

Abstract
We measured age-dependent effects of the human ApoE4 on cerebral blood flow (CBF) using ApoE4 transgenic mice compared to age-matched wild-type (WT) mice by use of [14C] iodoantipyrene autoradiography. ApoE4 associated factors reduce CBF gradually to create brain hypoperfusion when compared to WT and the differences in CBF are greatest as animals age from 6-weeks to 12-months. Transmission electron microscopy with colloidal gold immunocytochemistry showed structural damage in young and aged microvessel endothelium of ApoE4 animals extended to the cytoplasm of perivascular cells, perivascular nerve terminals and hippocampal neurons and glial cells. These abnormalities coexist with mitochondrial structural alteration and mitochondrial DNA overproliferation and/or deletion in all brain cellular compartments. Spatial memory and temporal memory tests showed a trend in improving cognitive function in ApoE4 mice fed selective mitochondrial antioxidants acetyl-L-Carnitine and R-Lipoic acid. Our findings indicate that ApoE4 genotype-induced mitochondrial changes and associated structural damage may explain age-dependent pathology seen in AD, indicating potential for novel treatment strategies in the near future.
Keywords: ApoE4, Alzheimer’s Disease, Cerebral Blood Flow, Electron Microscopy, Vascular and Mitochondrial Damage, Mitochondrial Antioxidant and Cognitive Performance
Introduction
A growing body of evidence suggests a common etiology for Alzheimer’s disease (AD) and cardiovascular disease (1–5). The E4 isoform of apolipoprotein E (ApoE) is involved in cardiovascular and cerebrovascular disorders and is the most prevalent risk factor for late onset or sporadic AD. ApoE facilitates transportation and metabolism of cholesterol and triglyceride in cells throughout the body (6, 7), promotes the normal metabolism of cholesterol by the liver, and aids in building and repairing neuronal processes in the brain as well as in the periphery (6–9). The genotype appears to be a determinant of brain amyloid-β (Aβ) burden in AD patients (10).
ApoE4 transgenic mice are appropriate models for studying the pathogenesis and preclinical treatment of ApoE-related cognitive deficits associated with late onset AD (11). They express human ApoE4 in glia and/or neurons in the brain depending on the promoter driving expression and exhibit accountable cognitive impairments and cerebrovascular and neuronal pathology.
An important factor in the pathogenesis of AD is hypoperfusion-induced oxidative stress, which is caused by disturbed cerebral blood flow (CBF) (22). AD patients exhibit decreased oxygen levels in the vasculature (23–25). Many studies finding chronic cerebral hypoperfusion in mild cognitive impairment (MCI) and AD have concluded that it is an initiator of the reduced supply of oxygen (8, 24–30). This suggests that low blood flow is a prominent feature of the brain during chronic hypoxia/hypoperfusion and possibly an initiating factor during the development of AD (2, 27–29, 31, 32).
The AD brain is characterized by the impairment of energy metabolism, indicating mitochondrial dysfunction (22, 29, 33, 34). These metabolic defects are present before AD symptoms develop in ApoE ε4 homozygotic patients (35, 36). In addition, it has been well documented that reduced resting global CBF is associated with cardiovascular diseases such as atherosclerosis, post-ischemic insult and heart failure (HF). A study by Alves and coworkers suggests that coexistence of blood flow reductions in HF patients with the functional deficit in these regions is relevant to the pathophysiology of the cognitive impairments presented by HF patients (37). De la Torre and colleagues proposed that advanced aging, along with a comorbid condition such as a vascular risk factor that further decreases cerebral perfusion, promotes a critically attained threshold of cerebral hypoperfusion. Studying the effect of aging as a main reason for chronic brain hypoperfusion (CBH) in oxidative stress induced cerebrovascular lesions and their relationship to MCI and AD could uncover the ultimate pathogenic mechanisms that lead to AD. We have previously shown that atherosclerotic lesions are associated with mitochondrial DNA deletions in brain microvessel endothelium and amyloid angiopathy in human AD (23), aged transgenic mice overexpressing amyloid beta precursor protein (AβPP) (34, 38), and two-vessel occlusion rat models of CBH (39). These studies suggest that cerebrovascular pathology may play a crucial role in predisposition to stroke and possibly MCI and AD (27–29).
Our previous studies found that treating aged rats with the selective mitochondrial antioxidants acetyl-l-Carnitine (ALCAR) and R-alpha-Lipoic acid (LA) restores cognitive performance and abolishes oxidative stress induced structural changes in brain parenchymal cells (neurons, vascular wall cells and glia) (40, 41). The effect of aging on CBF and brain parenchymal cell ultrastructure and the potential for treating these abnormalities by using selective mitochondrial antioxidants have not yet been fully explored. In the present study, we used the vascular dementia paradigm to analyze the effects of the selective mitochondrial antioxidants acetyl-Carnitine and R-Lipoic acid and ApoE4 on CBF, neuropathology, brain and vessel ultrastructural abnormalities, and behavior.
Materials and methods
For the blood flow and ultrastructural studies, Glial Fibrillary Acidic Protein (GFAP)-ApoE4 transgenic and wild-type C57BL/6 control mice were obtained from the Jackson Mouse Colony (Jackson, FL, USA). The GFAP promoter drives the expression of ApoE cDNA in glia, primarily in astrocytes. The ApoE-4 transgenic mice express no mouse ApoE and are on a C57BL/6 background. Animals were housed in 12 hours dark/light conditions and had unlimited access to food and water. All experimental procedures were performed according to NIH and International Guidelines for the use of animals in research, and appropriate protocols were approved by the relevant institutional committees. Six week- and six- and twelve-month-old (n=12/group) ApoE4 and wild-type C57BL/6 control mice were used for the blood flow study.
Cerebral blood flow measurement
Age-dependent effects of ApoE4 on CBF were measured using [14C] iodoantipyrene autoradiography in conjunction with a mathematical algorithm (42). Measurement of regional blood flow (rBF) was determined by a [14C] - iodoantipyrene (IAP, New England Nuclear) autoradiography technique modified for mice and described earlier by us (42, 43). This method measures local CBF by combining an intraperitoneal tracer with a single blood sampling from the heart. Mice were anesthetized by a halothane gas mixture (2% in 30/70% O2 and NO2), and then injected with IAP in normal saline (2.5 μCi in 200 μl) intraperitoneally. 60 seconds later the mice were frozen in liquid nitrogen and stored at −80°C. Brains and hearts were dissected in a cryotome (−20°C). Brains were thin-sectioned at the levels of atlas plate 13, 30 and 69 (described in detail by Puchowicz, et al.) and then placed on glass slides. The slides were placed on Amersham Hyperfilm β-Max autographic film along with calibrated standards (Amersham [14C] Micro-scales, RPA 504 and RPA 511) and then exposed for 3 months (42). The films were digitized using a BIOQUANT image analysis system (R & M Biometrics, INC) and background corrected. Optical densities were converted to nanocuries per gram using standard curves generated from the standards. The blood flow was calculated from the images and a reference blood sample using the equation below:
Blood Flow (ml/g/min) = Tissue (nCi/g)/[(reference blood (nCi)* Time (min)] Reference blood samples were prepared by pipetting 100 μl samples taken from the heart into a scintillation vial and then adding 15 ml of scintillation fluid (Aquasol, NEN Res. Products, DU PONT). After mixing, vials were counted on a γ-scintillation counter (dpm).
ALCAR and LA treatment
The Gladstone Institute, University of the California at San Francisco, provided Neuronal Specific Enolase (NSA)-ApoE4 transgenic mice for behavioral studies. These transgenic mice also are on a C57BL/6 background and have no mouse ApoE expression. Seven-month-old ApoE4 transgenic mice were randomly divided into two groups (n=4 per group): control and treated (0.2% ALCAR in drinking water and 0.15% dexlipotam, a tris-salt of LA, which is equal to 0.1% LA) as described by our group elsewhere (40). Wild-type C57BL/6 mice without treatment were used as controls. Mice were subjected to Morris water maze testing at the age of 12 months (following 5 months of treatment), again at 22 months (following 15 months of treatment) and to a Peak procedure test at the age of 13 months (after 6–7 months of treatment). At the end of the final cognitive tests all animals were perfusion fixed as described previously (31) for electron microscopic ultrastructural analysis, by using in situ hybridization techniques for mitochondrial DNA overproliferation determination, and deletion and immunogold decoration by using antibodies for protein immunoreactivity determination.
Morris water maze test of spatial memory
The Morris water maze task tests spatial memory by requiring mice to find a submerged platform in a pool of water using external visual cues as described previously (40, 44, 45). The time required for an individual mouse to find the platform was measured using a digital camera and a computer system to record movement (Columbus Instruments, VideoMex-V). Trials (4 consecutive days, 4 trials per day) were with the same hidden platform location, but with varied start locations. On day 5, the platform was removed from the pool for a probe test, (60 sec) and the time spent at the actual site where the platform was previously located was recorded. On day 6, the time required to reach a visible platform was measured to determine visual function and motor ability (40). In the reversal test, the platform was moved to the opposite quadrant of the previous test (4 trials/day and 120 s/trial).
Peak procedure test of temporal memory
Temporal memory, as assessed by the peak procedure, measures the function of the internal clock, learning process, attention, and exploratory behavior (40, 44, 46, 47). Mice were tested in 18 identical boxes that contain a light source and a speaker (for delivering light or noise signals) and a lever that dispenses single food (45 mg) pellets when pressed (BioServ mix T101). Prior to the test, the food supply was decreased to 85% of the free-feeding amount. In this test the animal is rewarded with one pellet only if the lever is pressed within 40 sec from the signal. In 20% of the tests no food was given, an empty trial, and the signal lasted 195 sec plus a geometrically distributed duration that averaged 50 sec. The results are presented as a sum of the two types of tests. The Peak rate, the maximum response rate in a given trial and a reflection of mouse choices and their motivation, was measured.
Perfusion fixation and ultrastructural analysis
Tissues from normal, non-transgenic wild-type C57BL/6 control mice and GFAP--ApoE4 transgenic mice were prepared at 6 weeks, 6, months and 12 months (n=6 for each age group) and 22 months (n=4 for each aged group: ApoE4 treated, ApoE4 non-treated and non-treated control wild-type mice) (31, 43). All animals received a standard laboratory diet ad libitum. Mice in the treatment group received the standard diet plus water enriched with ALCAR+LA (0.2% ALCAR in drinking water and 0.15% dexlipotam, a tris-salt of LA, which is equal to 0.1% LA). Wild-type C57BL/6 mice without treatment were used as a controls. The duration of the experiment was 22 months. Mice, under terminal anesthesia were perfusion fixed via the heart as described previously (31, 39). Tissues were processed for future analysis by EM and in situ hybridization for cytological detection of mtDNA. In situ hybridization was performed using human (normal and Δ5kb deleted) and mouse specific probes as described previously (22, 31, 38, 48). Finally, all the sections were exposed to OsO4 for 1 h at RT, rinsed, dehydrated and flat embedded in Spurr’s embedding media. Ultrathin sections were stained with uranyl acetate and lead citrate and viewed in a JEOL 100CX, Jeol 1200 or Jeol 1230 EX electron microscopes at 80 kV.
Statistical analysis
The statistical analysis for the data obtained from this study was performed using one way ANOVA with Newman-Keuls’ post hoc tests or Student’s t test as well as two-way ANOVA (F test). SPSS for Windows was used for the analyses.
Results
Blood flow
Compared to WT mice, CBF (mean ± SD, ml/g/min) was significantly lower (p < 0.05) in ApoE4 6-month (0.68 ± 0.21 vs. 0.98 ± 0.23) and 12-month (1.06 ± 0.09 vs. 1.54 ± 0.12) groups (see Fig. 1). ApoE4 6-week old mice had lower CBF (0.63 ± 0.15) compared to WT mice (0.82 ± 0.15) but the difference was not significant (Fig. 1). Our findings indicate for the first time that ApoE4 reduces CBF gradually to create brain hypoperfusion when compared to WT and that differences in CBF reduction are greatest as animals age from 6 weeks to 12 months (Fig. 2).
Figure 1.
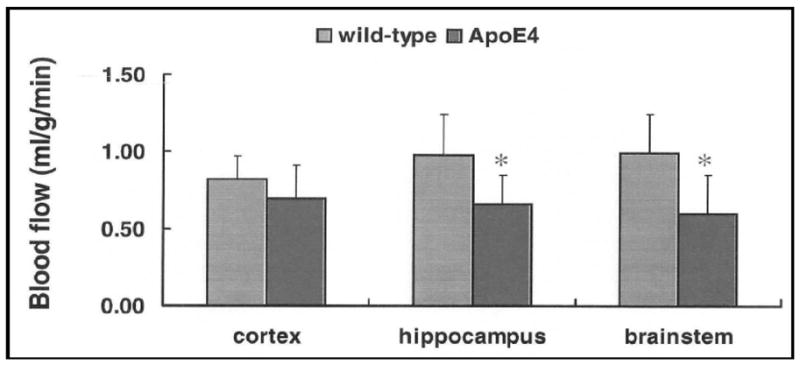
Regional blood flow in 6-week-old mice. ApoE4 is associated with decreased regional blood flow.
Figure 2.

Effect of age on cortical blood flow in wild-type and ApoE4 mice.
Ultrastructural analysis
In general, a heterogeneous morphology characterized the ultrastructure of the brain cellular compartments from young and old wild-type C57BL/6 control and ApoE4 transgenic mice. (Figs. 3–6). Brain tissues from young and old ApoE4 mice were distinguished by proliferation of abnormalities in the ultrastructure of brain microvessels and neurons (Figs. 3 and 6), in contrast to wild-type control mice, which generally lacked these abnormalities. Lipid granules in the cytoplasm of perivascular cells (Fig. 3) and neurons occasionally distinguished old from young wild-type mice.
Figure 3.
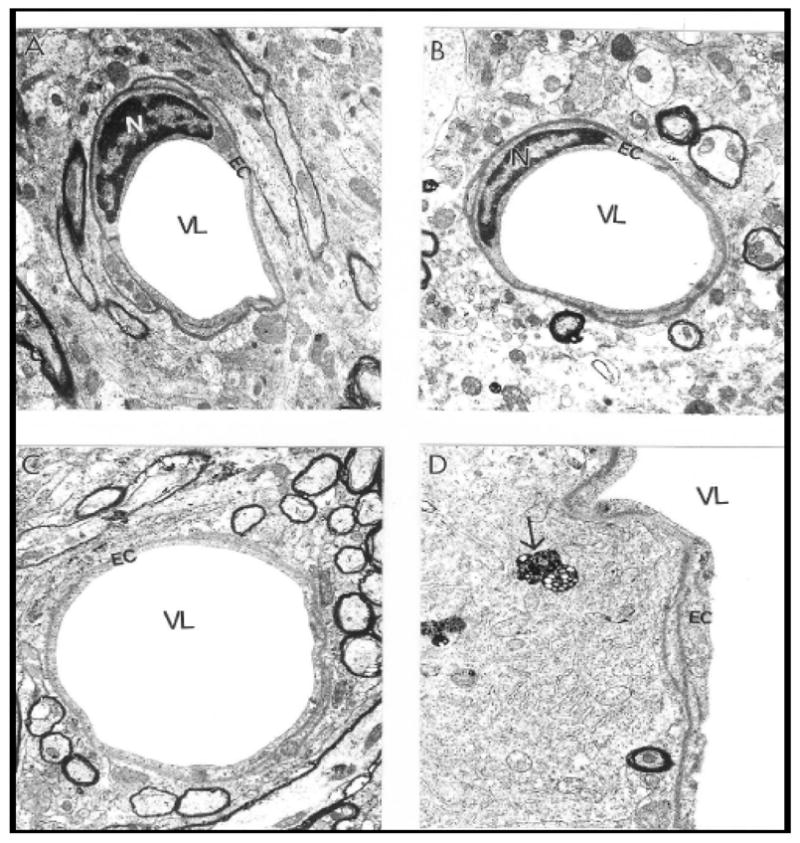
Vascular endothelium and perivascular cells from age-matched control mouse cortex and hippocampus did not show morphological changes in their ultrastructure. Only a single lipid droplet was seen in the matrix of a perivascular cells (indicated by single arrow). Magnification: A-X 10,000; B, C and D- X12, 000. Abbreviations used in figures: EC-Endothelial cells; N-Nucleus, VL-Vessel Lumen.
Figure 6.
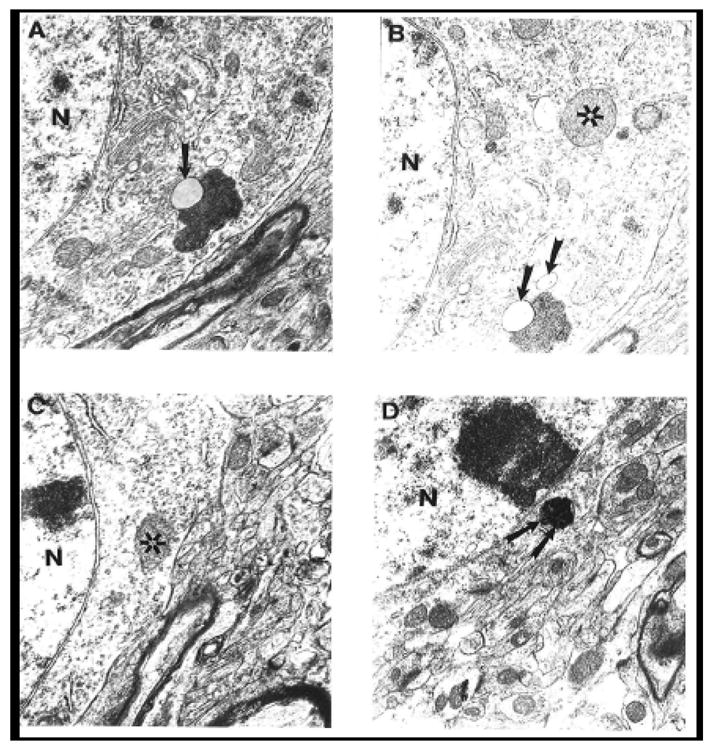
Ultrastructural feature of age-associated neuronal mitochondrial change in ApoE4 mice. Mitochondria-derived lysosome associated with lipofuscin appears to be the main feature of mitochondrial damage. Asterisk: normal mitochondria. Arrowhead: mitochondria-derived lysosomes. Double Arrowhead: hypoxic mitochondria. Magnification: A and B) 20,000; C) 25,000; D) 15,000. Abbreviations used in figures: N-Neuronal Nucleus.
Age-associated microvascular abnormalities characterized ApoE4 mouse brain tissue. “Vascular stress” reactions were early markers of endothelial damage (Fig. 4). Destructive changes were present in the cytoplasm of perivascular nerve terminals (Fig. 4) and in hippocampal neurons and glial cells.
Figure 4.

Microvessels from young (A, B) and aged (C–D) ApoE4 mice show the stress reaction of vascular endothelium. Destruction was also seen in the matrices of perivascular nerve terminals (arrow) and perivascular cells (double asterisk). Magnification: A) 25,000; B) 5,000; C) 10,000; D) 20,000. Abbreviations used in figures: EC-Endothelial cells; N-Nucleus, VL-Vessel Lumen.
In contrast to age-matched ApoE4 mice (Fig. 5), brain microvessels from 22 months old wild-type mice were characterized by minimal changes including edema in the cytoplasm of vascular endothelium and perivascular cells and occasionally the perivascular spaces. Nearly all mitochondria showed intact morphology. ApoE4 mouse microvessels display dystrophic changes in the vascular endothelium and the presence of an amyloid-like precipitate in the perivascular area which coexists with large vacuolar degenerative structures. Microvessels with atherosclerotic changes appeared to be permanent features of ApoE4 brain microvessels. Free lipid droplets in the cytoplasm of perivascular foam cells appeared to be one of the hallmarks of ApoE4 mouse brain. The dystrophic changes, especially mitochondrial, were predominately in the cytoplasm of vascular EC.
Figure 5.

Comparative characteristics of brain microvessels from 22-month-old wild- type C57BL/6 control (A) and ApoE4 transgenic (B–D) mice. Wild-type mouse microvessels reveal minimal edema in perivascular regions and relatively few mitochondrial abnormalities. ApoE4 microvessels display dystrophic changes in the vascular endothelium (B) and an amyloid-like precipitate in perivascular areas (double arrow) that coexist with large vacuolar structures (triple arrow). Microvessels with atherosclerotic changes (C) in the perivascular area contain large foam cells (asterisk) and lipid droplets (double arrow). Large lipid droplets (single asterisk) and amyloid deposits (double asterisk) in the cytoplasm of perivascular cells (D). EC display dystrophic changes in their organelles especially in the mitochondria.
Magnification: A) 12,000; B) 8,000; C) 6,000; D) 12,000. Abbreviations used in figures: EC-Endothelial cells; N-Nucleus, VL-Vessel Lumen.
Ultrastructural features of age-associated neuronal mitochondrial changes in ApoE4 mice were similar to our previous observations in human AD (22, 38, 48, 49), yeast artificial chromosome mice overexpressing of AβPP (31), and 2-vessel occlusion model of brain hypoperfusion (39). We demonstrated that the majority of the changes in neurons affected by age and/or disease occurred in the cell body but not in axonal or dendritic regions of the neurons (39, 48, 50). Mitochondria-derived lysosomes associated with lipofuscin appeared to be the main feature of mitochondrial damage in aged ApoE4 mice (Fig. 6).
Occasionally, hippocampal neuronal cell bodies in old C57BL/6 control mice mitochondria had transitional, minimal changes in their ultrastructure, such as intra-mitochondrial edema and electron-dense matrices (data not shown). The mitochondrial abnormalities, including the presence of giant mitochondria, clusters of mitochondria with electron-dense matrices (ED), mitochondria with partially and/or completely damaged cristae, and mitochondria with membrane disruptions were present throughout hippocampal and cortical areas (Fig. 6); however, a majority of neurons showed mitochondria with intact morphology, lacking visible and/or obvious ultrastructural alterations. Double membranes surrounding lysosomal or vacuolar degenerative structures indicate that these aberrant structures derived from mitochondria (Fig. 6) (23, 31, 34, 38, 51). The ED mitochondria were close to the perinuclear region of the cell body. ApoE4 mice treated with ALCAR+LA showed significant reductions in abnormal features in the mitochondria and more normal (intact) mitochondria similar to the morphology of young ApoE4 mice. Additionally, myelin-like structures with osmiphilic granules in mitochondrial matrices were occasionally seen (data not shown).
Neurons in the hippocampus of young and especially old ApoE4 mice were typically characterized by a range of mitochondrial alterations, although some mitochondria still show intact morphology. However, mitochondrial lesions (i.e., broken cristae, edema in the matrices, and mitochondria-derived lysosomal structures surrounded by double membranes) appeared to be permanent features of these neurons. Other characteristics of old ApoE4 hippocampi were giant mitochondria and lipofuscin and/or lysosomal structures with osmiphilic electron-dense matrices that often occupied much of the neuronal cell bodies. Treated ApoE4 mice showed patterns of mitochondrial recovery similar to those seen in our previous study of aged rats; the ALCAR+LA supplementation diet not only eliminated the mitochondrial damage, but also prevented the formation of lipofuscin and/or myelin-like structures in neurons (51). The majorities of mitochondria in nearly all neurons in old treated ApoE4 animals were healthier and seldom displayed lipofuscin granules in the cell bodies. In situ hybridization data showed that the majority of the mitochondria did not exhibit a positive signal for wild and/or deleted mtDNA (data not shown). Neuronal cell bodies from treated ApoE4 mice showed significantly fewer giant mitochondria. Treated ApoE4 hippocampal neurons generally lacked mitochondrial ultrastructural abnormalities, and most of the mitochondria appeared to be intact or with few alterations.
Spatial memory
The animals were tested during a 6-day period (4 swim trials/day and 120s/trial) after having received 10 days of training (4 trials/day and 60s/trial). The first test at about 12 months of age showed no difference between wild-type and ApoE4 control (untreated) animals. The treated mice, however, required a shorter time to find the hidden platform, compared with the wild-type and untreated transgenic animals (Fig. 7). The difference was not statistically significant, though there was a trend towards improvement. In a reversal test, the treated mice also performed better than the control animals in the first 4 days (Fig. 8), though again, without achieving statistical significance. The second test was carried out when the animals were 22 months old (Fig. 9). We found a statistically significant difference in cognitive function between wild-type and ApoE4 mice, and also an improvement with treatment. The most significant effects on preventing the decline of cognitive function are shown in Figures 9 and 10. In a reversal test, the treated animals found the target platform in a much shorter time than the untreated ApoE4 mice (Fig. 8).
Figure 7.

Average escape latency for 6 days (4 trials/day and 120s/trial) in the Morris water maze test. Data are mean±SEM of 4 transgenic mice in each group and 8 wild-type mice at age of 12 months (5-months of treatment).
Figure 8.
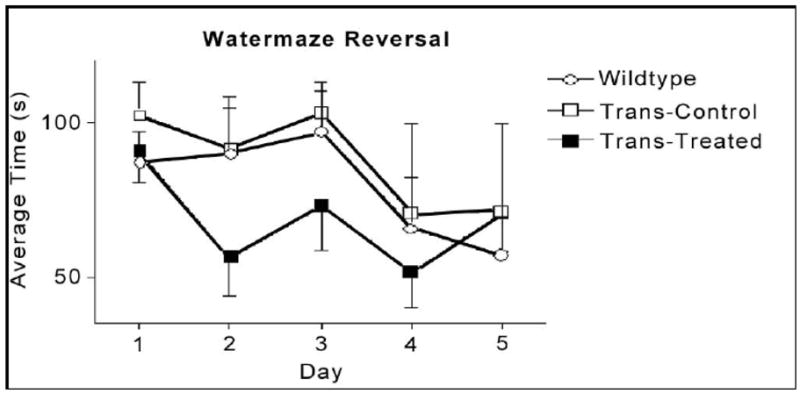
Average escape latency in a reversal test for 5 days. Data are mean±SEM of 4 transgenic mice in each group and 8 wild type mice at age of 12 months (5 months of treatment).
Figure 9.

Results of time to find hidden platform in the reversal test presented as the average of the 4 trials for 4 consecutive days. Data are mean±SEM of 3 transgenic mice in each group (one mouse in each transgenic group died by this time) and 4 wild type mice at age of 22 months (15 months of treatment).
###p<0.001 apoE4 control vs. wild type, and *p<0.05 apoE4 treated vs. apoE4 control.
Figure 10.
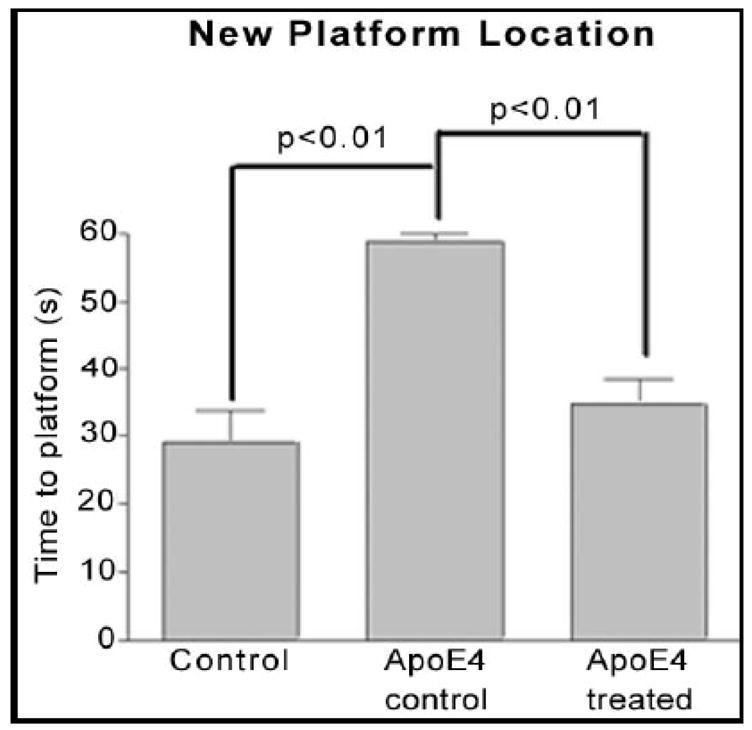
Results of time to find hidden platform in the reversal test presented as the average of the 4 days. Data are mean±SEM of 3 transgenic mice in each group and 4 wild type mice at age of 22 months (15 months of treatment).
Temporal memory
The peak performance procedure distinguished no difference between wild-type and ApoE4 mice (Figs. 10–11). Nevertheless, the 6–7-month treated mice had a higher response rate than the control animals (untreated ApoE4) mice suggesting the treatment slowed the temporal memory impairment (Fig. 10). The second test for temporary memory at the age of 22 months old was not carried out because of the extensive time requirements (about 2 months including a prior food restriction) and quick deterioration of health at old age. To avoid this problem, the authors suggest starting the peak procedure test at the age of 19–20 months (12–13 months of treatment) and then performing the Morris water maze test at the age of 21 months.
Figure 11.
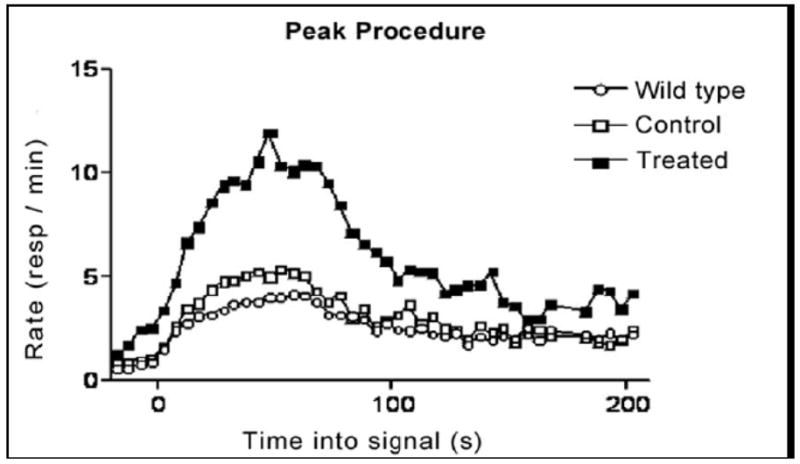
Response rates in the peak procedure test. Combining the sound signaled trials and the light signaled trials obtained during the last 20 days of the test plotted response rate. Data are mean±SEM of 4 mice in each transgenic group and 8 wild-type mice at age 12 months (6–7-months of treatment).
Discussion
The E4 allele of the human ApoE gene, which has previously been associated with increased risk of cardiovascular disease, is the best-validated susceptibility gene to date, with more wide spread effects than any other genetic factor implicated in the late-onset, sporadic form of AD (52). The E4 protein differs from those produced by the other common ApoE alleles only at two amino acids, 112 and 158. ApoE3 and ApoE2 have cysteines at one or both positions respectively whereas the ApoE4 allele encodes arginines at both positions. The change in charge alters its intra-domain interactions and molecular configuration (53) modifying its interactions with other molecules including lipids, receptors and cellular organelles. Our study for the first time demonstrates that ApoE4 reduces CBF gradually to create brain hypoperfusion when compared to WT and the differences in CBF are greatest as animals age from 6 weeks to 12 months. The structural damage of vascular wall cells, especially in their mitochondria, most likely plays a key role in the generation of reactive oxygen species (54) resulting in oxidative damage to neuronal cell bodies (22, 38, 48, 51), and therefore induces the brain pathology that appears to be a hallmark of AD (31, 39, 43, 48, 51, 55–60). Transmission electron microscopy and colloidal gold immunocytochemistry showed that structural damage in young and aged microvascular endothelium of ApoE4 animals extended to the matrices of perivascular cells, perivascular nerve terminals and to hippocampal neurons and glial cells. This evidence provides a basis for further examination of a role of ApoE4 in producing the age-dependent brain pathology seen in AD. Further examination of ultrastructural degeneration caused by aging, especially under hypoxic conditions, will likely contribute to our understanding of neurodegenerative etiology and indicates a new avenue of development for novel prophylactic and treatment strategies.
Acknowledgments
Supported by grants from the National Institute of Health, Alzheimer’s Association and NIGMS MBRS-RISE GM 60655.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.de la Torre JC. Critically attained threshold of cerebral hypoperfusion: the CATCH hypothesis of Alzheimer’s pathogenesis. Neurobiol Aging. 2000;21:331–42. doi: 10.1016/s0197-4580(00)00111-1. [DOI] [PubMed] [Google Scholar]
- 2.de la Torre JC, Stefano GB. Evidence that Alzheimer’s disease is a microvascular disorder: the role of constitutive nitric oxide. Brain Res Brain Res Rev. 2000;34:119–36. doi: 10.1016/s0165-0173(00)00043-6. [DOI] [PubMed] [Google Scholar]
- 3.Etiene D, Kraft J, Ganju N, Gomez-Isla T, Gemelli B, Hyman BT, et al. Cerebrovascular Pathology Contributes to the Heterogeneity of Alzheimer’s Disease. J Alzheimers Dis. 1998;1:119–34. doi: 10.3233/jad-1998-1205. [DOI] [PubMed] [Google Scholar]
- 4.Kalaria RN, Ballard C. Overlap between pathology of Alzheimer disease and vascular dementia. Alzheimer Dis Assoc Disord. 1999;13 (Suppl 3):S115–23. doi: 10.1097/00002093-199912003-00017. [DOI] [PubMed] [Google Scholar]
- 5.Kalaria RN. The blood-brain barrier and cerebrovascular pathology in Alzheimer’s disease. Ann N Y Acad Sci. 1999;893:113–25. doi: 10.1111/j.1749-6632.1999.tb07821.x. [DOI] [PubMed] [Google Scholar]
- 6.Mahley RW. Apolipoprotein E: cholesterol transport protein with expanding role in cell biology. Science. 1988;240:622–30. doi: 10.1126/science.3283935. [DOI] [PubMed] [Google Scholar]
- 7.Weisgraber KH, Pitas RE, Mahley RW. Lipoproteins, neurobiology, and Alzheimer’s disease: structure and function of apolipoprotein E. Curr Opin Struct Biol. 1994;4:507–15. [Google Scholar]
- 8.Poirier J, Davignon J, Bouthillier D, Kogan S, Bertrand P, Gauthier S. Apolipoprotein E polymorphism and Alzheimer’s disease. Lancet. 1993;342:697–9. doi: 10.1016/0140-6736(93)91705-q. [DOI] [PubMed] [Google Scholar]
- 9.Boyles JK, Zoellner CD, Anderson LJ, Kosik LM, Pitas RE, Weisgraber KH, et al. A role for apolipoprotein E, apolipoprotein A–I, and low density lipoprotein receptors in cholesterol transport during regeneration and remyelination of the rat sciatic nerve. J Clin Invest. 1989;83:1015–31. doi: 10.1172/JCI113943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schmechel DE, Saunders AM, Strittmatter WJ, Crain BJ, Hulette CM, Joo SH, et al. Increased amyloid beta-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90:9649–53. doi: 10.1073/pnas.90.20.9649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Holtzman DM, Bales KR, Tenkova T, Fagan AM, Parsadanian M, Sartorius LJ, et al. Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A. 2000;97:2892–7. doi: 10.1073/pnas.050004797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heeschen C, Jang JJ, Weis M, Pathak A, Kaji S, Hu RS, et al. Nicotine stimulates angiogenesis and promotes tumor growth and atherosclerosis. Nat Med. 2001;7:833–9. doi: 10.1038/89961. [DOI] [PubMed] [Google Scholar]
- 13.Walker LC, Parker CA, Pipinski WJ, Callahan MJ, Carroll RT, Gandy SE, et al. Cerebral Lipid Deposition in Aged Apolipoprotein-E-Deficient Mice. American Journal of Pathology. 1997;151:1371–7. [PMC free article] [PubMed] [Google Scholar]
- 14.Fisher A, Brandeis R, Chapman S, Pittel Z, Michaelson DM. M1 muscarinic agonist treatment reverses cognitive and cholinergic impairments of apolipoprotein E-deficient mice. J Neurochem. 1998;70:1991–7. doi: 10.1046/j.1471-4159.1998.70051991.x. [DOI] [PubMed] [Google Scholar]
- 15.Genis I, Michaelson DM. Hyperphosphorylation of Tau in Apolipoprotein E-deficient Mice. In: Fe, editor. Progress in Alzheimer’s and Parkinson’s Diseases. New York: Plenum Press; 1998. pp. 251–6. [Google Scholar]
- 16.Sparks DL. Coronary artery disease, hypertension, ApoE, and cholesterol: a link to Alzheimer’s disease? Ann N Y Acad Sci. 1997;826:128–46. doi: 10.1111/j.1749-6632.1997.tb48466.x. [DOI] [PubMed] [Google Scholar]
- 17.Fullerton SM, Shirman GA, Strittmatter WJ, Matthew WD. Impairment of the blood-nerve and blood-brain barriers in apolipoprotein e knockout mice. Exp Neurol. 2001;169:13–22. doi: 10.1006/exnr.2001.7631. [DOI] [PubMed] [Google Scholar]
- 18.Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. Jama. 1997;278:1349–56. [PubMed] [Google Scholar]
- 19.Trogan E, Choudhury RP, Dansky HM, Rong JX, Breslow JL, Fisher EA. Laser capture microdissection analysis of gene expression in macrophages from atherosclerotic lesions of apolipoprotein E-deficient mice. Proc Natl Acad Sci U S A. 2002;99:2234–9. doi: 10.1073/pnas.042683999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bales KR, Verina T, Cummins DJ, Du Y, Dodel RC, Saura J, et al. Apolipoprotein E is essential for amyloid deposition in the APP(V717F) transgenic mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A. 1999;96:15233–8. doi: 10.1073/pnas.96.26.15233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aliev G, Smith MA, Turmaine M, Neal ML, Zimina TV, Friedland RP, et al. Atherosclerotic lesions are associated with increased immunoreactivity for inducible nitric oxide synthase and endothelin-1 in thoracic aortic intimal cells of hyperlipidemic Watanabe rabbits. Exp Mol Pathol. 2001;71:40–54. doi: 10.1006/exmp.2001.2380. [DOI] [PubMed] [Google Scholar]
- 22.Aliev G, Smith MA, Obrenovich ME, de la Torre JC, Perry G. Role of vascular hypoperfusion-induced oxidative stress and mitochondria failure in the pathogenesis of Azheimer disease. Neurotox Res. 2003;5:491–504. doi: 10.1007/BF03033159. [DOI] [PubMed] [Google Scholar]
- 23.Aliev G, Smith MA, Seyidov D, Neal ML, Lamb BT, Nunomura A, et al. The role of oxidative stress in the pathophysiology of cerebrovascular lesions in Alzheimer’s disease. Brain Pathol. 2002;12:21–35. doi: 10.1111/j.1750-3639.2002.tb00419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jagust WJ, Eberling JL, Reed BR, Mathis CA, Budinger TF. Clinical studies of cerebral blood flow in Alzheimer’s disease. Ann N Y Acad Sci. 1997;826:254–62. doi: 10.1111/j.1749-6632.1997.tb48477.x. [DOI] [PubMed] [Google Scholar]
- 25.Jagust WJ, Friedland RP, Budinger TF, Koss E, Ober B. Longitudinal studies of regional cerebral metabolism in Alzheimer’s disease. Neurology. 1988;38:909–12. doi: 10.1212/wnl.38.6.909. [DOI] [PubMed] [Google Scholar]
- 26.Blain JF, Poirier J. Cholesterol homeostasis and the pathophysiology of Alzheimer’s disease. Expert Rev Neurother. 2004;4:823–9. doi: 10.1586/14737175.4.5.823. [DOI] [PubMed] [Google Scholar]
- 27.de la Torre JC. Vascular basis of Alzheimer’s pathogenesis. Ann N Y Acad Sci. 2002;977:196–215. doi: 10.1111/j.1749-6632.2002.tb04817.x. [DOI] [PubMed] [Google Scholar]
- 28.de la Torre JC. Alzheimer’s disease: how does it start? J Alzheimers Dis. 2002;4:497–512. doi: 10.3233/jad-2002-4606. [DOI] [PubMed] [Google Scholar]
- 29.de la Torre JC. Alzheimer disease as a vascular disorder: nosological evidence. Stroke. 2002;33:1152–62. doi: 10.1161/01.str.0000014421.15948.67. [DOI] [PubMed] [Google Scholar]
- 30.Lim GP, Calon F, Morihara T, Yang F, Teter B, Ubeda O, et al. A diet enriched with the omega-3 fatty acid docosahexaenoic acid reduces amyloid burden in an aged Alzheimer mouse model. J Neurosci. 2005;25:3032–40. doi: 10.1523/JNEUROSCI.4225-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aliyev A, Chen SG, Seyidova D, Smith MA, Perry G, de la Torre J, et al. Mitochondria DNA deletions in atherosclerotic hypoperfused brain microvessels as a primary target for the development of Alzheimer’s disease. J Neurol Sci. 2005;229–230:285–92. doi: 10.1016/j.jns.2004.11.040. [DOI] [PubMed] [Google Scholar]
- 32.Meguro K, Blaizot X, Kondoh Y, Le Mestric C, Baron JC, Chavoix C. Neocortical and hippocampal glucose hypometabolism following neurotoxic lesions of the entorhinal and perirhinal cortices in the non-human primate as shown by PET. Implications for Alzheimer’s disease. Brain. 1999;122 (Pt 8):1519–31. doi: 10.1093/brain/122.8.1519. [DOI] [PubMed] [Google Scholar]
- 33.Beal MF. Aging, energy, and oxidative stress in neurodegenerative diseases. Ann Neurol. 1995;38:357–66. doi: 10.1002/ana.410380304. [DOI] [PubMed] [Google Scholar]
- 34.Aliev G, Seyidova D, Neal ML, Shi J, Lamb BT, Siedlak SL, et al. Atherosclerotic lesions and mitochondria DNA deletions in brain microvessels as a central target for the development of human AD and AD-like pathology in aged transgenic mice. Ann N Y Acad Sci. 2002;977:45–64. doi: 10.1111/j.1749-6632.2002.tb04798.x. [DOI] [PubMed] [Google Scholar]
- 35.Markesbery WR. Oxidative stress hypothesis in Alzheimer’s disease. Free Radic Biol Med. 1997;23:134–47. doi: 10.1016/s0891-5849(96)00629-6. [DOI] [PubMed] [Google Scholar]
- 36.Reiman EM, Caselli RJ, Chen K, Alexander GE, Bandy D, Frost J. Declining brain activity in cognitively normal apolipoprotein E epsilon 4 heterozygotes: A foundation for using positron emission tomography to efficiently test treatments to prevent Alzheimer’s disease. Proc Natl Acad Sci U S A. 2001;98:3334–9. doi: 10.1073/pnas.061509598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Alves TC, Rays J, Fraguas R, Jr, Wajngarten M, Meneghetti JC, Prando S, et al. Localized cerebral blood flow reductions in patients with heart failure: a study using 99mTc-HMPAO SPECT. J Neuroimaging. 2005;15:150–6. doi: 10.1177/1051228404272880. [DOI] [PubMed] [Google Scholar]
- 38.Aliev G, Seyidova D, Lamb BT, Obrenovich ME, Siedlak SL, Vinters HV, et al. Mitochondria and vascular lesions as a central target for the development of Alzheimer’s disease and Alzheimer disease-like pathology in transgenic mice. Neurol Res. 2003;25:665–74. doi: 10.1179/016164103101201977. [DOI] [PubMed] [Google Scholar]
- 39.Obrenovich ME, Smith MA, Siedlak SL, Chen SG, de la Torre JC, Perry G, et al. Overexpression of GRK2 in Alzheimer disease and in a chronic hypoperfusion rat model is an early marker of brain mitochondrial lesions. Neurotox Res. 2006;10:43–56. doi: 10.1007/BF03033333. [DOI] [PubMed] [Google Scholar]
- 40.Liu J, Head E, Gharib AM, Yuan W, Ingersoll RT, Hagen TM, et al. Memory loss in old rats is associated with brain mitochondrial decay and RNA/DNA oxidation: partial reversal by feeding acetyl-L-carnitine and/or R-alpha -lipoic acid. Proc Natl Acad Sci U S A. 2002;99:2356–61. doi: 10.1073/pnas.261709299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Aliev G, Liu J, Shenk JC, Fischbach K, Pacheco GJ, Chen SG, et al. Neuronal mitochondrial amelioration by feeding acetyl-L-carnitine and lipoic acid to aged rats. J Cell Mol Med. 2009;13:320–333. doi: 10.1111/j.1582-4934.2008.00324.x. [Epub ahead of print, Mar 29, 2008] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Puchowicz MA, Radhakrishnan K, Xu K, Magness DL, LaManna JC. Computational study on use of single-point analysis method for quantitating local cerebral blood flow in mice. Adv Exp Med Biol. 2005;566:99–104. doi: 10.1007/0-387-26206-7_14. [DOI] [PubMed] [Google Scholar]
- 43.Aliev G, Puchowicz M, Xu K, Shenk J, Smith MA, Siedlak SL, et al. ApoE deficiency induces hypoperfusion that initiate age-dependent decreases in cerebral blood flow and microvessels lesions. 8th International Conference on Alzheimer’s and Parkinson’s disease; 2007; Salzburg, Austria. 2007. p. P545. [Google Scholar]
- 44.Schenk F, Morris RG. Dissociation between components of spatial memory in rats after recovery from the effects of retrohippocampal lesions. Exp Brain Res. 1985;58:11–28. doi: 10.1007/BF00238949. [DOI] [PubMed] [Google Scholar]
- 45.Morris R. Developments of a water-maze procedure for studying spatial learning in the rat. J Neurosci Methods. 1984;11:47–60. doi: 10.1016/0165-0270(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 46.Liu J, Killilea DW, Ames BN. Age-associated mitochondrial oxidative decay: improvement of carnitine acetyltransferase substrate-binding affinity and activity in brain by feeding old rats acetyl-L- carnitine and/or R-alpha -lipoic acid. Proc Natl Acad Sci U S A. 2002;99:1876–81. doi: 10.1073/pnas.261709098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Roberts S. Isolation of an internal clock. J Exp Psychol Anim Behav Process. 1981;7:242–68. [PubMed] [Google Scholar]
- 48.Hirai K, Aliev G, Nunomura A, Fujioka H, Russell RL, Atwood CS, et al. Mitochondrial abnormalities in Alzheimer’s disease. J Neurosci. 2001;21:3017–23. doi: 10.1523/JNEUROSCI.21-09-03017.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Aliev G, Castellani RJ, Petersen RB, Burnstock G, Perry G, Smith MA. Pathobiology of familial hypercholesterolemic atherosclerosis. J Submicrosc Cytol Pathol. 2004;36:225–40. [PubMed] [Google Scholar]
- 50.Aliev G, Smith MA, Obrenovich ME, Perry G. Role of Vascular Hypoperfusion-Induced Oxidative Stress and Mitochondrial Failure in the Pathogenesis of Alzheimer Disease. Neurotox Res. 2003;5:385–90. doi: 10.1007/BF03033159. [DOI] [PubMed] [Google Scholar]
- 51.Aliev G, Smith MA, de la Torre JC, Perry G. Mitochondria as a primary target for vascular hypoperfusion and oxidative stress in Alzheimer’s disease. Mitochondrion. 2004;4:649–63. doi: 10.1016/j.mito.2004.07.018. [DOI] [PubMed] [Google Scholar]
- 52.Causes, early detection, treatment, models, prevention, risk factors. 2002 [cited; Available from: http://www.alz.org/Researchers/RGP/Biological.htm.
- 53.Mahley RW, Weisgraber KH, Huang Y. Apolipoprotein E4: a causative factor and therapeutic target in neuropathology, including Alzheimer’s disease. Proc Natl Acad Sci U S A. 2006;103:5644–51. doi: 10.1073/pnas.0600549103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Perry G, Nunomura A, Hirai K, Zhu X, Perez M, Avila J, et al. Is oxidative damage the fundamental pathogenic mechanism of Alzheimer’s and other neurodegenerative diseases? Free Radic Biol Med. 2002;33:1475–9. doi: 10.1016/s0891-5849(02)01113-9. [DOI] [PubMed] [Google Scholar]
- 55.de la Torre JC, Aliev G. Inhibition of vascular nitric oxide after rat chronic brain hypoperfusion: spatial memory and immunocytochemical changes. J Cereb Blood Flow Metab. 2005;25:663–72. doi: 10.1038/sj.jcbfm.9600057. [DOI] [PubMed] [Google Scholar]
- 56.Aliev G, Perry G, Shenk J, Puchowicz M, Xu K, Siedlak SL, et al. The primary pathogenetic role of vascular hypoperfusion, and oxidative stress in Alzheimer disease. Atherosclerosis Supplements. 2007;8:105. [Google Scholar]
- 57.Aliyev A, Seyidova D, Rzayev N, Obrenovich ME, Lamb BT, Chen SG, et al. Is nitric oxide a key target in the pathogenesis of brain lesions during the development of Alzheimer’s disease? Neurol Res. 2004;26:547–53. doi: 10.1179/01610425017613. [DOI] [PubMed] [Google Scholar]
- 58.Castellani R, Hirai K, Aliev G, Drew KL, Nunomura A, Takeda A, et al. Role of mitochondrial dysfunction in Alzheimer’s disease. J Neurosci Res. 2002;70:357–60. doi: 10.1002/jnr.10389. [DOI] [PubMed] [Google Scholar]
- 59.Moreira PI, Siedlak SL, Wang X, Santos MS, Oliveira CR, Tabaton M, et al. Autophagocytosis of mitochondria is prominent in Alzheimer disease. J Neuropathol Exp Neurol. 2007;66:525–32. doi: 10.1097/01.jnen.0000240476.73532.b0. [DOI] [PubMed] [Google Scholar]
- 60.Moreira PI, Siedlak SL, Wang X, Santos MS, Oliveira CR, Tabaton M, et al. Increased Autophagic Degradation of Mitochondria in Alzheimer Disease. Autophagy. 2007:3. doi: 10.4161/auto.4872. [DOI] [PubMed] [Google Scholar]