

Abstract
Separase, an endopeptidase, plays a pivotal role in the separation of sister chromatids at anaphase by cleaving its substrate cohesin Rad21. Recent study suggests that Separase is an oncogene. Overexpression of Separase induces aneuploidy and mammary tumorigenesis in mice (Zhang et al., 2008, PNAS 105:13033). Separase is also overexpressed and mislocalized in a wide range of human cancers including breast, prostate and osteosarcoma. Currently, there is no quantitative assay to measure Separase enzymatic activity. To quantify Separase enzymatic activity, we have designed a fluorogenic assay in which 7-amido-4-methyl coumaric acid (AMC) conjugated Rad21 mitotic cleavage site peptide (Ac-Asp-Arg-Glu-Ile-Nle-Arg-AMC) is used as the substrate of Separase. We have used this assay to quantify Separase activity during cell cycle progression, and in a panel of human tumor cell lines as well as leukemia patient samples.
Keywords: Separase, Rad21, enzyme assay
Introduction
Accurate chromosomal segregation in each cell cycle relies on a conserved protein complex called cohesin and an endopeptidase called Separase. Separase is a CD clan endopeptidase (also known as Esp1 or Separin in yeast) 1-3, which is bound with its inhibitory chaperon Securin (also known as PTTG in human and Pds1 in yeast) prior to anaphase 1, 4, 5. In metaphase, ubiquitin-mediated degradation of the securin by Anaphase-Promoting Complex/Cyclosome (APC/C)-Cdc20 ubiquitin-ligase releases and activates Separase. The activated Separase proteolytically cleaves cohesin Rad21 at two sites, resulting in the removal of cohesin from chromosomes and separation of the sister chromatids 1, 6-9.
Recent studies demonstrate that overexpression of Separase induces aneuploidy (aberrant chromosome number) and mammary tumorigenesis in mice 10). siRNA mediated knockdown of Separase and Separase deficient mouse embryonic fibroblasts also results in genomic instability 11-13. Studies indicate that Separase protein is overexpressed and mislocalized in a wide range of human cancers including breast, prostate and osteosarcoma 10 (Meyer et al., Clinical Cancer Research, In Press). Furthermore, Separase overexpression strongly correlates with relapse, metastasis and lower 5 year overall survival rate in breast and prostate cancer patients (Meyer et al, Clinical Cancer Research, In Press).
Although Separase activity can be assessed by immunoblot 14, 15, there is currently no other method available to quantitatively estimate Separase enzymatic activity. We report the development of a simple, sensitive, robust quantitative assay for Separase using a fluorogenic peptide containing the Separase cleavage site of Rad21 as a substrate. This assay can be used specifically to measure Separase activity from cells and tumor specimens.
Materials And Methods
Preparation of cytostatic factor (CSF) extracts from Xenopus egg
CSF extracts were prepared as described 18. Briefly, frogs were induced to lay eggs with injection of 125U hCG to the dorsal sac. Eggs were collected the next morning and dejellied with 2% cysteine in 1× MMR (pH7.8). Eggs were packed evenly by a brief spin at 500 rpm followed by a second spin at 10,000 rpm for 10 min. Clear extract with floating membranes were collected by side puncture and collected in a fresh tube. Extracts were supplemented with CSF energy mix (40mM Phosphocreatine, 4mM ATP, 0.2mg/ml Creatine Phosphokinase), 34nM cyclinBΔ90 and rotated at room temperature for 15 min. Anaphase of the CSF was initiated by adding Ca2+ up to 0.6mM and incubating for 20 min at room temperature. Before used for activating Separase, the activity of CSF was confirmed using Securin degradation assay.
Overexpression, immunoprecipitation and activation of Separase
We obtained active Separase from human cells as previously described 14, 15. Briefly, ZZ TEV4-Separase (the construct was kindly provided by H. Zou, Southwest Medical Center, Dallas, TX) was expressed in 293T cells, and immunoprecipitated from exponentially growing cells using IgG conjugated Sepharose 6 (GE Healthcare). To release active Separase from its inhibitory chaperone Securin, we activated Separase by incubating IgG-Sepharose 6 containing ZZ TEV4-Separase with anaphase initiated Xenopus cytostatic factor (CSF) to degrade Securin at room temperature for 1h. Activated Separase protein was eluted from the beads using TEV protease (Invitrogen, Carlsbad, CA).
Separase Protease assay
Separase cleavage of Rad21 was performed as described 14 with minor modifications. Five microliters of activated Separase was mixed with 1μl recombinant Rad21 protein (expressed and purified from sf9 cells) and incubated in 20μl cleavage assay buffer (30mM HEPES-KOH pH 7.7, 50mM NaCl, 25mM NaF, 25mM KCl, 5mM MgCl2, 1.5mM ATP, 1mM EGTA) for 1hr at 37°C. The cleavage of Rad21 was detected by immunoblotting with Rad21 mAb.
For the fluorescence assays, same cleavage buffer was used in a 50μl reaction volume. Half to one microliter (∼33.5ng) of activated Separase was combined with 2μl of 10mM Rad21-MCA peptide (Ac-Asp-Arg-Glu-Ile-Nle-Arg-MCA) (Peptide International, Louisville, KY) and incubated for 3 hrs at 37°C. At the end of the reaction, fluorescence was measured by spectrofluorometry (model: Spectrafluorplus, TECAN, Mannedorf, Switzerland) at λex 405nm and λem 465nm.
Cell culture and synchronization
Mitotic cell lysates were prepared from HeLa cells according to Gaglio et al., 1995 with minor modifications 19. Briefly, HeLa cells were synchronized with a double thymidine block, and arrested at G2/M using nocodazole (40ng/ml final concentration) 6hr after release from the thymidine block. After 4hr of nocodazole arrest, mitotic cells were collected by mitotic shake off and released from nocodazole arrest. Typically 105 cells were collected at 0, 60, 80, 100, 120 and 140 min after nocodazole release. Cell lysate was prepared as described by Zhang et al.20.
Leukemic cell lines (ATCC, Manassas, VA) were cultured in RPMI 1640 supplemented with 10% bovine fetal serum (Invitrogen, Carlsbad, CA) to a density of 0.5-1.0×106 cells/ml. Ten million cells were pelleted, washed 3 times in cold PBS before making the protein lysates. Bone marrow aspirates from pediatric patients with newly-diagnosed AML were collected prior to administration of chemotherapy using an IRB-approved protocol according to institutional guidelines. Mononuclear cells were isolated using Lymphoprep (Axis-Shield, Oslo, Norway), washed twice with PBS, and 1×107 cells frozen as cell pellets at -80°C. Lysates were prepared as described previously20.
Results
Isolation and activation of Separase enzyme from human cells
Since the development of an in vitro Separase activity assay is limited by the availability of active enzyme, we used ZZ TEV4-Separase expressed in 293T cells as a source of Separase enzyme. Separase was immunoprecipitated using the IgG conjugated Sepharose 6 and eluted from the beads by cleaving the epitope using TEV protease. Because Separase is inactive when bound to its inhibitory chaperon Securin, the immunoprecipitated Separase was incubated in Xenopus egg extracts (pretreated with Ca2+ for APC activation) to degrade Securin and activating Separase. We resolved the activated Separase preparation on SDS-PAGE, and performed Coomassie staining (Figure 1A) as well as immunostaining with Separase mAb (Figure 1B) to estimate the concentration of Separase in the sample. According to the total immunoprecipitated protein loaded, and the intensity ratio of Separase bands to the total bands from the Coomassie-stained gel, the concentration of Separase was estimated to be ∼33.5ng per microliter of Separase preparation. Activated Separase was tested fore activity in a Rad21 cleavage assay (Fig. 1C), and used as an enzyme source in developing the fluoregenic assay. Activated Separase successfully cleaved full length recombinant Rad21 protein, generating expected mitotic cleavage fragments in vitro that were detected on immunoblot using Rad21 antibody (Fig 1C).
Figure 1.
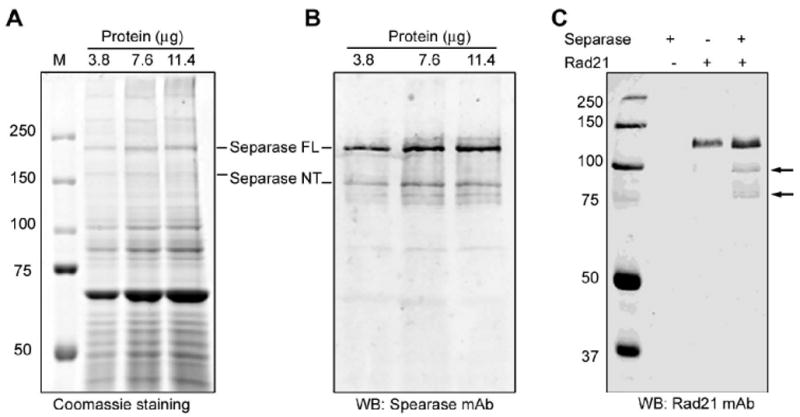
Isolation and activation of Separase. The Separase was immunoprecipitated from 293T and activated with Xenopus egg extract. (A-B). Estimation of active Separase protein concentration. Five, 10 and 15μl (equivalent to 3.8, 7.6 and 11.4μg input protein) of the activated Separase preparation were resolved on a 7% SDS-PAGE. The protein bands were visualized with Coomassie staining (A) and the Separase bands were probed with anti-Separase mAb on immunoblot (B). Full length and the autocleaved Separase bands vs. total band intensities on Coomassie gel were quantified using Imagequant software. Activated Separase protein concentration was calculated as a fraction of Separase band optical density over the total band intensities from the Coomassie gel and estimated to be ∼33.5ng/μl. (C). Immunoblot of Rad21 cleavage assay using activated Separase. The assay was performed at 37°C for 1h. The protein was resolved on 8% SDS-PAGE. Full length Rad21 and cleavage fragments (arrows) were visualized with anti-Rad21 monoclonal antibody.
Designing a synthetic fluorogenic Rad21 mitotic cleavage site peptide as Separase substrate
The specific mitotic Separase cleavage sites of cohesin Rad21 have been mapped, and it is known that active Separase cleaves Rad21 at two specific sites bearing the consensus Glu-X-X-Arg near the N-terminal and C-terminal end of the protein respectively 15. We have designed a Rad21 mitotic cleavage site peptide (Ac-Asp-Arg-Glu-Ile-Nle-Arg-MCA) conjugated to methyl coumaric acid (MCA) and tested this peptide as a substrate for Separase activity (Figure 2A).
Figure 2.
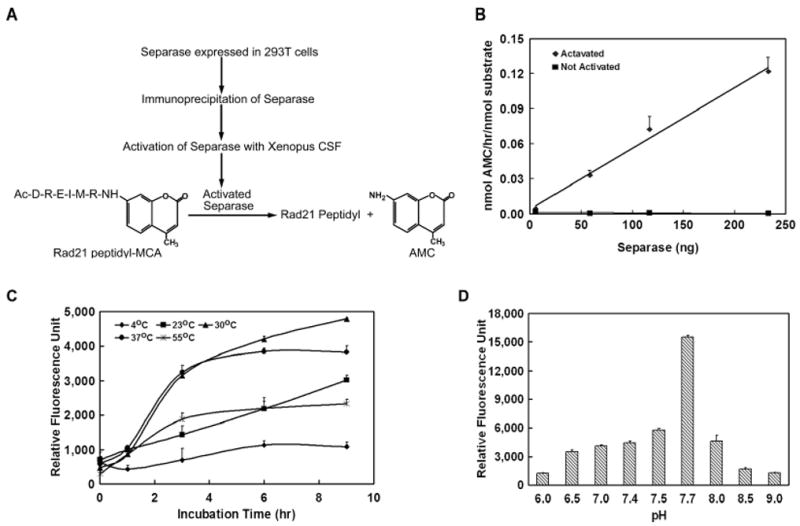
Development of Separase assay. (A). An outline of the fluorogenic assay to measure activated Separase in vitro. Activated Separase was used to cleave Rad21 peptidyl-MCA, and the amount of cleavage product was measured by fluorescence emission. (B). Effect of increasing concentration of activated Separase enzyme on the AMC release. Results obtained as relative fluorescence units after corrected for nonspecific background measure in the presence of substrate alone without the enzyme. The activity was calculated in terms of nanomole AMC released per hour per nanomole substrate (Rad21 peptidyl-MCA). ◆ = activated Separase, ▪ = not activated separase. Each data point is ±SEM of three observations. (C). Time and temperature dependence of AMC release after the Rad21-peptidyl MCA cleavage by activated Separase enzyme at 4, 23, 30, 37 and 55°C. 16.75ng of active enzyme and 2 nanomoles of Rad21 peptidyl-MCA were incubated in the presence of activated Separase enzyme at different temperature conditions over a period of 9h. Results are expressed as relative fluorescence units at indicated time points after corrected for nonspecific background measured in the absence of enzyme. Values are mean+/- SE of 3 observations. (D). AMC release after the Rad21-peptidyl MCA cleavage by activated Separase enzyme is a function of pH. Values are means +/- SE of 3 observations.
The standard curve of AMC fluorescence for the peptide-MCA showed a detectable difference in the concentration range of 11.0 to 300μM. 7-amido-4-methyl coumaric acid (AMC) fluorescence shows a concentration dependent linear increase in fluorescence at this range while the Rad21 peptide-MCA fluorescence did not increase over background (Fig. S1). This set of studies suggested that any product (AMC) formed due to the enzyme activity will be distinct and can be detected by fluorescence from the substrate once its concentration is greater than 11.0μM. Thus, any fluorescence produced in the reaction was due to AMC and not from the substrate peptide under these conditions.
Establishment of Separase activity assay using Rad21-peptidyl MCA as substrate
In a set of preliminary experiments, activated Separase was used to digest the synthetic Rad21 substrate, and fluorescence measured as the concentration of free AMC liberated, indicating cleavage of the scissile bond. Un-activated Securin-bound Separase preparation had no appreciable fluorescence activity in this assay (Figure 2B). We tested a range of enzyme and substrate concentrations to determine the minimum amount of enzyme and substrate required in a single reaction to obtain appreciable fluorescence intensity over the background. These studies indicated that a minimum of 0.5μl (16.75ng) of active enzyme (total 58μg IP protein equivalent) and 2 nanomoles Rad21-MCA peptide substrate in a 50μl reaction volume are necessary to distinguish fluorescence due to enzyme activity over the nonspecific background (data not shown).
Assay Optimization
To optimize the amount of active enzyme in a reaction, experiments were performed to determine the correlation between concentration of activated Separase and the formation of AMC. Twenty nanomoles of substrate was incubated in the presence and absence of increasing concentration of activated or inactive (Securin bound) Separase for 3h at 37°C. Fluorescence linearly correlated with activated Separase concentration with a positive correlation (r2 = 0.9894) over the range of 0-134 ng active protein. Securin-bound Separase demonstrated only background level of fluorescence with increased concentration (Figure 2B). The linear relationship between the amount of active Separase and the amount of fluorescence was used to calculate amount of active Separase in the range of 0–35,000 relative fluorescence units at excitation and emission wavelength of λex = 405nm, λem = 465nm, respectively. To produce an appreciable difference over the un-activated negative controls, a minimum of ∼33.5ng activated Separase/μl was required (Fig2B), and this was used as our standard experimental condition.
It can be noted that considerably less amount of activated enzyme preparation (∼5 fold) is required in the fluorescence assay to cleave Rad21 peptide compared to the amount necessary for cleaving the full length recombinant Rad21 in the conventional cleavage assay (Fig1C), indicating higher sensitivity of the fluorogenic assay compared to the conventional cleavage assay.
To further optimize the assay, time and temperature dependence of AMC release after the Rad21-peptidyl MCA cleavage by activated Separase enzyme were studied over a period of 9h at 4°C, 23°C, 30°C, 37°C and 55°C (Figure 2C). Although the optimal temperature for the assay was observed to be at physiological temperature of 37°C, the assay tolerated a wide range of temperature from 30°C – 55°C. The assay reaction proceeded at a slower kinetic rate at room temperature (23°C) and had no appreciable activity at 4°C (Figure 2C). Based on these observations, an incubation time of 3h at 37°C was used as standard experimental condition for subsequent studies.
Optimal cleavage activity in terms of released AMC was studied over a pH range of 6.0 – 9.0. Maximum AMC release was obtained at pH of 7.7 (Figure 2D).
Enzyme Kinetics
Activated Separase enzyme was incubated with increasing concentration of Rad21 peptidyl-MCA (100-3200μM) for a period of 3h at 37°C. AMC release was corrected for nonspecific background measured in the absence of the enzyme. The ability of Separase to cleave the synthetic peptide in vitro follows distinct Michaelis-Menten kinetics and was saturated at a substrate concentration of >1000μM Rad21-MCA (Figure 3A). We calculated a maximum velocity Vmax of 1.25 nmoles per hour and a KM of 2.93 × 10-4 M (Figure 3B).
Figure 3.
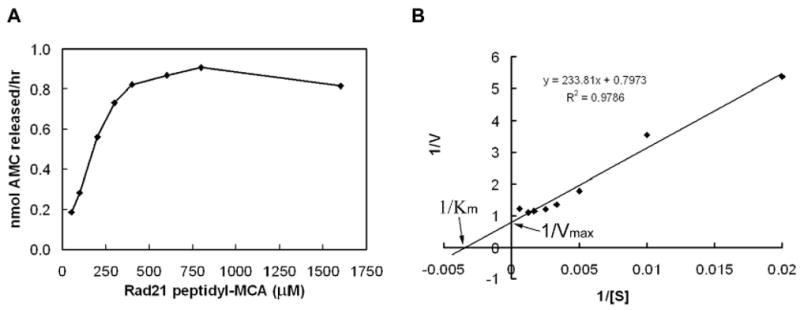
Characterization of Separase enzyme kinetics. (A). Cleavage reaction was carried out in the presence of increasing concentration of the Rad21-peptidyl MCA (50-1600μM) and the activated Separase enzyme (33.5ng/μl) at 37°C for 3h. Results are expressed as nanomole AMC release/hr after corrected for nonspecific background measured in the absence of the enzyme. Saturation kinetics is plotted as Michaelis-Menten curve. (B). Vmax and KM were calculated using Lineweaver-Burk plot.
Enzyme stability
We tested the stability of the activated enzyme using freshly prepared as well as several storage conditions, including multiple freeze-thaw cycles, to assess enzyme stability. Allowing the activated enzyme to stay for 48hrs at room temperature (23°C) reduced its activity by approximately 17% whereas activity reduced only 12% when stored at 4°C. However, storage below -20°C for a week had no significant effect on Separase activity. The activated enzyme could tolerate up to three freeze-thaw cycles without significant loss in activity (Figure S2A).
Assay variability
To assess inter-assay and intra-assay variability significant difference between the positive and negative controls was calculated using Z′ values (Fig S2B). Z′ compares the mean value of the maximum signal control to the mean value of the minimum control, and will have a higher value when (a) there is a wide separation band between maximum and minimum controls and (b) the standard deviations are low (Zhang et al. 1999). Both the substrate and enzyme alone as well as the buffer yielded Z′ values greater than 0.5 (Z'substrate only = 0.6513, Z′Separase only = 0.7141, Z′buffer = 0.7314) when compared with positive control, indicating acceptable variability.
Validation of the Separase activity assay
Specificity of the assay
To validate Separase activity assay is substrate specific, we compared Rad21-peptidyl MCA and LEHD-CHO-MCA. The latter is a Caspase 9/6 specific substrate. Incubating under similar conditions in the presence and absence of activated Separase, the caspase substrate LEHA-AMC did not produce any detectable fluorescence over the negative control, indicating Separase could not cleavage LEHA-AMC (Figure S3).
Separase activity at the metaphase to anaphase transition
To measure the Separase enzymatic activity during mitotic progression of the cell cycle, we synchronized HeLa cells with double thymidine block followed by nocodazole arrest. Separase activity significantly increased after 60 minutes of nocodazole release and then declined in a graded fashion over next 40 min and remained stable thereafter (Figure 4A). To corroborate Separase activation at the metaphase-anaphase transit with other cell cycle events, we examined the Securin and cyclinB levels in these cells at the same time points after nocodazole release (Figure 4B). As expected, Western blot analysis showed a time dependent degradation of Securin and cyclin B starting 60 min after release, and was nearly undetectable by 100 min. Therefore, the fluorogenic assay can successfully detect Separase in these synchronized cells when most of the cells are at metaphase to anaphase transition.
Figure 4.
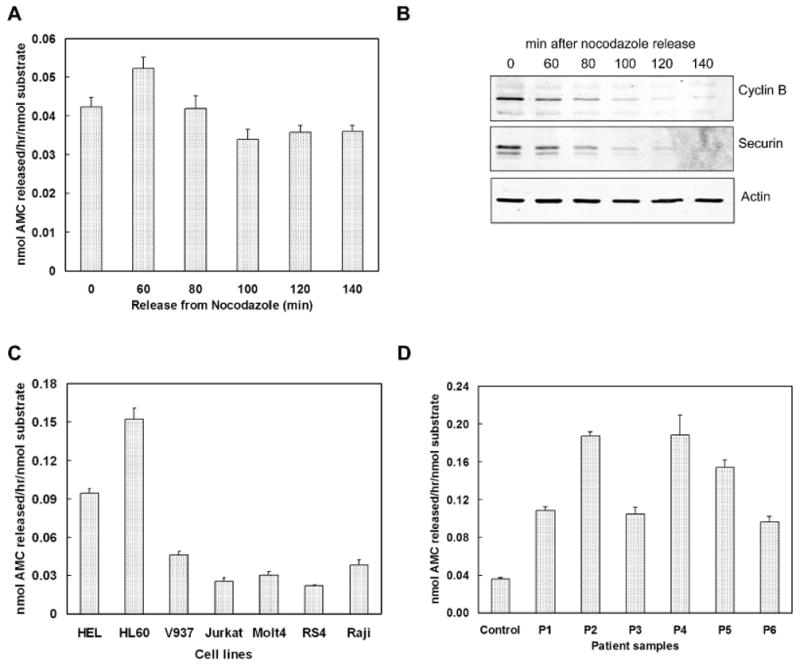
Determination of Separase activity in cells and leukemia samples. (A). Separase activity during mitotic progression. HeLa cells were synchronized with double thymidine and arrested at G2/M with nocodazole. Metaphase arrested cells were harvested for lysis at 0, 60, 80, 100, 120 and 140min after release from nocodazole block. Separase activity assay was performed with 20μg protein from each time point. Each time point is mean ± SEM of three observations. (B). Immunoblotting of Cyclin B and Securin levels in HeLa cells after release from nocodazole arrest. (C). Separase activity assay in a panel of leukemic cell lines. 20μg total protein from each line was analyzed. (D). Separase activity in pediatric AML samples. Peripheral blood cells from normal individual were used as control. 25μg total protein for each sample was analyzed.
Because we have recently demonstrated that Separase is overexpressed in many tumor samples, we used several malignant leukemic cell lines to measure Separase activity (Figure 4C). A basal level of Separase activity was expected since they are rapidly dividing cells. However, significantly higher Separase activity could be detected in two (HEL and HL60) of the seven cell lines tested, indicating increased Separase activity in acute myeloid leukemia (AML) cells. We extended our assay to quantify Separase activity in six pediatric acute myelogenous leukemia (AML) patient samples. Compared to normal individuals, the AML samples studied showed significantly greater Separase activity than the control (Figure 4D).
Discussion
Although attempts have been made to quantify Separase from cells and tissues, those approaches mostly relied on modifications of Separase protein (eg phosphorylation) and were unable to measure its enzymatic activity 16. Our fluorescence based assay is a simple, sensitive, specific, inexpensive and user friendly method. Importantly the assay is a robust assay that tolerates a wide range of physical parameters and can effectively detect a wide range of Separase activity. The KM value (a measure of the affinity of the enzyme for its substrate) of Separase was comparable to that as observed for other enzymes belonging to the same family eg. Caspase 317.
Separase has been identified as an important regulator of metaphase-anaphase transition in vertebrate cells. We have shown recently that overexpression of Separase leads to aneuploidy and tumor development in murine mammary glands 10. We also found Separase is overexpressed in a number of human cancers including breast, prostate and osteosarcoma (Meyer et al., Clinical Cancer Research, in Press). Here we show higher Separase enzymatic activity in leukemia samples. Given the tight regulatory mechanisms that lead to a very fine-tuned protease activity of the protein for a brief window during the cell cycle, it would be worth looking at the activity profile of this protease in different tumors. Separase activity profile in tumors may provide insights into the process of chromosomal instability, tumorigenesis and tumor progression. In addition to its potential clinical diagnostic and prognostic tool to assess tumor status, Separase assay can also be used to develop a high throughput screen for inhibitors of Separase. Separase assay could be an important tool for researchers as well as clinicians studying sister chromatid cohesion, aneuploidy and cancer.
Supplementary Material
Acknowledgments
This study was supported by Award Number 1RO1 CA109330 from the National Cancer Institute to D. Pati.
Abbreviations
- AMC
7-amido-4-methyl coumaric acid
- CSF
Xenopus cytostatic factor
- MCA
methyl coumaric acid
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Ciosk R et al. An ESP1/PDS1 complex regulates loss of sister chromatid cohesion at the metaphase to anaphase transition in yeast. Cell. 1998;93:1067–1076. doi: 10.1016/s0092-8674(00)81211-8. [DOI] [PubMed] [Google Scholar]
- 2.Uhlmann F, Lottspeich F, Nasmyth K. Sister-chromatid separation at anaphase onset is promoted by cleavage of the cohesin subunit Scc1. Nature. 1999;400:37–42. doi: 10.1038/21831. [DOI] [PubMed] [Google Scholar]
- 3.Uhlmann F, Wernic D, Poupart MA, Koonin EV, Nasmyth K. Cleavage of cohesin by the CD clan protease separin triggers anaphase in yeast. Cell. 2000;103:375–386. doi: 10.1016/s0092-8674(00)00130-6. [DOI] [PubMed] [Google Scholar]
- 4.Zou H, McGarry TJ, Bernal T, Kirschner MW. Identification of a vertebrate sister-chromatid separation inhibitor involved in transformation and tumorigenesis. Science. 1999;285:418–422. doi: 10.1126/science.285.5426.418. [DOI] [PubMed] [Google Scholar]
- 5.Leismann O, Herzig A, Heidmann S, Lehner CF. Degradation of Drosophila PIM regulates sister chromatid separation during mitosis. Genes Dev. 2000;14:2192–2205. doi: 10.1101/gad.176700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cohen-Fix O, Peters JM, Kirschner MW, Koshland D. Anaphase initiation in Saccharomyces cerevisiae is controlled by the APC-dependent degradation of the anaphase inhibitor Pds1p. Genes Dev. 1996;10:3081–3093. doi: 10.1101/gad.10.24.3081. [DOI] [PubMed] [Google Scholar]
- 7.Funabiki H et al. Cut2 proteolysis required for sister-chromatid seperation in fission yeast. Nature. 1996;381:438–441. doi: 10.1038/381438a0. [DOI] [PubMed] [Google Scholar]
- 8.Jallepalli PV, Lengauer C. Chromosome segregation and cancer: cutting through the mystery. Nat Rev Cancer. 2001;1:109–117. doi: 10.1038/35101065. [DOI] [PubMed] [Google Scholar]
- 9.Zhan Q et al. The gadd and MyD genes define a novel set of mammalian genes encoding acidic proteins that synergistically suppress cell growth. Mol Cell Biol. 1994;14:2361–2371. doi: 10.1128/mcb.14.4.2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang N et al. Overexpression of Separase induces aneuploidy and mammary tumorigenesis. Proc Natl Acad Sci U S A. 2008;105:13033–13038. doi: 10.1073/pnas.0801610105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Waizenegger I, Gimenez-Abian JF, Wernic D, Peters JM. Regulation of human separase by securin binding autocleavage. Curr Biol. 2002;12:1368–1378. doi: 10.1016/s0960-9822(02)01073-4. [DOI] [PubMed] [Google Scholar]
- 12.Kumada K et al. The selective continued linkage of centromeres from mitosis to interphase in the absence of mammalian separase. J Cell Biol. 2006;172:835–846. doi: 10.1083/jcb.200511126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wirth KG et al. Separase: a universal trigger for sister chromatid disjunction but not chromosome cycle progression. J Cell Biol. 2006;172:847–860. doi: 10.1083/jcb.200506119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stemmann O, Zou H, Gerber SA, Gygi SP, Kirschner MW. Dual inhibition of sister chromatid separation at metaphase. Cell. 2001;107:715–726. doi: 10.1016/s0092-8674(01)00603-1. [DOI] [PubMed] [Google Scholar]
- 15.Hauf S, Waizenegger IC, Peters JM. Cohesin cleavage by separase required for anaphase and cytokinesis in human cells. Science. 2001;293:1320–1323. doi: 10.1126/science.1061376. [DOI] [PubMed] [Google Scholar]
- 16.Gerber SA, Rush J, Stemman O, Kirschner MW, Gygi SP. Absolute quantification of proteins and phosphoproteins from cell lysates by tandem MS. Proc Natl Acad Sci U S. 2003;100:6940–6945. doi: 10.1073/pnas.0832254100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Karki P, Lee J, Shin SY, Cho B, Park IS. Kinetic comparison of procaspase-3 and caspase-3. Arch Biochem Biophys. 2005;442:125–132. doi: 10.1016/j.abb.2005.07.023. [DOI] [PubMed] [Google Scholar]
- 18.Murray A. Cell cycle checkpoints. Curr Opin Cell Biol. 1994;6:872–876. doi: 10.1016/0955-0674(94)90059-0. [DOI] [PubMed] [Google Scholar]
- 19.Gaglio T, Saredi A, Compton DA. NuMA is required for the organization of microtubules into aster-like mitotic arrays. J Cell Biol. 1995;131:693–708. doi: 10.1083/jcb.131.3.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang N et al. A handcuff model for the cohesin complex. J Cell Biol. 2008;183:1019–1031. doi: 10.1083/jcb.200801157. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.