RM, Taraseviciene-Stewart L, Le Cras TD, Abman S, Hirth PK, Waltenberger J, Voelkel NF. Inhibition of VEGF receptors causes lung cell apoptosis and emphysema. J Clin Invest 2000;106:1311–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Vandivier RW, Fadok VA, Hoffmann PR, Bratton DL, Penvari C, Brown KK, Brain JD, Accurso FJ, Henson PM. Elastase-mediated phosphatidylserine receptor cleavage impairs apoptotic cell clearance in cystic fibrosis and bronchiectasis. J Clin Invest 2002;109:661–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Pryor WA, Stone K. Oxidants in cigarette smoke: radicals, hydrogen peroxide, peroxynitrate, and peroxynitrite. Ann N Y Acad Sci 1993;686:12–27. [DOI] [PubMed] [Google Scholar]
- 96.MacNee W. Oxidants/antioxidants and COPD. Chest 2000;117:303S–317S. [DOI] [PubMed] [Google Scholar]
- 97.Gutteridge JM. Lipid peroxidation and antioxidants as biomarkers of tissue damage. Clin Chem 1995;41:1819–1828. [PubMed] [Google Scholar]
- 98.Pratico D, Basili S, Vieri M, Cordova C, Violi F, FitzGerald GA. Chronic obstructive pulmonary disease is associated with an increase in urinary levels of isoprostane F2α-III, an index of oxidant stress. Am J Respir Crit Care Med 1998;158:1709–1714. [DOI] [PubMed] [Google Scholar]
- 99.Morrow JD, Frei B, Longmire AW, Gaziano JM, Lynch SM, Shyr Y, Strauss WE, Oates JA, Roberts LJ. Increase in circulating products of lipid peroxidation (F2-isoprostanes) in smokers: smoking as a cause of oxidative damage. N Engl J Med 1995;332:1198–1203. [DOI] [PubMed] [Google Scholar]
- 100.Rahman I, Skwarska E, MacNee W. Attenuation of oxidant/antioxidant imbalance during treatment of exacerbations of chronic obstructive pulmonary disease. Thorax 1997;52:565–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Morrison D, Rahman I, Lannan S, MacNee W. Epithelial permeability, inflammation, and oxidant stress in the air spaces of smokers. Am J Respir Crit Care Med 1999;159:473–479. [DOI] [PubMed] [Google Scholar]
- 102.Nowak D, Antczak A, Krol M, Pietras T, Shariati B, Bialasiewicz P, Jeczkowski K, Kula P. Increased content of hydrogen peroxide in the expired breath of cigarette smokers. Eur Respir J 1996;9:652–657. [DOI] [PubMed] [Google Scholar]
- 103.Ludwig PW, Hoidal JR. Alterations in leukocyte oxidative metabolism in cigarette smokers. Am Rev Respir Dis 1982;126:977–980. [DOI] [PubMed] [Google Scholar]
- 104.Hoidal JR, Fox RB, LeMarbe PA, Perri R, Repine JE. Altered oxidative metabolic responses in vitro of alveolar macrophages from asymptomatic cigarette smokers. Am Rev Respir Dis 1981;123:85–89. [DOI] [PubMed] [Google Scholar]
- 105.Hoidal JR, Fox RB, LeMarbre PA, Takiff HE, Repine JE. Oxidative metabolism of alveolar macrophages from young asymptomatic cigarette smokers: increased superoxide anion release and its potential consequences. Chest 1980;77:270–271. [DOI] [PubMed] [Google Scholar]
- 106.Schaberg T, Haller H, Rau M, Kaiser D, Fassbender M, Lode H. Superoxide anion release induced by platelet-activating factor is increased in human alveolar macrophages from smokers. Eur Respir J 1992;5:387–393. [PubMed] [Google Scholar]
- 107.Wesselius LJ, Nelson ME, Skikne BS. Increased release of ferritin and iron by iron-loaded alveolar macrophages in cigarette smokers. Am J Respir Crit Care Med 1994;150:690–695. [DOI] [PubMed] [Google Scholar]
- 108.Duthie GG, Arthur JR, Beattie JA, Brown KM, Morrice PC, Robertson JD, Shortt CT, Walker KA, James WP. Cigarette smoking, antioxidants, lipid peroxidation, and coronary heart disease. Ann N Y Acad Sci 1993;686:120–129. [DOI] [PubMed] [Google Scholar]
- 109.Chow CK, Thacker RR, Changchit C, Bridges RB, Rehm SR, Humble J, Turbek J. Lower levels of vitamin C and carotenes in plasma of cigarette smokers. J Am Coll Nutr 1986;5:305–312. [DOI] [PubMed] [Google Scholar]
- 110.Rahman I, Morrison D, Donaldson K, MacNee W. Systemic oxidative stress in asthma, COPD, and smokers. Am J Respir Crit Care Med 1996;154:1055–1060. [DOI] [PubMed] [Google Scholar]
- 111.Linden M, Hakansson L, Ohlsson K, Sjodin K, Tegner H, Tunek A, Venge P. Glutathione in bronchoalveolar lavage fluid from smokers is related to humoral markers of inflammatory cell activity. Inflammation 1989;13:651–658. [DOI] [PubMed] [Google Scholar]
- 112.Rahman I, Li XY, Donaldson K, Harrison DJ, MacNee W. Glutathione homeostasis in alveolar epithelial cells in vitro and lung in vivo under oxidative stress. Am J Physiol 1995;269:L285–L292. [DOI] [PubMed] [Google Scholar]
- 113.Rahman I, Antonicelli F, MacNee W. Molecular mechanism of the regulation of glutathione synthesis by tumor necrosis factor-α and dexamethasone in human alveolar epithelial cells. J Biol Chem 1999;274:5088–5096. [DOI] [PubMed] [Google Scholar]
- 114.Rahman I, Smith CA, Antonicelli F, MacNee W. Characterisation of γ-glutamylcysteine synthetase-heavy subunit promoter: a critical role for AP-1. FEBS Lett 1998;427:129–133. [DOI] [PubMed] [Google Scholar]
- 115.Li XY, Donaldson K, Rahman I, MacNee W. An investigation of the role of glutathione in increased epithelial permeability induced by cigarette smoke in vivo and in vitro. Am J Respir Crit Care Med 1994;149:1518–1525. [DOI] [PubMed] [Google Scholar]
- 116.Lannan S, Donaldson K, Brown D, MacNee W. Effect of cigarette smoke and its condensates on alveolar epithelial cell injury in vitro. Am J Physiol 1994;266:L92–100. [DOI] [PubMed] [Google Scholar]
- 117.Jones JG, Minty BD, Lawler P, Hulands G, Crawley JC, Veall N. Increased alveolar epithelial permeability in cigarette smokers. Lancet 1980;1:66–68. [DOI] [PubMed] [Google Scholar]
- 118.Tuder RM, Zhen L, Cho CY, Taraseviciene-Stewart L, Kasahara Y, Salvemini D, Voelkel NF, Flores SC. Oxidative stress and apoptosis interact and cause emphysema due to vascular endothelial growth factor receptor blockade. Am J Respir Cell Mol Biol 2003;29:88–97. [DOI] [PubMed] [Google Scholar]
- 119.MacNee W, Wiggs B, Belzberg AS, Hogg JC. The effect of cigarette smoking on neutrophil kinetics in human lungs. N Engl J Med 1989;321:924–928. [DOI] [PubMed] [Google Scholar]
- 120.Drost EM, Selby C, Lannan S, Lowe GD, MacNee W. Changes in neutrophil deformability following in vitro smoke exposure: mechanism and protection. Am J Respir Cell Mol Biol 1992;6:287–295. [DOI] [PubMed] [Google Scholar]
- 121.Klut ME, Doerschuk CM, van Eeden SF, Burns AR, Hogg JC. Activation of neutrophils within pulmonary microvessels of rabbits exposed to cigarette smoke. Am J Respir Cell Mol Biol 1993;9:82–89. [DOI] [PubMed] [Google Scholar]
- 122.Keatings VM, Collins PD, Scott DM, Barnes PJ. Differences in interleukin-8 and tumor necrosis factor-α in induced sputum from patients with chronic obstructive pulmonary disease or asthma. Am J Respir Crit Care Med 1996;153:530–534. [DOI] [PubMed] [Google Scholar]
- 123.Drost EM, Skwarski KM, Sauleda J, Soler N, Roca J, Agusti A, MacNee W. Oxidative stress and airway inflammation in severe exacerbations of COPD. Thorax 2005;60:293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Rahman I, Gilmour PS, Jimenez LA, MacNee W. Oxidative stress and TNF-α induce histone acetylation and NF-κB/AP-1 activation in alveolar epithelial cells: potential mechanism in gene transcription in lung inflammation. Mol Cell Biochem 2002;234–235:239–248. [PubMed] [Google Scholar]
- 125.Rahman I, MacNee W. Role of transcription factors in inflammatory lung diseases. Thorax 1998;53:601–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Antonicelli F, Parmentier M, Drost EM, Hirani N, Rahman I, Donaldson K, MacNee W. Nacystelyn inhibits oxidant-mediated interleukin-8 expression and NF-κB nuclear binding in alveolar epithelial cells. Free Radic Biol Med 2002;32:492–502. [DOI] [PubMed] [Google Scholar]
- 127.Wright DT, Fischer BM, Li C, Rochelle LG, Akley NJ, Adler KB. Oxidant stress stimulates mucin secretion and PLC in airway epithelium via a nitric oxide-dependent mechanism. Am J Physiol 1996;271:L854–L861. [DOI] [PubMed] [Google Scholar]
- 128.Takeyama K, Dabbagh K, Jeong SJ, Dao-Pick T, Ueki IF, Nadel JA. Oxidative stress causes mucin synthesis via transactivation of epidermal growth factor receptor: role of neutrophils. J Immunol 2000;164:1546–1552. [DOI] [PubMed] [Google Scholar]
- 129.Adler KB, Holden-Stauffer WJ, Repine JE. Oxygen metabolites stimulate release of high-molecular-weight glycoconjugates by cell and organ cultures of rodent respiratory epithelium via an arachidonic acid-dependent mechanism. J Clin Invest 1990;85:75–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Hubbard RC, Ogushi F, Fells GA, Cantin AM, Jallat S, Courtney M, Crystal RG. Oxidants spontaneously released by alveolar macrophages of cigarette smokers can inactivate the active site of α1-antitrypsin, rendering it ineffective as an inhibitor of neutrophil elastase. J Clin Invest 1987;80:1287–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kramps JA, Van Twisk C, Klasen EC, Dijkman JH. Interactions among stimulated human polymorphonuclear leucocytes, released elastase and bronchial antileucoprotease. Clin Sci (Lond) 1988;75:53–62. [DOI] [PubMed] [Google Scholar]
- 132.Owen CA, Campbell EJ. Extracellular proteolysis: new paradigms for an old paradox. J Lab Clin Med 1999;134:341–351. [DOI] [PubMed] [Google Scholar]
- 133.Massaro GD, Massaro D. Retinoic acid treatment abrogates elastase-induced pulmonary emphysema in rats. Nat Med 1997;3:675–677. [DOI] [PubMed] [Google Scholar]
- 134.Nakamura Y, Romberger DJ, Tate L, Ertl RF, Kawamoto M, Adachi Y, Mio T, Sisson JH, Spurzem JR, Rennard SI. Cigarette smoke inhibits lung fibroblast proliferation and chemotaxis. Am J Respir Crit Care Med 1995;151:1497–1503. [DOI] [PubMed] [Google Scholar]
- 135.Cantral DE, Sisson JH, Veys T, Rennard SI, Spurzem JR. Effects of cigarette smoke extract on bovine bronchial epithelial cell attachment and migration. Am J Physiol 1995;268:L723–L728. [DOI] [PubMed] [Google Scholar]
- 136.Wang H, Liu X, Umino T, Skold CM, Zhu Y, Kohyama T, Spurzem JR, Romberger DJ, Rennard SI. Cigarette smoke inhibits human bronchial epithelial cell repair processes. Am J Respir Cell Mol Biol 2001;25:772–779. [DOI] [PubMed] [Google Scholar]
- 137.Laurent P, Janoff A, Kagan HM. Cigarette smoke blocks cross-linking of elastin in vitro. Am Rev Respir Dis 1983;127:189–192. [DOI] [PubMed] [Google Scholar]
- 138.Betsuyaku T, Fukuda Y, Parks WC, Shipley JM, Senior RM. Gelatinase B is required for alveolar bronchiolization after intratracheal bleomycin. Am J Pathol 2000;157:525–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Juvelekian GS, Stoller JK. Augmentation therapy for α1-antitrypsin deficiency. Drugs 2004;64:1743–1756. [DOI] [PubMed] [Google Scholar]
- 140.Dhami R, Gilks B, Xie C, Zay K, Wright JL, Churg A. Acute cigarette smoke-induced connective tissue breakdown is mediated by neutrophils and prevented by α1-antitrypsin. Am J Respir Cell Mol Biol 2000;22:244–252. [DOI] [PubMed] [Google Scholar]
- 141.Wright JL, Farmer SG, Churg A. Synthetic serine elastase inhibitor reduces cigarette smoke-induced emphysema in guinea pigs. Am J Respir Crit Care Med 2002;166:954–960. [DOI] [PubMed] [Google Scholar]
- 142.Churg A, Zay K, Shay S, Xie C, Shapiro SD, Hendricks R, Wright JL. Acute cigarette smoke-induced connective tissue breakdown requires both neutrophils and macrophage metalloelastase in mice. Am J Respir Cell Mol Biol 2002;27:368–374. [DOI] [PubMed] [Google Scholar]
- 143.Martin RL, Shapiro SD, Tong SE, van Wart H. Macrophage metalloelastase inhibitors. In: Hansel TBP, editor. Progress in respiratory research., vol. 31: New drugs for asthma, allergy and COPD. Basel, Switzerland: Karger; 2001. pp. 177–180.
- 144.Rahman I, MacNee W. Role of oxidants/antioxidants in smoking-induced lung diseases. Free Radic Biol Med 1996;21:669–681. [DOI] [PubMed] [Google Scholar]
- 145.Daga MK, Chhabra R, Sharma B, Mishra TK. Effects of exogenous vitamin E supplementation on the levels of oxidants and antioxidants in chronic obstructive pulmonary disease. J Biosci 2003;28:7–11. [DOI] [PubMed] [Google Scholar]
- 146.MacNee W, Bridgeman MM, Marsden M, Drost E, Lannan S, Selby C, Donaldson K. The effects of N-acetylcysteine and glutathione on smoke-induced changes in lung phagocytes and epithelial cells. Am J Med 1991;91:60S–66S. [DOI] [PubMed] [Google Scholar]
- 147.Dekhuijzen PN. Antioxidant properties of N-acetylcysteine: their relevance in relation to chronic obstructive pulmonary disease. Eur Respir J 2004;23:629–636. [DOI] [PubMed] [Google Scholar]
- 148.Bridgeman MM, Marsden M, Selby C, Morrison D, MacNee W. Effect of N-acetyl cysteine on the concentrations of thiols in plasma, bronchoalveolar lavage fluid, and lung tissue. Thorax 1994;49:670–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Gillissen A, Jaworska M, Orth M, Coffiner M, Maes P, App EM, Cantin AM, Schultze-Werninghaus G. Nacystelyn, a novel lysine salt of N-acetylcysteine, to augment cellular antioxidant defence in vitro. Respir Med 1997;91:159–168. [DOI] [PubMed] [Google Scholar]
- 150.Linden M, Wieslander E, Eklund A, Larsson K, Brattsand R. Effects of oral N-acetylcysteine on cell content and macrophage function in bronchoalveolar lavage from healthy smokers. Eur Respir J 1988;1:645–650. [PubMed] [Google Scholar]
- 151.Jankowska R, Passowicz-Muszynska E, Medrala W, Banas T, Marcinkowska A. [The influence of N-acetylcysteine on chemiluminescence of granulocytes in peripheral blood of patients with chronic bronchitis].Pneumonol Alergol Pol 1993;61:586–591. [PubMed]
- 152.Gerrits CM, Herings RM, Leufkens HG, Lammers JW. N-Acetylcysteine reduces the risk of re-hospitalisation among patients with chronic obstructive pulmonary disease. Eur Respir J 2003;21:795–798. [DOI] [PubMed] [Google Scholar]
- 153.Lundback B, Linstrom M, Andersson S, Nystrom L, Rosenhall L, Stjernberg N. Possible effect of acetylcysteine on lung function. Eur Respir J 1995;5:895–985. [Google Scholar]
- 154.British Thoracic Society Research Committee. Oral N-acetylcysteine and exacerbation rates in patients with chronic bronchitis and severe airways obstruction. Thorax 1985;40:823–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Boman G, Backer U, Larsson S, Melander B, Wahlander L. Oral acetylcysteine reduces exacerbation rate in chronic bronchitis: report of a trial organized by the Swedish Society for Pulmonary Diseases. Eur J Respir Dis 1983;64:405–415. [PubMed] [Google Scholar]
- 156.Aylward M, Maddock J, Dewland P. Clinical evaluation of acetylcysteine in the treatment of patients with chronic obstructive bronchitis: a balanced double-blind trial with placebo control. Eur J Respir Dis Suppl 1980;111:81–89. [PubMed] [Google Scholar]
- 157.Tsan MF, Phillips PG. l-2-Oxothiazolidine-4-carboxylate protects cultured endothelial cells against hyperoxia-induced injury. Inflammation 1988;12:113–121. [DOI] [PubMed] [Google Scholar]
- 158.Giembycz MA. Phosphodiesterase 4: selective and dual specificity inhibitors for the therapy of COPD. Am J Respir Crit Care Med (In press) [DOI] [PubMed]