

Abstract
Hydroxyl radicals generated from a variety of methods, including not only synchrotron radiation but also Fenton reactions involving chelated iron, have become an accepted macromolecular footprinting tool. Hydroxyl radicals react with proteins via multiple mechanisms that lead to both polypeptide backbone cleavage events and side chain modifications (e.g., hydroxylation and carbonyl formation). The use of site-specifically tethered iron chelates can reveal protein-protein interactions, but the interpretation of such experiments will be strengthened by better understanding of how hydroxyl radicals produced at a point on a protein react with other protein sites. We have developed methods to monitor carbonyl formation on proteins as a function of distance from a hydroxyl generator, iron-(S)-1-[p-(bromoacetamido)benzyl]EDTA, “FeBABE”, conjugated to an engineered cysteine residue. After activation of the chelated iron with ascorbate and peroxide produces new protein carbonyl groups, their positions can be identified using element-coded affinity tagging (ECAT), with carbonyl-specific tags (e.g., rare earth chelates of ((S)-2-(4-(2-aminooxy)acetamidobenzyl)-1, 4, 7, 10-tetraazacyclododecane-N, N′, N″, N‴-tetraacetic acid, “AOD”) that allow for affinity purification, identification, and relative quantitation of oxidation sites using mass spectrometry. Intraprotein oxidation of single-cysteine mutants of E. coli σ70 by tethered FeBABE was used to calibrate the reach of hydroxyl radical by comparison to the crystal structure; the application to protein-protein interactions was demonstrated using the same σ70 FeBABE conjugates in complexes with RNA polymerase core enzyme. The results provide fundamental information for interpreting protein footprinting experiments in other systems.
The use of hydroxyl radicals as probes of protein structure has been developed to a high level by Chance and co-workers (e.g., 1, reviewed in 2), who irradiate protein solutions with x-ray pulses and identify oxidized residues by mass spectrometry. The recent work of Hahn and co-workers (3, 4) is an alternative use of chemistry in this context. They use the tethered FeBABE reagent (Scheme 1 (5)) as a localized source to cleave peptide backbones in transcription complexes, and analyze the polypeptide fragments by gel electrophoresis and western blots to identify protein-protein interactions, analogous to the methodology of Meares and co-workers (6). The promise of tethered FeBABE chemistry is that it can serve a purpose similar to chemical cross-linking, but in a more generalized way, providing proximity information about sites on proteins, nucleic acids, or other organic molecules in macromolecular complexes. Unlike cross-linking, a nucleophilic or electron-rich side chain on the target molecule is not required. In a typical experiment, a cysteine residue on a protein is modified with FeBABE to form a macromolecular probe. This probe is allowed to interact with binding partners to form stable complexes, and Fe redox chemistry is stimulated by the addition of ascorbate and peroxide. A variety of products are formed on neighboring moieties in the complex, including not only breaks in the polypeptide backbone but also oxidatively modified side chains.
Scheme 1.
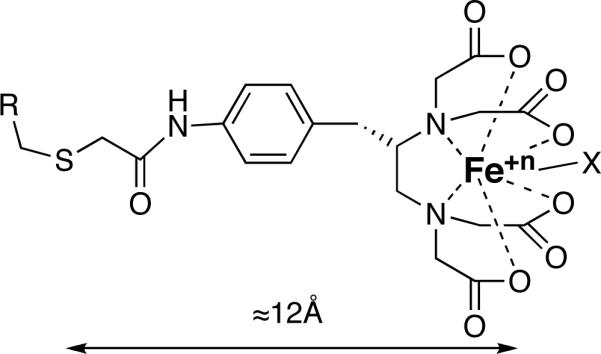
FeBABE attached to a Cys side chain on a protein R. X is an activated oxygen ligand, which may produce a hydroxyl radical or other reactive species, even including a nucleophile (5). Rotation relative to the polypeptide backbone is possible around several single bonds. When the FeBABE is extended, the distance from the Cys sulfur to Fe is approximately 12Å.
The results of FeBABE chemistry are frequently attributed to oxidation by hydroxyl radical, though alternatives involving iron-oxygen complexes exist (7). The interpretation of experiments using tethered iron complexes to study protein-protein interactions is affected by this, because while the direct attack of iron-oxygen complexes on proteins must occur adjacent to the site of attachment (6), the diffusible nature of the hydroxyl radical can greatly extend the reaction range. We have found a way to measure the reach of hydroxyl radicals generated by the FeBABE reagent under commonly used conditions, and we propose a framework to rationalize the results.
As reviewed by Chance (2), many factors influence the reaction of hydroxyl radical with an amino-acid residue, including the residue's intrinsic chemical reactivity and its solvent accessibility. Thus if the molecular information available from a point source of hydroxyl radicals is to be utilized in more than a yes-or-no way, it must be validated by comparison to known structures. Here we provide two examples, which show the essential features.
Because they are well-known products of hydroxyl radicals (8), we studied the formation of aldehyde and ketone groups on the protein subunits of E. coli RNA polymerase by the FeBABE reagent tethered to engineered Cys residues (9) on the σ70 protein. Self-oxidation of single-cysteine mutants of E. coli σ70 by tethered FeBABE was used to initially calibrate the method by comparison to the crystal structure of a large fragment of the protein. This allowed us to examine the hydroxyl-radical oxidation pattern that occurs when FeBABE is positioned at different sites on the surface of a single protein. Finally, we applied the method to complex protein-protein interactions by using FeBABE σ70 conjugates as probes in complexes with E. coli RNA polymerase core enzyme.
EXPERIMENTAL PROCEDURES
Materials
Harvested and frozen E. coli MRE600 log phase cells were purchased from Woody Robbins Fermentation Facility, U. Alabama at Birmingham. Rare earth hexahydrate salts and bovine serum albumin (minimum 98% electrophoresis) were purchased from Sigma Aldrich (St. Louis, MO). The anti-DOTA-chelate antibody 2D12.5 was a generous gift from Dr. David Goodwin. Modified sequencing grade trypsin was purchased from Promega (Madison, WI). Sep-Pak Plus C-18 cartridges were purchased from Waters (Milford, MA). Gentle Ag/Ab elution buffer, Aminolink Plus resin, and biotin-LC-hydrazide were purchased from Pierce Biotechnology (Rockford, IL). The elution buffer was purified prior to use with a Sep-Pak Plus C-18 cartridge to remove any dissolved hydrophobic polymers resulting from the manufacturer's packaging and prolonged storage (50 mL of buffer per cartridge). PD-10 desalting columns were purchased from GE Healthcare (Fairfield, CT). Micro Bio-spin columns were purchased from Bio-Rad Laboratories (Hercules, CA). CPM, (N-(4-(7-diethylamino-4-methylcoumarin-3-yl)phenylmaleimide) was purchased from Molecular Probes (Eugene, OR). (S)-2-(4-aminobenzyl)-DOTA•(HCl)4 was purchased from Macrocyclics (Dallas, TX). (S)-2-(4-(2-bromo-acetamido)-benzyl)-EDTA was purchased from Toronto Research Chemicals. (S)-2-(4-(2-aminooxy)acetamidobenzyl)-1, 4, 7, 10-tetraazacyclododecane-N, N′, N″, N‴-tetraacetic acid (AOD) was synthesized and metallated with various lanthanide salts as described previously (21). Pure water (18 MΩ cm) was used throughout.
Protein Preparation
Single-Cys σ70 mutants were overexpressed and purified as described previously (9). Briefly, pGEMD132C, pGEMD376C, pGEMD396C, pGEMD422C, pGEMD517C and pGEMD581C are overexpression plasmids of mutant σ70 subunits, which contain a single cysteine residue at position 132, 376, 396, 422, 517, or 581, respectively. Each plasmid was transformed into chemically competent E. coli BL21(DE3) cells. Transformed cells were grown in LB medium containing 200 μg/mL carbenicillin at 37 °C until OD600 = 0.4. Expression was induced by adding isopropyl thiogalactoside (IPTG, 0.5 mM final), and cells were allowed to grow at 37 °C for an additional 3 h before harvesting. Induction and expression was confirmed using SYPRO Ruby stained SDS-PAGE.
RNAP core was purified from MRE600 cells using established protocols (10, 11).
FeBABE conjugation to single-Cys σ70 mutants and BSA
Conjugation of BABE to each single-Cys σ70 mutant was carried out by adding 20mM BABE in DMSO (final 1mM) to the protein (final ∼20 μM) in 0.1 M sodium phosphate, pH 8.0/1mM EDTA, followed by incubation for 3 h at 37 °C. A control aliquot was treated with DMSO in the same manner. FeBABE conjugation yields were determined to be 132C, 25%; 376C, 26%; 396C, 20%; 422C, 50%; 517C, 41%; 581C, 40%. Lysine-conjugated σ70-2IT-FeBABE was prepared by sequentially adding BABE in DMSO and freshly prepared 2IT in water to σ70 517C (final BABE concentration 1.4 mM, final 2IT concentration 0.7 mM, final protein concentration ≈20 μM), followed by incubation for 1 h at 37 °C. Identical reaction conditions were used to prepare BSA-2IT-FeBABE as a control. The reactions were quenched by adding an excess of 2-mercaptoethanol (to 10 mM), and unreacted reagent was removed via dialysis into 0.1 M sodium phosphate, pH 7.0. BABE metallation with iron was carried out by adding 2.5 mol excess (over total protein) of freshly prepared 10 mM aqueous FeSO4 solution and allowing the solution to stand at room temperature for 25 min. Free iron was also added to the control, unconjugated protein. To scavenge excess metal, 0.5 M EDTA was added to a final concentration of 6 mM and the solution was allowed to stand for an additional 25 min at room temperature before being loaded on a PD-10 column pre-equilibrated in 50 mM Hepes, pH 7.0/10% v/v glycerol/1mM EDTA. The FeBABE conjugation yield was determined using either a fluorometric CPM thiol assay to detect the presence of free Cys sulfhydryl or a quantitative CHA255 ELISA to count the number of FeBABE molecules per molecule of protein (12).
Self-oxidation of FeBABE single-Cys σ70 conjugates
Approximately 10 nmol of FeBABE-conjugated protein in 50 mM Hepes, pH 7.0/10% v/v glycerol/1mM EDTA was used for each experiment. Hydroxyl radical formation was initiated by the rapid, sequential addition of stock solutions of ascorbate and freshly prepared hydrogen peroxide in the same buffer (final ascorbate concentration 10 mM, final hydrogen peroxide concentration 10 mM, final protein concentration 5 μM). FeEDTA in the same buffer was added to the unconjugated control (final concentration 20 mM). In addition, pre-existing and background oxidation due to sample handling or adventitious iron bound to protein was studied by conducting experiments without BABE or FeEDTA added. The oxidation reaction was allowed to proceed for 10 min at 25 °C before the protein was precipitated using ice-cold aqueous trichloroacetic acid (final concentration 5 % (w/v)). The precipitation was allowed to proceed for an additional 20 min on ice before centrifugation at 16,000 × g for 10 min. The supernatant was discarded and the pellet was washed with 0.5 mL of cold absolute ethanol and dried in a speed-vac.
Protein-protein interaction studies with RNAP core and FeBABE single-Cys σ70 mutants
Using gel filtration, RNAP core in storage buffer1 was exchanged into cleavage buffer. FeBABE σ70-RNAP holoenzyme was constituted by mixing the appropriate FeBABE σ70 conjugate with RNAP core in a 1:1 molar ratio (≈5 nmol of each), and incubating at 30 °C for 20 min. FeBABE was activated by the rapid, sequential addition of ascorbate and hydrogen peroxide (final ascorbate concentration 5 mM, final hydrogen peroxide concentration 5 mM, final protein concentration ≈4 μM). Oxidation was allowed to proceed for 15 min at 30 °C prior to quenching with aqueous TCA as described above.
Carbonyl tagging with O-ECAT
FeEDTA-oxidized σ70 and FeBABE-oxidized σ70 were tagged with two different MAOD, e.g. M = Pr or Ho. The MAOD (final 1 mM) in tagging buffer was added directly to the precipitated protein. The mixture was sonicated and vortexed repeatedly for 5 min to fully redissolve the oxidized protein and allowed to stand for 24 h at 25 °C. Unreacted MAOD was removed and the buffer was changed by gel filtration using Micro Bio-spin 6 columns, pre-equilibrated with 50 mM NH4HCO3, pH 7.8, containing 1 M urea.
Oxidation of RNAP core in the presence of 2IT-FeBABE BSA, FeBABE σ70 376C, FeBABE σ70 396, FeBABE σ70 517C, or FeBABE σ70 581C was carried out as above. Each sample was tagged with a different MAOD tag (TbAOD, PrAOD, and HoAOD, respectively). All tagged tryptic peptides were pooled prior to affinity purification with a 2D12.5-Aminolink column.
Trypsin digestion of modified protein
Just prior to trypsin digestion, thiourea and calcium chloride were added to 125 mM and 1 mM, respectively. A stock solution of trypsin was added to a final ratio of 1:50 (w/w) and incubated at 37 °C for 24 h. The peptides were diluted at least 10× into HBS, and PMSF was added to a final concentration of 1 mM to inhibit trypsin prior to application to a 2D12.5-Aminolink affinity column.
Affinity Purification of O-ECAT peptides
To isolate tagged peptides, we used an affinity column composed of the MAOD-binding 2D12.5 mAb conjugated to Pierce Aminolink Plus resin (21). The mAb-resin coupling was conducted as previously described (13). Neutral pH elution conditions were developed to minimize hydrolysis of the Schiff base linkage associated with the MAOD tag (Scheme 2). Following digestion with trypsin, the MAOD tagged peptides that were going to be quantified were combined and loaded onto 400 μL of 2D12.5-Aminolink resin that had been washed with Pierce gentle Ag/Ab elution buffer and equilibrated with HBS. The column was washed extensively with HBS (minimum 20 CV). To elute, a single CV of room temperature Ab/Ag gentle elution buffer was applied to the column and incubated for 15 min at room temperature before an additional 6 CV was applied. Eluted fractions were combined and purified on Sep-Pak Plus C-18 cartridges. Each cartridge was prepared for use by treatment with 10 mL of CH3CN followed by 5 mL of deionized water and 5 mL of Gentle Ag/Ab elution buffer. The ECAT peptides (in gentle Ag/Ab elution buffer) were loaded onto the cartridge and washed with 10 mL of deionized water. The peptides were eluted with 2 mL of a 1:1 CH3CN/water solution and dried using a speed-vac.
Scheme 2.
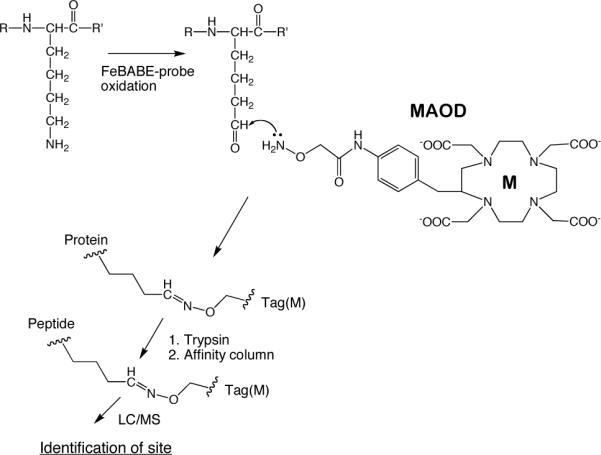
Carbonyl affinity tag MAOD, where M is a monoisotopic rare earth, showing how it is used to tag carbonyl groups produced by hydroxyl radical chemistry and identify their location in the protein sequence (13, 21).
NanoLC-MS/MS ECAT peptides
A high-resolution mass spectrometer (LTQ-FT; Thermo Finnigan), coupled with a NanoAcquity UPLC system (Waters, Milford, MA), was used to acquire LC-MS/MS data sets. A Fourier Transform survey scan at resolution 100,000 and MS/MS acquisition for the top four precursor ions were acquired during a 90-min-long gradient (1−7% Buffer B for 5 min, 7−35% Buffer B for 65 min, 35−70% Buffer B for 5 min, hold at 70% Buffer B for 1 min, and regenerate the column at 1% Buffer B for 14 min). Two buffers used for the RP chromatography were 0.1% HCOOH (buffer A) and 0.1% HCOOH in 100% CH3CN (buffer B). Dynamic exclusion of 3 min was used with early expiration turned on if signal-to-noise ratio became lower than 2. Preview mode for FTMS master scan was turned on and singly charged ion was rejected for MS/MS.
Database searching to aid in peptide sequencing
SEQUEST searches (14) were performed for MS/MS data, against a database containing sequences of each of the σ70 single-Cys mutants. The following differential modifications were allowed: oxidation on all 20 standard amino acids (+16 Da), oxidation on methionine, tryptophan, cysteine, tyrosine, and phenylalanine (+32 Da), and the MAOD tag on the N-terminus and on various side chains. The predicted mass changes were based on the most common oxidation products observed during synchrotron radiolysis and previous metal-catalyzed oxidation studies (2, 21). Oxidation to a ketone or aldehyde and subsequent MAOD tagging of lysine, arginine, proline, glutamic acid, aspartic acid, threonine, and histidine side chains were checked. To allow for oxidative backbone cleavage, the search was not restricted to tryptic peptides. Correlation values (Xcorr) of 1.5, 2.0, and 2.5 were used for singly, doubly, and triply charged peptide ions, respectively. All spectra were verified manually.
Quantitation of O-ECAT peptides
As a control, a FeBABE/ascorbate (final 1 mM)/hydrogen peroxide (final 10 mM) oxidized 376C sample was tagged with equimolar PrAOD and HoAOD and the identified oxidized and tagged peptides were quantitated. Following ECAT peptide identification and verification, the ion chromatogram was extracted from the narrow region of the monoisotopic peak of each parent and the integrated ion signals were used to quantify O-ECAT peptide pairs. An example of this is shown in Figure 1, with the O-ECAT peptide (R)VRTQER, where the tag is on the bold residue. In the control experiment seven O-ECAT peptide pairs were identified. The average ratio was 1.03 ± 0.13; the expected value was 1.00. The remaining control data can be found in Table s1 of Supporting Information.
Figure 1.

Example of O-ECAT quantitation. During this experiment the oxidized protein was tagged with equimolar HoAOD and PrAOD so the theoretical HoAOD/PrAOD ratio was 1. First, the identity of the tagged peptide was manually confirmed after initially using Sequest (e.g. see panel A for the identification of the HoAOD tagged peptide). Next, the ion chromatogram from the narrow region of the monoisotopic peak of each parent was extracted (B and C). The intensities of the integrated ion signals were used to quantify O-ECAT peptide pairs. In this example, the HoAOD peptide has a signal intensity of 1.43E4 (B), while the PrAOD peptide has a signal of 1.53E4 (C), so the ratio of the intensities is 0.93.
Numerical methods
The average movement of a molecule, such as a hydroxyl radical, in solution is described by the three-dimensional diffusion equation , where P(r,t) is the probability that the diffusing molecule will be located at a distance r from its source after a time t has elapsed, is the Laplacian operator and D is the diffusion coefficient of the molecule. Solving the three-dimensional diffusion equation for a point source of hydroxyl radicals (15) leads to
| (1) |
In equation 1, N is the amount of hydroxyl radical produced at the FeBABE; D ≅ 2.8 × 10−5 cm2s−1 is the diffusion coefficient of the radical (16).
So far we have not included the chemical reactions of the radical. For example, hydroxyl radical readily reacts with organic molecules such as glycerol. This can be accounted for by inserting the factor exp(–kt) into equation 1 (see, e.g., 17), to yield the diffusion-reaction equation
| (2) |
When glycerol is added to the solution as a quencher to limit the range of the radical, the second-order rate constant for its reaction with hydroxyl radical in dilute solution is (18) k2 = 1.8 × 109 M−1s−1, so the pseudo-first-order rate constant k in exp(–kt) is k = k2[glycerol] = (1.8 × 109 M−1s−1)[glycerol]. If the concentration of glycerol is much greater than that of the radical, [glycerol] will be constant, making k constant. The molecular weight of glycerol is 92g/mol and its density is 1.26g/mL, so for a 10% v/v solution as used in the experiments described here, [glycerol] = 1.37M. However, the viscosity of this solution is approximately 1.36 × that of water, which will reduce the rate constant (and also the diffusion coefficient) proportionately, so k ≅ 1.8 × 109s−1 and D ≅ 2.0 × 10−5 cm2s−1 under these conditions. Reactions of the radical with biological molecules at low concentrations in the solution do not materially deplete the radical relative to what occurs in the presence of 10% glycerol. In what follows, glycerol is considered part of the solvent, and attention is directed to sites on solute molecules such as proteins.
FeBABE experiments are generally performed over several minutes, allowing all the radicals to be consumed. To obtain the total probability of the hydroxyl radical being consumed at each distance r from the source at any time from t = 0 to ∞, we must numerically integrate equation 2 with respect to time:
| (3) |
Mass spectrometry is particularly good for identifying modifications of different amino-acid residues in their sequence contexts, but less facile at quantitatively measuring the amount of one peptide relative to a different peptide in a large set without synthesis of an impractically large set of tagged peptides as authentic calibration standards. Instead of measuring yields, we used the natural strength of mass spectrometry to identify and characterize a number of different tagged peptide oxidation products, and then observed the frequency distribution of distinct products as a function of r.
As an approximation of the surroundings of a tethered FeBABE over the course of its active lifetime, consider a solution containing a uniform concentration of distinguishable peptide sites. As the radical diffuses radially from the FeBABE, it encounters different potential reaction partners in numbers that increase as the square of distance, or more precisely as 4πr2, so the average number of distinguishable reaction events as a function of r is related to
| (4) |
RESULTS AND DISCUSSION
We evaluated W (r) from equation 4 numerically using Mathematica 6 and 7 software; it is plotted for three different glycerol concentrations in Figure 2.
Figure 2.
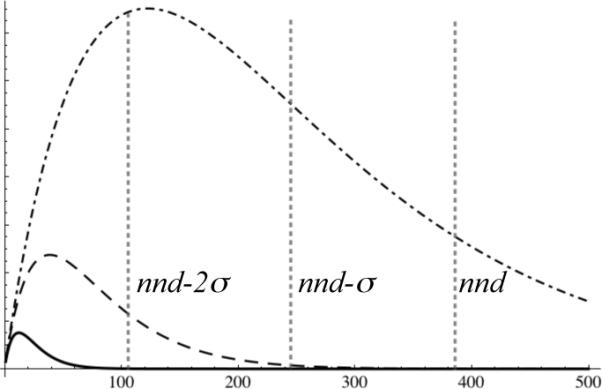
The relative number of different potential reaction events, , among solute molecules that may be encountered by hydroxyl radicals as a function of distance (Å) from a point source, in 10% v/v glycerol (solid curve at lower left corner), 1% v/v glycerol (dashed curve), and 0.1% v/v glycerol (dot-dashed curve). The vertical scale is arbitrary. Glycerol is considered part of the solvent. Also shown, for a 5 μM protein solution, are vertical lines marking the average protein-protein nearest-neighbor distance nnd = 384Å, and distances one standard deviation (139Å) closer, nnd – σ, and two standard deviations closer, nnd – 2σ. For a 5 μM solution containing 10% v/v glycerol, hydroxyl radical encounters are effectively confined to the protein complex from which the radical originates.
In order to map the components in a macromolecular complex, it is essential that FeBABE experiments be designed to avoid the migration of hydroxyl radicals from one complex to another. The diffusion coefficient of a typical protein is <1% that of hydroxyl radical, so to a reasonable approximation we can consider the protein stationary.
The separation between different macromolecular complexes in dilute solution can be described using a model of complete spatial randomness (19), which when derived in three dimensions (20) leads to a nearest-neighbor distance nnd with standard deviation σ of
where [M] is the molar concentration of the macromolecule. For a 1μM solution, nnd = 656Å and σ = 238Å, and less than 1.5% of the nearest-neighbor distances lie within nnd – 2σ = 180Å. The cube-root dependence on concentration is relatively weak: for a 5μM protein solution, nnd = 384Å and σ = 139Å, so nnd – 2σ = 106Å. Figure 2 shows that 10% v/v glycerol is an effective solvent for most situations encountered in biochemistry, while smaller glycerol concentrations could be problematic.
The plots in Figure 2 provide insight into what may be expected when a hydroxyl radical is produced at FeBABE and diffuses away from the tethered protein into the solution. The number of distinguishable reaction events is expected to initially increase with distance because of the 4πr2 factor in equation 4, then pass through a maximum, and finally decline. The factor exp(–kt) in equation 2 controls the decline by reducing the lifetime of the radical. The strength of this effect can be adjusted by changing the concentration of glycerol in the buffer, which will alter the value of the pseudo-first-order rate constant k as described above. The same formalism could be used to describe other diffusible reactive species, and other quenchers besides glycerol.
There is sufficient information in the literature for quantitatively describing the reaction-diffusion behavior of hydroxyl radicals in aqueous glycerol solutions, but not for hydroxyl radicals in a protein. For a reproducible methodology that detects the products of hydroxyl radical chemistry but is simple enough for use in very complex systems, we chose a carbonyl-reactive element-coded affinity tagging method (O-ECAT) for analysis of protein-bound carbonyl groups following oxidation (21). (A similar method using biotin tags has also been reported (22, 23).) O-ECAT uses an aminooxy functionalized DOTA derivative, MAOD, to tag protein-bound carbonyls for later purification (Scheme 2), with different chelated rare earth ions serving as mass tags (13). This was inspired by Aebersold's isotope-coded affinity tag methodology (ICAT), the proteomics method designed to compare relative protein abundance between two samples (24). Stable chemical tagging of carbonyl products is followed by trypsin digestion and affinity capture of tagged peptides, simplifying identification of modified amino-acid residues by LC/MS. It should be noted that carbonyl products are produced by hydroxylation of a carbon atom attached to an electronegative atom, and therefore are a minority of the total products of hydroxyl radical chemistry. However, they are weighted toward surface-accessible residues such as lysine and arginine, and so may give a useful picture of protein proximity in the way that chemical cross-linking does, but with the added feature of precise identification of the residues and positions involved.
Sigma self-oxidation
Including Pro, Arg, Lys, Thr, His, and Glu without taking into account solvent accessibility, σ70 has 217 potential side chain oxidation sites leading to carbonyl formation. It also has a total of 612−19 = 593 non-proline residues that might be oxidatively cleaved in the backbone to yield new N-terminal carbonyl products, plus the methionine Cα at the original N terminus. The oxidized σ70 peptides that we tagged and identified are listed in Supporting Information Table s2, and the data that could be mapped onto the crystal structure 1SIG of the σ70 fragment are shown in Figure 3. Background oxidation due to sample processing and adventitious iron binding to sites on the protein was also investigated on samples in the absence of FeBABE and FeEDTA, and if there were peptides in common between the background and the FeBABE samples, they were considered background unless the FeBABE sample was at least 5× more intense.
Figure 3.

Oxidized and tagged amino-acid residues mapped onto the crystal structure of a major fragment of σ70 (PDB file: 1SIG) for three single-Cys mutants (132C, 376C, 422C). On the left side, the red sphere in each image represents the α-carbon of the single cysteine residue. The α-carbons of oxidized and tagged side chains are shown as green spheres, while the α-carbons of the oxidized and tagged N terminal residues resulting from backbone cleavage are shown as orange spheres. The identity of each amino acid residue is displayed, followed by the distance in Å from the α-carbon of the Cys residue to the α-carbon of the oxidized and tagged amino acid residue. Backbone cleavage determined from western blots (25) is shown as yellow ribbon (±3 residue std dev). On the right side, the space-fill images include information regarding the ratio of intensities of oxidation produced by FeBABE to that produced by FeEDTA -- e.g., 7.1× indicates that the FeBABE cleavage produced a 7.1× higher yield of that peptide -- for those residues where data are available. The sites efficiently oxidized by tethered FeBABE are colored blue, while the sites obscured from FeBABE but readily accessible to FeEDTA in solution are rendered in purple. If the ratio was not determined, colors match the images on the left side. The figures are rotated so that all the residues of interest can be seen in the space-fill model.
Random FeEDTA protein oxidation was compared to tethered FeBABE-mediated self-oxidation using a library of single-cysteine mutants of σ70. Oxidation using FeEDTA was studied with 3 mutants (132C, 376C, and 422C) and analyzed simultaneously with FeBABE oxidation data using O-ECAT. This provided an interesting index of the efficiency with which hydroxyl radicals propagate from the tethered FeBABE to a site of oxidation: radicals produced by dissolved FeEDTA encounter the surface of the protein uniformly, while radicals produced at a single site on the protein surface may be consumed by reacting with polypeptide loops that lie between the FeBABE and other sites that are readily oxidized by FeEDTA. Thus the FeBABE/FeEDTA carbonyl production ratio is high for unobstructed sites close to FeBABE but low for others. Sites that could be placed on the crystal structure are indicated in Figure 3; the entire list is given in Supporting Information Table s2.
The distances between the α–carbon of each oxidized and tagged amino acid, and the cysteine α–carbon of the appropriate FeBABE-conjugated σ70, were determined using the E. coli σ70 crystal structure 1SIG where possible (26). The crystal structure consists of residues 113−446 of the 613-residue protein, at a resolution of 2.6Å. There are three 3 N-terminal residues, three internal regions (168−172, 192−211, and 238−241), and two C-terminal residues 447−448 in the protein fragment that are not resolved in the structure. The dimensions of the fragment are approximately 75Å × 50Å × 30Å.
For carbonyl formation reactions involving three single-Cys σ70 mutants (132C, 376C, 422C), FeBABE oxidation led to a total of 31 distinct α–carbon - α–carbon distances that could be measured from the 1SIG crystal structure. The average Cα-Cα distance from an engineered Cys to a distinct oxidized residue was 27.2 Å, and the shortest and longest observed distances were 8.61Å and 58.0Å. Fifty-three percent of the distinguishable Cα-Cα distances were within 25Å, and 84% were within 40Å.
Equations 1-4 lead to derivation of the probability density function W (r) for distinguishable reaction products, which we can use to calculate the average distance from a point source to react with a solute molecule:
Further, the fraction of reactions with solutes within 19Å of a point source is obtained by integrating W (r) from 0 to 19Å and dividing by the normalization factor
and the fraction of reactions within 34Å of a point source is
The theoretical treatment leading to Figure 2 applies strictly to hydroxyl radicals diffusing through an aqueous glycerol solution of distinguishable peptide sites, and we have no detailed model for hydroxyl radical transport through or around a protein. However, it is interesting to note that if we apply the same equations to estimate Cα-Cα distances from the FeBABE-tagged cysteine to the oxidized amino-acid residue, there is a parallel between the observed (27.2Å) and calculated (21.4Å) average Cα-Cα distances, as well as the observed (25Å) and calculated (19Å) Cα-Cα distance within which 53−54% of the reactions will occur, and the observed (40Å) and calculated (34Å) Cα-Cα distance within which 83−84% of the reactions will occur.
Of course, the source of the radicals is not located at the Cα of the cysteine residue, but at FeBABE on the side chain. Presumably, the fact that the observed distances above exceed the calculated numbers by a consistent ≈ 6Å may be attributed to the average additional separation due to the size and conformational freedom of FeBABE.
This parallel between experimental observations and calculations provides insight into the behavior of the two reactants on the protein. The FeBABE group is attached to a cysteine side chain, and has rotational freedom about several single bonds (Scheme 1); all allowed orientations of FeBABE relative to the protein are present within the collection of molecules in the sample, so that the results represent an average of hydroxyl radical production within a small three-dimensional region adjacent to the cysteine. Data from three different single-Cys mutants of σ70 were used, further averaging the individual effects of a single site. Most of the sites of oxidation are located at the ends of flexible side chains on residues such as lysine and arginine, which will also have a range of orientations. Apparently the short-range structural disorder of both reactants in the sample, in the presence of the longer-range effects of hydroxyl radical diffusion, leads to Cα-Cα distance results in reasonable agreement with the diffusion-reaction calculations. Continuing these calculations to longer distances in 10% glycerol, 99% of the distinguishable reactions are predicted to occur within 71+ 6 = 77Å of the tagged Cys.
There were 62 total oxidation events identified between the three single-Cys mutants. Side chain arginine and lysine carbonyls were most commonly observed (37% and 27%, respectively), followed by N-terminal carbonyls (13%), proline (13%), and glutamate (8%). Approximately half of the total identified oxidation sites could be mapped to the crystal structure.
As with protein cross-linking, there is the possibility that in a different system an individual mutant might deviate from average behavior. If we consider the three Cys mutants individually, the number of events is smaller and the statistics suffer accordingly, but the site-to-site variation is illustrated. When FeBABE was attached to 132C, there were 17 total oxidation sites (8 arginine side chain, 5 lysine side chain, 2 proline, and 2 N-terminal carbonyls), 10 of which could be mapped to the crystal structure. The Cα-Cα distances ranged from 11.1−36.8Å with the average distance being 22.3 ± 7.9Å. For FeBABE-376C, there were 16 out of 27 total oxidation sites (10 arginine side chain, 7 lysine side chain, 3 proline, 2 glutamic acid side chain, 1 threonine side chain, and 4 N-terminal carbonyls) that could be mapped to the crystal structure. The Cα-Cα distances ranged from 8.61−54.2Å with the average distance being 29.8 ± 15.4Å. For FeBABE-422C, there were 10 out of 18 total oxidation sites (5 arginine side chain, 5 lysine side chain, 3 proline, 3 glutamic acid side chain, and 2 N-terminal carbonyls) that could be mapped to the crystal structure. The Cα-Cα distances ranged from 9.5−58.0Å with the average being 29.0 ± 14.7Å.
Oxidation of RNAP Core subunits by FeBABE-conjugated σ70
When three of the FeBABE conjugated σ70 mutants (396C, 517C, and 581C) were bound to the RNAP core enzyme, they were observed to oxidatively modify both subunits β and β′. When FeBABE was attached to 376C, only oxidized β residues were identified. None of the FeBABE conjugates modified the α subunit or ω subunit detectably. No carbonyls were identified when FeBABE was attached to the non-binding control protein BSA via 2-iminothiolane. This was an essential control experiment to identify any pre-existing RNAP core carbonyls, demonstrate the specificity of oxidation of RNAP core by FeBABE-conjugated σ70 mutants under these conditions, and quantitatively compare oxidation by FeBABE- σ70 conjugation to non-specific and background oxidation due to sample processing. Because of the complexity of experiments involving RNAP core, detecting carbonyls was more difficult than for the self-oxidation by FeBABE- σ70. Nonetheless, some oxidized residues were reliably identified. There was a single carbonyl modification that was common to all of the FeBABE conjugated σ70 mutant experiments—a carbonyl on 1070H of β. It is possible that this was an artifact, and we do not attempt to interpret it.
Because no crystal structure is available for the E. coli holoenzyme, the tagged and identified carbonyls on E. coli RNAP were mapped to the Thermus Aquaticus RNAP holoenzyme structure solved by Darst and co-workers (Figure 4) (27). Residues in E. coli regions that lacked sequence homology with Taq according to ClustalW software were not considered. Otherwise, it was assumed that conserved regions of the solution structure of E.coli RNAP and Taq RNAP in the crystal share similar 3D structures. It is certainly possible that the details of the structure of E. coli RNA polymerase in solution differ from the Taq crystal structure, but displaying the data in this way provides a useful framework for comparison with earlier experiments (6). Table s3 lists the residues that could be mapped onto the Taq structure.
Figure 4.

Oxidized and tagged residues mapped onto the crystal structure of to Thermus Aquaticus RNA polymerase holoenzyme (PDB file: 1L9U). The β subunit is colored blue, the β′ subunit is colored pink, the α2 and ω subunits are gray, and the σ70 subunit is green. The single cysteine side chain is shown in red. Oxidized and tagged side chains are shown as gray spheres. Oxidized and tagged N termini are shown as orange spheres. Previously determined backbone cleavage data (strong cuts) are shown as 20-residue segments of small spheres (uncertainty due to assignment by electrophoretic mobility) (6).
In contrast to the self-oxidation of σ70-FeBABE conjugates, the other subunits of RNAP are offset from the FeBABE source of hydroxyl radicals on the bound σ70 conjugate. This is manifest in the data: for example, the average inferred Cα -Cα distance to oxidized β and β′ residues from studies with FeBABE-517C was ≈ 31Å, and the average Cα -Cα distance to oxidized β and β′ residues for FeBABE-581C was ≈ 63Å.
For the reasons noted above, this method will be sensitive to polypeptide loops that are present in E. coli RNAP but absent in Taq. For example, while E. coli and Taq RNAP molecules share essential conserved sequences, the overall number of residues for each subunit is quite different: E. coli σ70 has 613 amino acids, while Taq σA has 438; E. coli β 1342, Taq β 1118; E. coli β′ 1407, Taq β′ 1524 (6). By inference from the Taq RNAP crystal structure, none of the FeBABE-σ70 mutants had α or ω atoms within ; no carbonyls were identified on either subunit. Sigma residue 517C is inferred to have ∼4000 RNAP atoms within , 581C ∼800, 396C ∼3500, and 376C ∼2000.
When FeBABE was attached to 376C, located in the conserved σ70 region 2.1, besides H1070 on β there was only one oxidized and tagged residue, and it was the side chain of T1163 on β′. In comparison, earlier backbone-cleavage experiments for the same mutant resulted in two moderately intense western-blot bands, for cleavage sites assigned to residues 319 and 330 on β′, with estimated standard deviation 16 residues (9).
When FeBABE was attached to 396C, near the C-terminal end of conserved σ70 region 2.2, besides H1070 on β a single oxidized and tagged residue was identified on β′ (P41). Earlier experiments observed backbone cleavage at β (residues 383, 391, 467, 490) and β′ (307, 322, 344), with estimated standard deviation 10 residues for β and 16 residues for β′ (9).
No sites were identified when FeBABE was attached to 422C; earlier experiments identified rather weak bands on western blots for this mutant (9). Problems with sample preparation led to no ECAT results for the 496C mutant.
Compared with 376C and 396C, there were significantly more oxidized and identified β and β′ residues identified when FeBABE was attached to 517C, in σ70 region 3.2: besides β H1070 there were 7 tagged residues in each of β (K191, R233, R470, R530, P1044, K1065, F1323) and β′ (K2, P27, P41, R47, R339, R392, E1188), and 4 on σ70 (R232, R339, K418, R441). Earlier experiments with 517C observed backbone cleavage at β (residues 493, 523, 550, 858, 913, 1020) and β′ (276, 294, 328, 434, 461) (9). Note that, unlike the other mutants, for 517C there is some overlap between the two data sets obtained by different techniques; however, side chain oxidation here is related to backbone cleavage in the earlier experiments (e.g., β R530 vs 523±10; β′ R339 vs 328±16), and most data points in one experiment do not have obvious links to the other.
When FeBABE was attached to 581C, in conserved σ70 region 4.2, 2 β residues (H1070, D1241), 5 β′ residues (P27, P41, R47, R270, L282), and 5 σ70 residues (R232, R339, K371, R554, R588) were tagged. Earlier experiments with 581C observed backbone cleavage at β (residues 477, 501, 527, 554, 875, 913, 1022) and β′ (278, 301, 328) (9). Here there is one correlation between backbone cleavages observed in the two experiments (β′ L282 vs 278±16).
As shown in Figure 4, the sites identified in both types of experiment are located plausibly close in space to the σ-tethered FeBABE group even though there is very little sequence overlap. Since hydrolysis of the polypeptide backbone does not produce a carbonyl group, it would not yield products detectable by the O-ECAT methods used here.
The sets of Cα-Cα distances obtained for σ70 and inferred for RNAP holoenzyme show that, even though the reach of hydroxyl radical is flexible beyond that of a chemical cross-linking reagent, its reactions with protein sites can be correlated with expectations based on its known properties in solution. As demonstrated by the striking differences between the results of the four FeBABE-σ70 conjugates in RNAP complexes, moving the FeBABE site has an important effect. The lack of modification of the two alpha subunits of RNAP by any of the four FeBABE-σ70 conjugates shows that components of a large complex may lie in a steric shadow, with intervening polypeptide chains depleting hydroxyl radicals so that no oxidation can be detected.
As with cross-linking, interpreting the results of FeBABE mapping requires control experiments carried out under the same conditions. Negative controls include omitting FeBABE but including everything else, replacing the specific probe protein with an irrelevant protein having a similar size, etc. Positive controls include self-oxidation of the probe. Besides details of methodology, the main distinction relative to cross-linking is that, as shown in Figure 2, there is not a sharp minimum or maximum reach for hydroxyl radical. Nonetheless, the results with RNAP demonstrate clearly which subunits are adjacent to the FeBABE-σ70 sites and which are not, and there is reason to expect this approach will be useful for more complex systems.
CONCLUSIONS
Analysis of specific products of hydroxyl-radical oxidation shows how the diffusion of radicals from a point source is reflected in the oxidation of individual amino-acid residues in proteins. This provides a better description of the distance dependence of protein-mapping experiments, experimentally showing that the hydroxyl radical can reliably provide local information even though its reach is flexible. The minimal overlap between polypeptide backbone cleavage sites produced by hydroxyl radicals and those produced by other mechanisms points out the advantage of selectively analyzing the products in order to better understand the relationship between source and site.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr. Young Jin Lee, Dr. Rich Eigenheer, Dr. Brett Phinney, and Michelle Salemi of the UC Davis Genome Center Proteomics Core for mass spectrometry analysis and assistance with data analysis. We also thank the members of the Meares lab for helpful discussions.
Footnotes
Supported by NIH Grant GM025909.
ABBREVIATIONS
2D12.5, antibody that binds metal-DOTA chelates; 2IT, 2-iminothiolane; AOD, ((S)-2-(4-(2-aminooxy)acetamidobenzyl)-1, 4, 7, 10-tetraazacyclododecane-N, N′, N″, N‴-tetraacetic acid; BABE, (S)-2-(4-(2-bromo-acetamido)-benzyl)-ethylenediaminetetraacetic acid; BSA, bovine serum albumin; cleavage buffer, 50 mM Hepes, pH 7.0/10% v/v glycerol/1mM EDTA; CPM, (N-(4-(7-diethylamino-4-methylcoumarin-3-yl)phenylmaleimide); CV, column volume; DOTA, 1, 4, 7, 10-tetraazacyclododecane-N, N′, N″, N‴-tetraacetic acid; ECAT, element-coded affinity tag; FeBABE, iron complex of BABE; FeEDTA, iron-ethylenediaminetetraacetate; FT, Fourier transform; HBS, 50 mM Hepes, 100 mM NaCl, pH 7.4; ICAT, isotope-coded affinity tag; MAOD, metal-AOD complexes; mAb, monoclonal antibody; MS, mass spectrometry; nanoLC-MS, nanoflow liquid chromatography-mass spectrometry; O-ECAT, carbonyl-reactive element-coded affinity tag; PMSF, phenylmethanesulfonyl fluoride; RNAP, RNA polymerase; RNAP core, complex of subunits α2ββ′; RP, reverse phase; SDS-PAGE, sodium (lithium) dodecylsulfate-polyacrylamide gel electrophoresis; storage buffer, 10 mM Tris-HCl pH 7.9, 100 mM NaCl, 0.1 mM EDTA, 50% glycerol; tagging buffer, 100 mM KH2PO4 pH 6.5/0.5 M NaCl/5 M urea/1.8 M thiourea/1mM EDTA; TBS, 24.8mM Tris, 137mM NaCl, 0.269mM KCl, pH 7.4; TBST, TBS with 0.05% Tween-20; TCA, trichloroacetic acid.
SUPPORTING INFORMATION AVAILABLE
Example MS/MS of O-ECAT peptides; Determination of ratios of (M1)AOD and (M2)AOD tagged peptides using mass spectrometry; Sigma self-oxidation data; Sigma-RNAP oxidation data. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Bohon J, Jennings LD, Phillips CM, Licht S, Chance MR. Synchrotron protein footprinting supports substrate translocation by ClpA via ATP-induced movements of the D2 loop. Structure. 2008;16:1157–1165. doi: 10.1016/j.str.2008.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xu G, Chance MR. Hydroxyl Radical-Mediated Modification of Proteins as Probes for Structural Proteomics. Chem. Rev. 2007;107:3514–3543. doi: 10.1021/cr0682047. [DOI] [PubMed] [Google Scholar]
- 3.Chen H-T, Hahn S. Mapping the Location of TFIIB within the RNA Polymerase II Transcription Preinitiation Complex: A Model for the Structure of the PIC. Cell. 2004;119:169–180. doi: 10.1016/j.cell.2004.09.028. [DOI] [PubMed] [Google Scholar]
- 4.Miller G, Hahn S. A DNA-tethered cleavage probe reveals the path for promoter DNA in the yeast preinitiation complex. Nat Struct Mol Biol. 2006;13:603–10. doi: 10.1038/nsmb1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rana TM, Meares CF. Transfer of Oxygen from an Artificial Protease to Peptide Carbon During Proteolysis. Proc. Natl. Acad. Sci. U.S.A. 1991;88:10578–10582. doi: 10.1073/pnas.88.23.10578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Meares CF, Datwyler SA, Schmidt BD, Owens JT, Ishihama A. Principles and methods of affinity cleavage in studying transcription. Methods Enzymol. 2003;371:82–106. doi: 10.1016/S0076-6879(03)71006-4. [DOI] [PubMed] [Google Scholar]
- 7.Datwyler SA, Meares CF. Protein-protein interactions mapped by artificial proteases: where σ factors bind to RNA polymerase. Trends Biochem. Sci. 2000;25:408–414. doi: 10.1016/s0968-0004(00)01652-2. [DOI] [PubMed] [Google Scholar]
- 8.Stadtman ER. Oxidation of Free Amino Acids and Amino Acid Residues in Proteins by Radiolysis and by Metal-Catalyzed Reactions. Ann. Rev. Biochem. 1993;62:797–821. doi: 10.1146/annurev.bi.62.070193.004053. [DOI] [PubMed] [Google Scholar]
- 9.Owens JT, Miyake R, Murakami K, Chmura AJ, Fujita N, Ishihama A, Meares CF. Mapping the sigma70 subunit contact sites on Escherichia coli RNA polymerase with a sigma70-conjugated chemical protease. Proc. Natl. Acad. Sci. U.S.A. 1998;95:6021–6026. doi: 10.1073/pnas.95.11.6021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hager DA, Jin DJ, Burgess RR. Use of Mono Q High-Resolution Ion-Exchange Chromatography to Obtain Highly Pure and Active Escherichia coli RNA Polymerase. Biochemistry. 1990;29:7890–7894. doi: 10.1021/bi00486a016. [DOI] [PubMed] [Google Scholar]
- 11.Burgess RR, Jendrisak JJ. Procedure for the rapid, large-scale purification of Escherichia coli DNA-dependent RNA polymerase involving polymin P precipitation and DNA-cellulose chromatography. Biochemistry. 1975;14:4634–4638. doi: 10.1021/bi00692a011. [DOI] [PubMed] [Google Scholar]
- 12.Schmidt BD, Meares CF. Proteolytic DNA for mapping protein-DNA interactions. Biochemistry. 2002;41:4186–4192. doi: 10.1021/bi015582r. [DOI] [PubMed] [Google Scholar]
- 13.Whetstone PA, Butlin NG, Corneillie TM, Meares CF. Element-coded affinity tags for peptides and proteins. Bioconjugate Chem. 2004;15:3–6. doi: 10.1021/bc034150l. [DOI] [PubMed] [Google Scholar]
- 14.Eng JK, McCormack AL, Yates JR., III An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 15.Carslaw HS, Jaeger JC. Conduction of Heat in Solids. 2nd Edition Oxford University Press; London: 1959. pp. 256–257. [Google Scholar]
- 16.Nikjoo H, O'Neill P, Goodhead DT, Terrissol M. Computational modelling of low-energy electron-induced DNA damage by early physical and chemical events. Int. J. Radn. Biol. 1997;71:467–483. doi: 10.1080/095530097143798. [DOI] [PubMed] [Google Scholar]
- 17.Huang R-C, Gillette R. Kinetic Analysis of cAMP-activated Na+ Current in the Molluscan Neuron: A Diffusion-Reaction Model. J. Gen. Physiol. 1991;98:835–848. doi: 10.1085/jgp.98.4.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Willson RL, Greenstock CL, Adam GE, Wageman R, Dorfman LM. The Standardization Of Hydroxyl Radical Rate Data From Radiation Chemistry. Int. J. Radiat. Phys. Chem. 1971;3:211–220. [Google Scholar]
- 19.Diggle PJ. Statistical Analysis of Spatial Point Patterns. Academic Press; San Diego: 1983. [Google Scholar]
- 20.Scheuerell MD. Quantifying Aggregation and Association in Three-Dimensional Landscapes. Ecology. 2004;85:2332–2340. [Google Scholar]
- 21.Lee S, Young NL, Whetstone PA, Cheal SM, Benner WH, Lebrilla CB, Meares CF. Method to site-specifically identify and quantitate carbonyl end products of protein oxidation using oxidation-dependent element coded affinity tags (O-ECAT) and nanoliquid chromatography Fourier transform mass spectrometry. J. Proteome Res. 2006;5:539–547. doi: 10.1021/pr050299q. [DOI] [PubMed] [Google Scholar]
- 22.Mirzaei H, Regnier FE. Affinity Chromatographic Selection of Carbonylated Proteins Followed by Identification of Oxidation Sites Using Tandem Mass Spectrometry. Anal. Chem. 2005;77:2386–2392. doi: 10.1021/ac0484373. [DOI] [PubMed] [Google Scholar]
- 23.Ahrends R, Pieper S, Kühn A, Weisshoff H, Hamester M, Lindemann T, Scheler C, Lehmann K, Taubner K, Linscheid MW. A metal-coded affinity tag approach to quantitative proteomics. Mol. Cell. Proteomics. 2007;6:1907–16. doi: 10.1074/mcp.M700152-MCP200. [DOI] [PubMed] [Google Scholar]
- 24.Gygi SP, Rist B, Gerber SA, Turecek F, Gelb MH, Aebersold R. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat. Biotechnol. 1999;17:994–999. doi: 10.1038/13690. [DOI] [PubMed] [Google Scholar]
- 25.Miyake R, Owens JT, Xu DD, Jackson WM, Meares CF. Site-directed photocleavage for mapping protein architecture. J. Am. Chem. Soc. 1999;121:7453–7454. [Google Scholar]
- 26.Malhotra A, Severinova E, Darst SA. Crystal structure of a sigma 70 subunit fragment from E. coli RNA polymerase. Cell. 1996;87:127–136. doi: 10.1016/s0092-8674(00)81329-x. [DOI] [PubMed] [Google Scholar]
- 27.Murakami KS, Masuda S, Darst SA. Structural basis of transcription initiation: RNA polymerase holoenzyme at 4 Å resolution. Science. 2002;296:1280–1284. doi: 10.1126/science.1069594. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.