

Abstract
Bacteriophages of the Podoviridae family use short non-contractile tails to inject their genetic material into Gram-negative bacteria. In phage P22, the tail contains a thin needle, encoded by the phage gene 26, which is essential both for stabilization and ejection of the packaged viral genome. Bio-informatic analysis of the N-terminal domain of gp26 (residues 1–60) led us to identify a family of genes encoding putative homologues of the tail needle gp26. To validate this idea experimentally and to explore their diversity, we cloned the gp26-like gene from phages HK620, Sf6, HS1, and characterized these gene products in solution. All gp26-like factors contain an elongated α-helical coiled-coil core consisting of repeating, adjacent trimerization heptads and form trimeric fibers with length ranging between about 240Å to 300Å. Gp26-tail needles display high structural stability in solution, with Tm (temperature of melting) between 85–95°C. To determine how the structural stability of these phage fibers correlates with the length of the α-helical core, we investigated the effect of insertions and deletions in the helical core. In P22 tail needle, we identified an 85-residue long helical domain, termed MiCRU (Minimal Coiled-coil Repeat Unit), that can be inserted in frame inside gp26 helical core, preserving the straight morphology of the fiber. Likewise, we were able to remove three quarters of the helical core of HS1 tail needle minimally decreasing the stability of the fiber. We conclude that in the gp26 family of tail needles, structural stability increases non-linearly with the length of the α-helical core. Thus, the overall stability of these bacteriophage fibers is not solely dependent on the number of trimerization repeats in the α-helical core.
Keywords: Podoviridae, bacteriophage P22, HK620, Sf6, HS1, tail needle gp26, protein fiber, α-helical coiled-coil
Introduction
Fibrous proteins are widespread in nature. They can be built by coiled-coil α-helices, such as those found in myosin and keratin, left-handed polyproline II-type helices, as in collagen or β-sheets, as in amyloid fibers and silks.1; 2; 3 In higher organisms, protein fibers can serve as structural material to support, sustain and reinforce skin, tendons, muscles and bones, and play a critical role in cell motility and contraction. In viruses and bacteriophages, protein fibers are often associated to the machinery responsible for host attachment and virus infection. The rod-shaped shafts of Adenovirus virions4, for instance, occupy the twelve vertices of the icosahedral capsid, where they expose a distal C-terminal knob responsible for correct trimerization and for cellular receptor binding. In bacteriophages, protein fibers are usually found protruding from the tail apparatus, where they can play a sensory function, as in phage T4 fibritin5; 6, or mediate virion attachment to the surface receptor of the host.
The tail needle of bacteriophage P22 is encoded by its gene 26, and is a well-characterized α-helical protein fiber. Gp26 forms a 240Å long trimeric coiled-coil fiber located at the distal tip of the P22 central tail axis.7; 8; 9; 10 The needle seals the portal vertex structure to trap the highly condensed genome inside the capsid.11 After injection, gp26 is found to have been released from virions suggesting a critical role of this protein in mediating P22 genome ejection into Salmonella enterica.12 The domain organization of gp26 is known from deletion analysis.13 The N-terminal 60 amino acids of the needle bind gp10 and form the plug that closes the portal vertex channel through which DNA is packaged and injected.9; 13 The ‘plugging-tip’ of the tail needle extends to the trimeric α-helical coiled-coil core, which spans the middle three quarters of gp26 length. The C-terminus of gp26 consists of a short triple β-helix connected to an inverted helical coiled-coil. This represents the distal tip of the virion tail and has been hypothesized to be involved in cell envelope penetration.8 Overall, the gp26 fiber is distinctly thinner than most known trimeric coiled-coil structures, with an average diameter of 25Å.8 This is due to the peculiar tighter twist of gp26 helices, which make a complete revolution around each other every 92 amino acids, as opposed to most coiled-coils, where a complete revolution is observed every 150–200 amino acids.8
Similar to many other helical fibers, the tail needle gp26 is characterized by remarkable structural stability. The trimeric fiber remains folded in the absence of water, or 10% SDS at room temperature and denatures in solution with an apparent midpoint of guanidine half concentration (Cm) of 6.4M.13 This is clearly explained at the three dimensional level, by the numerous contacts observed between the three gp26 protomers.8 At least three types of interactions stabilize gp26 helical core: hydrophobic contacts mediated by heptad repeats, salt bridges latching the three helices laterally, and polar buried contacts inside the helical core mediated by calcium and chloride ions.14 Heptad repeats (abcdefg) have hydrophobic residues at position a and d of one helix that fill the interface on the surface of another helix. As predicted by Crick15, hydrophobic residues protruding on the surface of an α-helix may cement into another helix as “knobs-into-holes” to form a tight interdigitation (Fig. 2B). This inter-helical mutual interaction causes heptad-based coiled-coils to spiral around each other to generate a left-handed supercoil.16 Although the most important determinant for gp26 stability is in the long α-helical core, the two domains near the C-terminus enhance the stability of the fiber, likely by knotting the helical core.13 Replacement of the C-terminal residues 141–233 with phage T4 “foldon” domain (only 27 amino acids in length; from T4 fibritin protein) preserves gp26 stability while rendering gp26 unfolding completely reversible.17
Figure 2. Sequence alignment of gp26-like tail needle homologues.
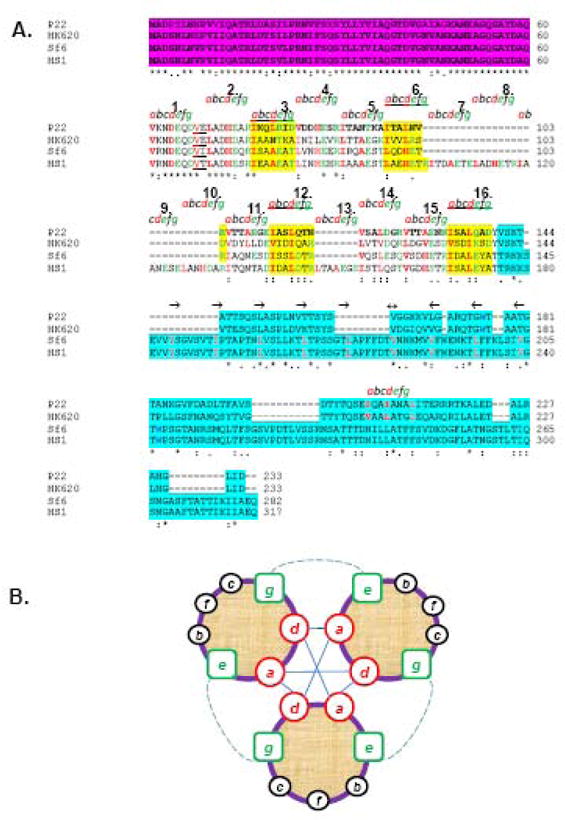
(A) The ClustalW alignment of gp26 homologues from bacteriophage P22, HK620, Sf6 and HS1. Primary sequence of gp26 can be divided into three functional regions, namely, the gp10 binding motif (in magenta), a central helical core containing heptad repeats (labeled as letter a, b, c, d, e, f and g), and the C-terminal domain involved in cell wall penetration (aqua blue). (B) Helical wheel diagram of three-stranded coiled-coils, showing the possible locations of the heptad positions in the central helical core of gp26 structures. Classically, position a and d are occupied by non-polar core residues forming hydrophobic interactions (straight line) whereas e and g are solvent exposed polar residues known to provide specificity between strands through electrostatic interactions (dotted line).
In this report, we identify and characterize a family of gp26-like tail needles in the P22-like phages. All these tail needles share similar N-terminal and trimeric α-helical coiled-coil core domains, but the C-terminal domain can be highly divergent. Using the structure of phage P22 and HS1 tail needles as a molecular framework, we have studied how insertions and deletions in the α-helical core correlate with the structural stability of the fiber.
Results
The family of bacteriophage P22-like tail needle proteins
Our analysis has identified complete or partially deleted genome sequences of numerous members of the P22-like subgroup of the Podoviridae (short tailed dsDNA bacteriophages). These sequences represent either phages that infect Enterobacteriacaea and related insect endosymbiont bacteria, or prophages we detect in related bacterial genome sequences. Thirty-three of these sequences have intact or largely intact virion morphogenetic gene clusters. The details of the bio-informatic analysis of these phage genomes will be described elsewhere, but we note here that phages P22,18 Sf6,19 CUS-3,20 φSG121; 22 and APSE-123 are prototypic of five subgroups within the P22-like phages that have, for example, very large differences among their procapsid assembly and terminase protein (S. Casjens, in preparation). The 14% identity between the coat proteins of phages P22 and Sf6, for example, demonstrates that the genes of these five types have been diverging for a very long time. In spite of this large diversity within the group of P22-like phages, all current evidence and argument strongly indicate that each of these genomes carries a cluster of fourteen syntenic and largely homologous genes that encode proteins with parallel functions in the assembly of these virions (S. Casjens, unpublished). Analysis of the proteins encoded by genes that lie in the position parallel to that of P22 gene 26 in each of these thirty-three phages showed that all have substantial sequence similarity; table I lists these gp26 homologues. Their lengths range from 118 to 317 AAs, and their sequence similarities in pair wise comparisons range from 17% to 100% identity (as aligned by DNA Strider).24 The gp26s of the phages that infect the Enterobacteriacaea are moderately diverse in sequence (maximum of about 60% different within group), while those that infect insect endosymbionts (APSE-1/APSE-2 and φSG1) form two substantially different sequence types. BLAST searches with these proteins or “parts of them” do not reveal any additional convincing homologues as of this writing (April, 2009).
Table I.
P22 tail needle homologues.
| Phage | Gene or locus_tag2 | Length (AA) | N- term type 3 | Number of heptads3 | C- term type3 | Host species |
|---|---|---|---|---|---|---|
| P221 | 26 | 233 | 1a | 11 | 1a | Salmonella enterica |
| Sari1 | SARI_02643 | 233 | 1a | 11 | 1a | S. enterica |
| Shei1 | SeHA_C0407 | 233 | 1a | 11 | 1a | S. enterica |
| Shei2 | SeHB_A0362 | 233 | 1a | 11 | 1a | S. enterica |
| Schwartz1 | SeSB_B0124 | 233 | 1a | 11 | 1a | S. enterica |
| ε341 | 9 | 233 | 1a | 11 | 1c | S. enterica |
| SE11 | 26 | 233 | 1a | 11 | 1c | S. enterica |
| Sdub1 | SeD_A0645 | 233 | 1a | 11 | 1c | S. enterica |
| ST1041 | 26 | 233 | 1a | 11 | 1c | S. enterica |
| Shad1 | SeH_A0882 | 212 | 1b | 8 | 1b | S. enterica |
| ParaC3 | SPC_1303 | 212 | 1b | 8 | 1b | S. enterica |
| Scho1 | SC060 | 212 | 1b | 8 | 1b | S. enterica |
| ST64T1 | 26 | 212 | 1b | 8 | 1b | S. enterica |
| L1 | 26 | 212 | 1b | 8 | 1b | S. enterica |
| Para1 | –4 | 212 | 1b | 8 | 1b | S. enterica |
| Para2 | SSPA2232a | 212 | 1b | 8 | 1b | S. enterica |
| Sf61 | 9 | 282 | 1c | 11 | 2 | Shigella flexneri |
| B7A1 | EcB7A_4183 | 282 | 1c | 11 | 2 | Escherichia coli |
| Flex301 | SF0222 | 282 | 1c | 11 | 2 | S. flexneri |
| FLex8401 | SFV_0226 | 282 | 1c | 11 | 2 | S. flexneri |
| Flex2457 | S0244 | 282 | 1c | 11 | 2 | S. flexneri |
| Eco82-1 | LF82_p094-140 | 317 | 1c | 16 | 2 | E. coli |
| HS1 | EcHS_A0316 | 317 | 1c | 16 | 2 | E. coli |
| HK6201 | hkbW | 233 | 1c | 11 | 1d | E. coli |
| Eco989-1 | EC55989_2607 | 282 | 1c | 11 | 2 | E. coli |
| Ferg1 | EFER_2038 | 282 | 1c | 11 | 2 | Escherichia fergusonii |
| CUS-31 | 14 | 233 | 1d | 11 | 1d | E. coli |
| UT1 | UTI89_C2647 | 233 | 1d | 11 | 1d | E. coli |
| APEC1 | APECO1_4207 | 233 | 1d | 11 | 1d | E. coli |
| EcoS88-1 | ECS88_2510 | 233 | 1d | 11 | 1d | E. coli |
| APSE-1 | P30 | 118 | 2 | 3 | 1e | Hamiltonella defensa |
| APSE-2 | P30 | 176 | 2 | 3 | 1e | H. defensa |
| φSG1 | SGPHI_0017 | 177 | 3 | 3 | 1f | Sodalis glossinidius |
Shown to be a functional phage. The others were identified as P22-like prophages in the bacterial genome sequence. Additional details of the prophages will be published later. Some prophages such as APSE-1 and APSE-2 produce virion-like particles that have not yet been propagated.
The phage gene names are shown, and the GenBank locus_tags are given for the prophages. Locus-tags allow the reader to access the gene and protein sequences.
The N-terminal and C-terminal domain boundaries were estimated as described in the legend for figure 1. The number of heptads was determined by homology with the P22 gp26 protein and by inspection of fit to the consensus.
Gene not annotated in accession number CP000026.
The P22 gp26 can be described as being made up of four domains that are linearly arrayed along the protein; the N-terminal 60 AAs form the domain that binds to gp10 of the virion and plugs the portal vertex channel (where DNA enters and exits), the middle coiled-coil rod, a β-helix section, and finally a C-terminal distal tip that is a tight trimer of α-helical hairpins. The N-terminal domain displays a short external region of pseudo-6 fold symmetry (Fig. 1A), possibly to match the hexameric quaternary structure of gp10, to which it binds.13; 25 The N-terminal domains (AAs 1-60) of the thirty-three P22 gp26 homologues form three major sequence types typified by P22, APSE-1 and φSG1; these domain types are clearly homologous, but only about 40% identical in sequence (Fig. 1C). Type 1 N-terminal domains are in turn robustly divided into four sub-types exemplified by phages P22, ST64T, HK620 and CUS-3, which are 75–85% identical to one another (the bootstrap value of group 1a and 1b separation is less than fully convincing, but the P22 and ST64T clusters form a very robust sub-group within each branch). The middle sections of the gp26 homologues have high coiled-coil prediction26 but are more diverse in sequence. The diversity and variable lengths of these central regions (they contain 3, 8, 11 or 16 heptad repeats) makes convincing alignment difficult, but there are at least five sequence types in this region that are less than about 30% identical. Finally, the C-terminal domains downstream of the heptad repeat region fall into two major sequence types, 1 and 2 typified by P22 and Sf6, respectively, whose AA sequences are not recognizably similar (Fig. 1D). The C-terminal domain of type 1 (typified by P22 gp26) is in turn divided into a number of subtypes that range from about 60 to 80% identical. Among the thirty-three proteins there are eight different combinations of N- and C-terminal domain types and within two of these (branches 1e and 2 in Fig. 1D) there are additional length differences; these findings are summarized in figure 1B. Clearly, the sequence relationships among the these proteins show a strong correspondence with the known domain structure of the P22 needle, and the two major C-terminal region types likely correspond to the different distal end shapes we observe on the purified needles by electron microscopy below.
Figure 1. Phage P22-like tail needle diversity and evolution.

(A) End-on view of N-terminal domain of P22-gp26. Three gp26 protomers fold into helical hairpins, which form a pseudo-hexameric structure with equivalent symmetry positions indicated as 1, 3, 5 and pseudo(ps)2, ps4, ps6. (B) Each rectangular box represents one of the ten gp26 tail needles with different domain combinations. Colors indicate domain “sequence types” as defined in the text and figure 1C and D below. Domain name types are indicated on the rectangle for the N- and C-terminal domains, and the number of heptads in the central coiled-coil domain (some of the latter that have identical numbers of heptads are quite different in AA sequence). Sequences of the gp26 homologous proteins can be obtained from GenBank with the gene names and locus tags in Table I. Below, a ribbon diagram is shown for the structure of P22 gp26.8 (C) Neighbor-joining tree of the N-terminal 60 AA domain of all known gp26 homologues constructed by CLUSTAL-X249 numbers above the lines are bootstrap values out of 1000 trials and numbers below are distances in changes per site. Colored boxes indicate branches (types) discussed in the text. (D) Neighbor-joining tree of the C-terminal domain of all known gp26 homologues displayed as in (C). C-terminal “domain” AA sequences were defined as everything C-terminal to the central run of tandem coiled-coil heptads.
Characterization of gp26-like tail needles in phages HK620, Sf6 and HS1
To learn more about the structural diversity among the P22-like tail needle proteins and to validate the idea that the P22 26-like genes identified bioinformatically encode tail needles, we have characterized three additional gp26 proteins. The gp26s from phages HK620 and Sf6 were chosen for study because they have substantial AA sequence differences from P22 (Figs. 1C, 1D and 2A). A number of the thirty-three P22-like “phage” genomes mentioned above are prophages in bacterial genome sequences, and among these is prophage HS1 in the genome of E. coli strain HS.27 This prophage includes HS chromosomal genes EcHS_A0272 through A0328 (S. Casjens, unpublished), and although its functionality has not been studied, it contains apparently intact homologues of all fourteen P22-like virion assembly genes. Since, the HS1 homologue of P22 gene 26 is the longest of the known 26-like genes, and yet contains N- and C-terminal domains that are very closely related to the Sf6 protein, it was also chosen for study. The HK620 and P22 gp26s are 233 AA long, and the Sf6 and HS1 homologues are 282 and 317 AAs long, respectively.
Each of the above three gp26-like genes was cloned in an expression vector plasmid, and the recombinant tail needles were expressed in E. coli and purified as described in the Experimental Procedures. In SDS-PAGE, all three gp26-like tail needles migrate as SDS-resistant oligomers at room temperature. In each case prolonged boiling in 0.1% SDS disrupts the trimeric quaternary structure yielding monomers of ~25–35kDa (Fig. 3A). The oligomeric state of the P22 gp26 was previously shown to be a trimer by sedimentation equilibrium,10 and here all four proteins were investigated by sedimentation rate analysis. In all cases, the sedimentation boundary of putative gp26-tail needles exhibited monophasic behavior, which is indicative of a single major component migrating with sedimentation coefficients of 2.5, 3.1, 3.15 and 3.1S for P22, HK620, Sf6 and HS1, respectively (Fig. 3B). Using program SEDFIT,28 conversion of the distribution of the apparent sedimentation coefficients to molecular mass for three independent runs revealed a molecular weight consistent with trimeric fibers of 83–106 kDa, which agrees well with the molecular weights expected for homotrimers. The gp26-tail needles’ trimerization was concentration-independent under the range of concentrations tested (<10 μM), suggesting a low trimerization constant.
Figure 3. Gp26-like tail needles fold into homotrimers in solution.
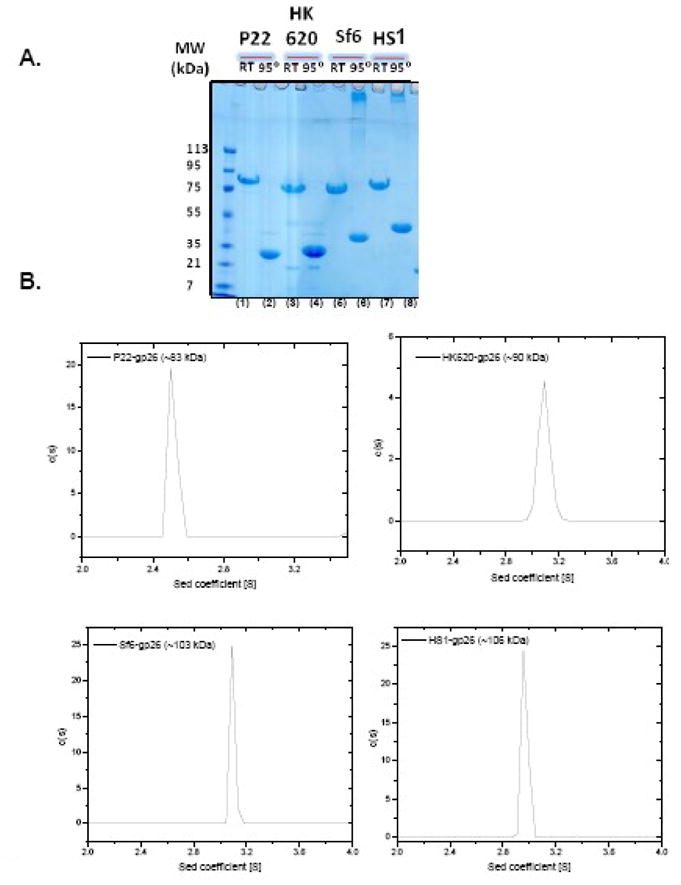
(A) SDS-PAGE analysis of purified untagged tail needles from P22, HK620, Sf6 and HS1. Prior to loading, sample was either boiled for 5 min at 95°C (denoted as “95°”) or incubated 5 min at 22°C (RT). All gp26 homologous proteins migrated as SDS-resistant trimers at RT (lane 1, 3, 5, and 7). (B) Sedimentation velocity analysis of gp26 homologues performed in 0.02 M Tris, pH 8.0, 0.15 M sodium chloride. The approximate molecular weight for gp26 homologue estimated from the sedimentation coefficient is indicated in each panel.
Secondary structure prediction with PredictProtein Server29 suggests the tail needles of phages HK620 and Sf6 contain an uninterrupted α-helical central core of 114 AA in length, which equal in length to that of P22 gp26. In contrast, the HS1 tail needle is significantly longer, with as many as 149 predicted AAs in helical conformation (Figs. 1B and 2A). In each of the proteins, the primary sequence downstream of residues 60 reveals a pattern of tandemly repeated, adjacent putative trimerization heptad sequences. We previously interpreted this region as being composed of octads8, however, reinvestigation of the gp26 primary sequence suggests that the P22 gp26 helical core can be thought as being formed by 11 contiguous AA heptads (except for two AAs between heptads 1 and 2, underlined in figure Fig. 2A). P22 and HK620 tail needles are 60% identical overall and each contain 11 such heptads. The phage Sf6 helical central region is also formed by 11 consecutive heptads, but in contrast to the α-helical enriched C-terminal domain of P22 and HK620, the Sf6 and HS1 C-terminal domains are predicted to be highly β-stranded (PredictProtein Server;29 data not shown). In HS1 the helical core is significantly longer than in P22/HK620 or in Sf6. HS1 tail needle has 16 predicted heptads, where four additional heptads (compared to P22) appear to be present between positions 103–131 in figure 2A and one additional heptad is at position 146–154. The pattern of adjacent heptads is conserved in all gp26-like tail needles, and reinforces the idea that all these proteins share a common architecture, and likely function, even though the phages that encode them infect different Gram-negative bacteria.
Gp26-like tail needles fold into elongated fibers of high structural stability
To visualize the morphology of gp26-like tail needles at the single molecule level, purified protein samples were spotted on a carbon-coated copper grid, negative stained with 1% uranyl formate and visualized by transmission electron microscopy (TEM) (Fig. 4A, B, D and E). Despite being only 75–102 kDa in MW (Table II), gp26-like tail needles share a characteristic rod-like morphology. The crystal structure of phage P22 gp26 is known from crystallographic data,8 and it forms a thin rod 240Å in length and 20–30Å in diameter, which matches well the dimensions seen on grid (Fig. 4A). HK620 tail needle is 60% identical to P22 gp26 in AA sequence and also contains 233 amino acids. At the resolution of TEM, both tail needles form thin sticks where N- and C-terminal tips are indistinguishable from each other (see topological diagram in Fig. 4C). In contrast, Sf6 and HS1 tail needles appear as longer rods (~280Å and 320Å, respectively), and have a larger diameter “knob” at one tip. Because the N-terminal and central regions of all these proteins are homologous, but the C-terminal regions of P22/HK620 and Sf6/HS1 are not recognizably similar, we suggest that the knob of the latter is formed by the C-terminal domain, which is predicted to be rich in β-strands. In P22 the distal tip is an inverted coiled-coil that connects to the helical core by a short triple β-helix,8 and sequence homology suggests that HK620 has a similar structure. We suggest that in Sf6/HS1 needles the helical core connects to the knob, which occupies the distal tip of the needle. Based on this morphology of the C-terminal domain, gp26-like tail needles can be divided into two subfamilies, whose domain organizations is shown in Fig. 4C and F.
Figure 4. Gp26-tail needles form elongated fibers.

(A, B, D, E ) Electron micrographs of negatively stained gp26 fibers from bacteriophage P22, HK620, Sf6 and HS1 showing rod-like structures. In Sf6 and HS1 a prominent globular knob-like domain is seen at one tip of the fiber. In all images scale bar corresponds to 50 nm. (C, F) Topological diagram for the two sub-classes of gp26 homologues. In (C) is the P22/HK620 topology where a short β-helix connects the main α-helical core to a short inverted C-terminal coiled-coil domain. In (F) is the putative topology of Sf6/HS1 tail needle, which presents a large globular knob, likely C-terminal of the helical core.
Table II.
Thermodynamic parameters measured for gp26-like tail needles.
| Tail Needle | Family | Host | # aa’s | MWTrimers kDa | # Heptads | Tm (°C) | Cm GdnHCl (M) |
|---|---|---|---|---|---|---|---|
| P22 | Podoviridae | Salmonella enterica | 233 | 74.2 | 11 | 85 | 6.4 |
| HK620 | Podoviridae | E. Coli | 233 | 75.2 | 11 | 90 | 7.4 |
| Sf6 | Podoviridae | Shigella flexneri | 282 | 91.8 | 11 | 88 | 7.4 |
| HS1 | Podoviridae | E. Coli | 317 | 102 | 16 | 88 | 7.4 |
| MiniHS1 | - | - | 241 | 77 | 5 | 82 | 6.8 |
Despite the different topologies, do gp26-like tail needles have comparable structural stability? To answer this question experimentally, we measured guanidine hydrochloride (GdnHCl) and temperature-induced equilibrium unfolding curves for purified untagged tail needles. As shown in figure 5, all tail needles in this study displayed highly cooperative two-state unfolding transitions, from fully folded trimers to unfolded monomers. The half denaturant concentrations and melting temperatures measured for gp26 homologues were even higher than the previously reported for P22 gp26 (6.4M/85°C).13 The HK620, Sf6 and HS1 homologues gave astonishing apparent Cm and Tm values of ~7.4M and/88–90°C, respectively (Table II). Likewise, the apparent Cm and Tm did not change over a concentration range between 1–200 μM (data not shown), suggesting a very low trimerization constant. All refolding attempts led to severe aggregation, suggesting that, similar to P22 gp26, the other tail needles unfolding transition are also strictly irreversible.
Figure 5. Gp26-tail needles have extraordinary high structural stability.

Stability of gp26-homologues was measured against GdnHCl (A) and thermal denaturation (B). All experiments were performed in 0.02 M sodium phosphate buffer (pH 8), 0.15 M sodium chloride at 20 °C. Unfolding curves were recorded by measuring changes in ellipticity intensity at 222 nm using a final protein concentration ~6 μM. All unfolding curves in (A) and (B) were found to be irreversible.
In frame extension of P22 gp26 helical core yields fibers of enhanced stability
To better understand the relationship among gp26 tail needle primary sequence, stability and quaternary structure, we investigated whether the tail needle α-helical core can be extended by in frame (with respect to the heptads) addition of defined heptad containing modules. Since P22 gp26 is the only tail needle for which a crystal structure is available, we began our analysis with this protein, which also happens to be the least stable of the studied tail needles. As previously reported, the eleven tandemly repeated heptads between residues 60–140 of P22 gp26 are critically important for the fiber self-assembly and stability.8; 13 However, the conservation of hydrophobic residues at position “a” and “d” (of an “abcdefg” heptad) varies greatly among the heptads. We name the heptads in P22 gp26 1 through 16 according to the longest tail needle, that of HS1, as indicated in Fig. 2A. While heptads 3, 6, 12 and 16 (highlighted in yellow in Fig. 2A) contain conserved Ile and Leu at positions “a” and “d”, heptads 1–2, 4–5, 11, 14–15 have different, sometimes non-hydrophobic residues at these two positions.
To begin to determine if the helical core of gp26 could be modularly extended while preserving its stability and stiffness, we noted that at the three dimensional level, heptad 3 (residues 77–83) and 16 (residues 133–139) superimpose well in P22 gp26. Between residues 77–133, each the three identical gp26 helices makes a ~90º revolution around the super-helical axis of the fiber (Fig. 6A). Likewise, both heptads 3 and 16 terminate with Asp in position “d” (Fig. 2A). Since heptads 3 and 16 are structurally superimposable, we hypothesized that P22 gp26 helical core might be successfully extended by insertion of heptads 4–16 (residues 85–140) immediately downstream of heptad 3. This region of the tail needle gp26 will be referred to as MiCRU, or Minimal Coiled-coil Repeat Unit. One MiCRU contains eight trimerization heptads (4 through 16) (underlined in Fig. 2A). The crystal structure suggests that it should be about 85Å in length and 25Å in diameter (Fig. 6A). We therefore introduced one MiCRU unit between residues 84–85, downstream of heptad 3. This insertion generates a protein (named gp26-2Mwt) that is expected to be longer than gp26 core by ~85Å, assuming the fiber remains straight. P22 gp26-2Mwt was expressed in E. coli and purified to homogeneity. On SDS-PAGE, boiled monomeric P22 gp26-2Mwt displayed slightly lower electrophoretic mobility than wild type gp26, consistent with the insertion of 55 residues (~6kDa in mass) (Fig. 6B). Like wild type gp26, gp26-2Mwt was found to be resistant to 10% SDS at room temperature (data not shown). Denaturation studies revealed that gp26-2Mwt melts irreversibly with a Tm ~5°C higher than wild type gp26 (93°C versus 88°C) (Fig. 6C) and a Cm ~7M GdnHCl, which is ~0.5M higher than the concentration of denaturant used to unfold wild type gp26 (Fig. 6D). Thus, insertion of one MiCRU inside gp26 helical core appears to be compatible with native folding of the protein and results in a protein fiber of enhanced structural stability.
Figure 6. Defining and inserting the MiCRU (Minimal Coiled-coil Repeat Unit).

(A) Ribbon diagram of P22 gp26 (pdb ID: 2poh) with the MiCRU shown in purple. (B) SDS-PAGE analysis of gp26wt and the chimeric gp26-2Mwt, which contains an additional MiCRU between residues 85–140 of gp26 helical core. The gel clearly demonstrates the increase in the molecular size of engineered gp26-2Mwt in comparison to gp26wt (lane 1 and 2 versus 3 and 4). Gp26-2Mwt fibers show a slight increase in both Gdn-HCl (C) and thermal stability (D) as compared to gp26wt.
Quantitative analysis of MiCRU insertions in P22 gp26 helical core
If insertion of one MiCRU yields a protein of enhanced stability, will insertion of additional MiCRUs further increase the structural stability of the tail needle? In other words, does the structural stabilization of gp26 α-helical core continue to increase with length? To answer these questions, we used the approach described in the previous paragraph, but using as template for MiCRU insertion the previously characterized chimera of P22 gp26 fused to the phage T4 foldon domain. This chimeric-fiber, which will be referred to as gp26-1M-F, contains gp26 residues 1–140 fused to a C-terminal foldon domain, and so contains one MiCRU repeat.17 As previously reported, gp26-1M-F has wild type stability, but unlike the wild type protein it has a completely reversible unfolding profile. This makes it an ideal subject for quantitative folding studies. Inserting a second MiCRU motif inside gp26-1M-F yielded fiber gp26-2M-F. This fiber was used as template for a third MiCRU to yield fiber gp26-3M-F. Assuming the in frame addition of MiCRU elements is tolerated and the engineered fiber remains straight, it is possible to rationally predict the length of gp26-(n)M-F fibers, as shown in Table III. Gp26-(n)M-F fibers with one, two and three tandem MiCRUs were expressed in E. coli and purified from the soluble fraction. On SDS-PAGE, all fibers were SDS resistant at room temperature (22ºC). The gp26-2M-F and gp26-3M-F fibers gave a mixture of trimer and monomer population even after boiling at 95ºC in 0.1% SDS (Fig. 7A, lane 6 and 8, respectively), suggesting an even faster refolding kinetics for longer fibers after boiling.17 It is very unlikely that the trimeric species seen on gel after boiling are covalently cross-linked, as all gp26 fibers lack cysteines in their primary sequence. Consistent with this SDS-resistance, all gp26-(n)M-F fibers were extremely stable. Wild type gp26 and gp26-1M-F denature with an apparent midpoint of guanidine half concentration (Cm) and thermal melting (Tm) of 6.4M/85°C, respectively (Fig. 7B–C, and Table III). Fibers of gp26-2M-F and gp26-3M-F both had Cm and Tm values of 7.1M and 90°C, respectively, which suggests that insertion of one MiCRU motif enhanced the Tm by ~5°C, but insertion of a third MiCRU did not further increase the stability. Thus, the stabilization of gp26 α-helical core plateaus when two or more MiCRU units are present.
Table III.
Topological and thermodynamic parameters predicted or experimentally determined for engineered fibers containing P22 gp26-(n)M-Foldon.
| Fiber | Length (Å) | # MiCRU | # Heptads | # Residues | Tm (°C) | Cm GdnHCl (M) |
|---|---|---|---|---|---|---|
| gp26wt | 240 | 1 | 11 | 233 | 85 | 6.4 |
| gp26-2Mwt | 325 | 2 | 19 | 298 | 88 | 6.8 |
| gp26-1M | 165 | 1 | 11 | 140 | 75 | 4.7 |
| gp26-1M-F | 185 | 1 | 11 | 167 | 85 | 6.4 |
| gp26-2M | 250 | 2 | 19 | 205 | 84 | 6.4 |
| gp26-2M-F | 270 | 2 | 19 | 232 | 90 | 7.1 |
| gp26-3M-F | 355 | 3 | 27 | 297 | 90 | 7.1 |
Figure 7. In frame insertion of MiCRUs in gp26-Foldon chimeric fibers.
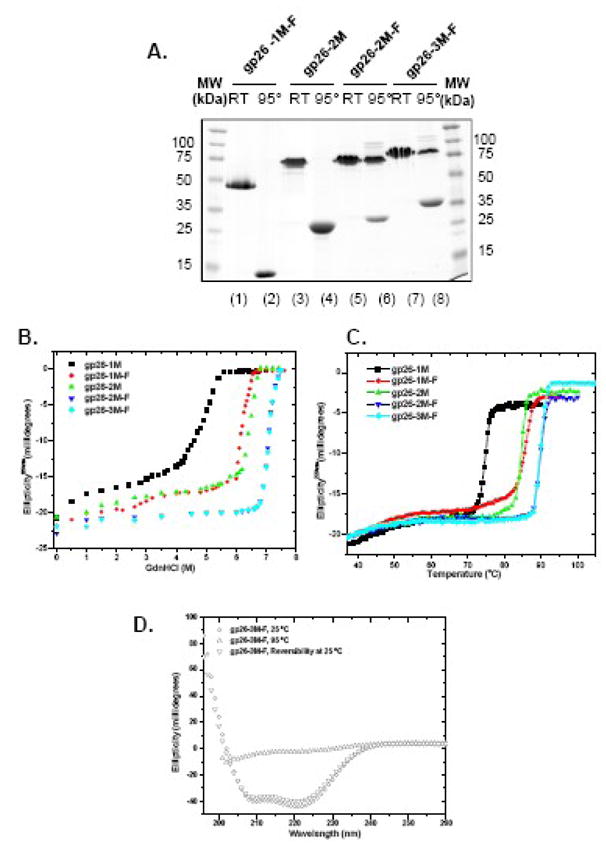
(A) SDS-PAGE analysis of gp26-MiCRU foldon fibers. “95°” and “RT” denote samples that were either boiled for 5 min or incubated 5 min at 22°C prior to be separated on gel. Stability of gp26-F-fibers was also measured independently against GdnHCl (B) and thermal (C) denaturation. (D) Reversibility of gp26-3M-F fiber was assessed by circular dichroism. The CD spectrum of fiber was measured between 197–260 nm in 20 mM sodium phosphate buffer pH 8 and 170 mM sodium chloride. Circular dichroism spectra was measured at 25°C, 95°C and after 30 minutes of cooling from 95 to 25°C (lower triangle). gp26-3M-F fiber refolded immediately after cooling. The protein concentration used in these experiments was 6μM.
Notably, the unfolding profile of longer gp26-(n)M-F fibers was also completely reversible as was seen for gp26-1M-F.17 Refolding experiments in which the longer fibers were first thermally unfolded at 95°C for five minutes and then quickly cooled back to 25°C revealed complete reversibility, as determined by monitoring the real time variations in the ellipticity at 220 nm during the thermal transition. Under these experimental conditions, refolding of thermally unfolded gp26-3M-F fiber resulted in ~99% of the initial helical signal (Figure 7D), suggesting complete refolding. Thus, the ability of the foldon domain to induce reversible unfolding of gp26 helical core is independent of the length of α-helical core.
To determine whether the engineered fibers are straight or bent, we characterized the morphology of gp26-(n)M-F fibers by using negative electron microscopy analysis (Fig. 8). The left panels A-C of figure 8 shows sections of negative stained micrographs fibers containing one, two and three MiCRUs. All three fibers are both straight and monodisperse. Projection averages (Fig. 8A–C, right panel) of each of them were computed by averaging 50 individual fibers. The improved signal to noise allowed better measurement of the length of individual fibers. The gp26-1M-F was ~180Å in length, while the single and double insertion of MiCRU resulted in an approximate length of ~265 and 355Å, respectively. These values are in agreement with the estimated insertion of one and two ~85Å MiCRU motifs, confirming the in frame addition of MiCRU units yields straight fibers whose length experimentally measured matches accurately the expected length predicted in Table III.
Figure 8. Morphology of gp26-MiCRU-foldon fibers.
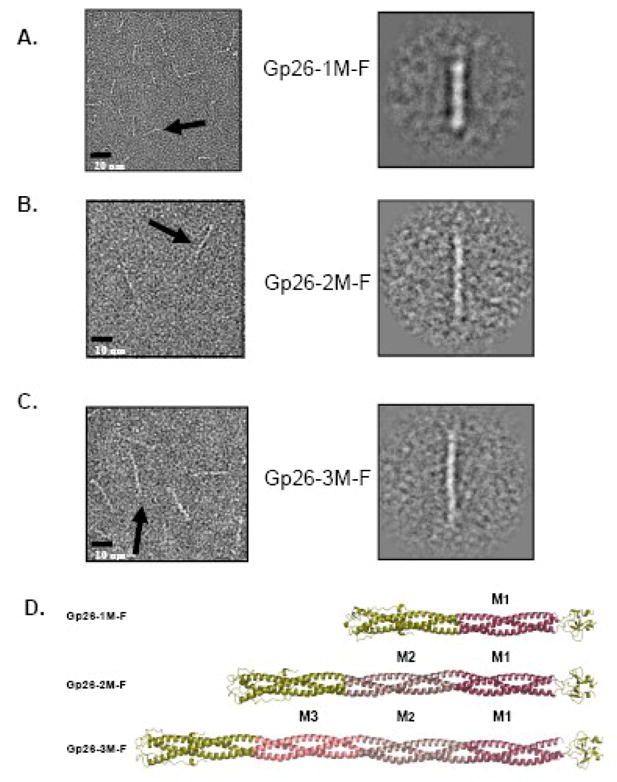
(A, B, C) Left panels show representative micrographs of purified gp26-1M-F, gp26-2M-F and gp26-3M-F stained with 1% uranyl formate. Right panels show relative projection averages for each of gp26-(n)M-F fibers. The approximate length estimated from the projection averages is 180, 265 and 355Å, for gp26-1M-F,17 gp26-2M-F and gp26-3M-F, respectively. (D) Ribbon models for gp26-1M-F, gp26-2M-F and gp26-3M-F with MiCRUs labeled as M1, M2, M3 and color-coded in red, magenta and pink, respectively.
Deletion of eleven heptads in HS1tail needle results in a fiber of high stability
To determine whether removing portions of the tail needle α-helical core would reduce the fiber stability, we turned to the phage HS1 tail needle, which contains 16 heptads and is the longest and most stable tail needle characterized in this study. We removed residues 77–153, corresponding to heptads 3 through 13 (as named in Fig. 2A), and the resulting construct was named mini-HS1, which contains only five heptads (numbers 1, 2, 14, 15 and 16) (Fig. 9A). Such a large deletion reduced the length, but surprisingly did not have a large effect on the stability of the fiber. Mini-HS1 remained trimeric on SDS-PAGE at room temperature (Fig. 9B) and denatured with an apparent midpoint of guanidine half concentration (Cm) and thermal melting (Tm) of 6.8M and 82°C, respectively (Fig. 9C–D and Table II). This stability is lower than that of the full length HS1 fiber (~approximately 8°C drop in Tm), but is comparable to wild type P22 gp26, which contains 11 heptads. Thus, removing nearly three quarters of phage HS1 α-helical core (11 of 16 heptads) only slightly reduced the fiber’s structural stability. This suggests the stability of the HS1 fiber is greatly influenced by a minimum of five heptads (1–2 and 14–16) as well as by the C-terminal knob domain, which is highly enriched in β-sheets.
Figure 9. Mini-HS1 tail needle retains high structural stability.
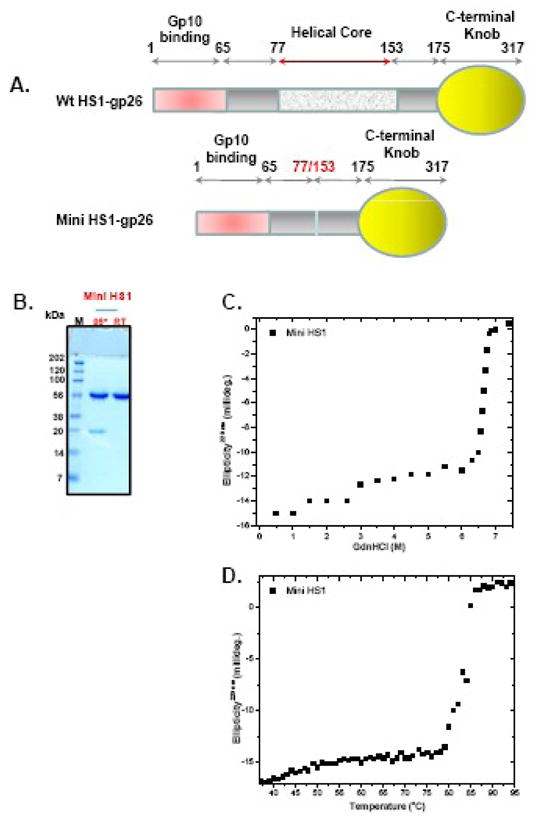
(A) Topological representation of HS1 gp26 and mini-HS1, which lacks residues 77–153 of the helical core. (B) SDS-PAGE analysis of MiniHS1 fiber. (C-D) Gdn-HCl and thermal denaturation of mini-HS1 tail needle measured using a final protein of ~ 6μM.
Discussion
α-coiled-coil structures in nature
α-helical coiled-coil structures are widespread in nature. It has been estimated that approximately 3–5% of all amino acids in proteins exist in a coiled-coil conformation.30 Helical domains hidden in the hydrophobic core are important for the native folding of certain proteins; surface exposed helical domains commonly function as a protein-protein interaction scaffold to assemble macromolecular complexes. In eukaryotic cells, the cytoskeleton is composed in large part by α-helical fibrous proteins, which act as mechanic stress absorbers and allow for motility.31; 32 Similarly, viruses and bacteriophages use helical fibers exposed on the surface of the virion to mediate essential interactions with the host cell surface. For instance, the helical core of influenza haemagglutinin forms an ~80Å long metastable stem at neutral pH, that extends into a 135Å coiled-coil rod at pH 5 to promote cell entry and membrane fusion.33; 34; 35; 36 Another well characterized example, fibritin, is a ~530Å trimeric segmented coiled-coil protein that forms the “whiskers” of bacteriophage T4 and is thought to function as a rudimentary environment-sensing device.5; 6 In biotechnology, isoleucine zippers (coiled-coils) derived from GCN4 have been extensively used in biology as oligomerization domain to induce dimerization of exogenous proteins.37 Likewise, α-helical peptide or protein building block containing heptad motifs have been widely exploited in synthetic biology for the de novo design of self assembling units (or tectons), which can interact to build self-assembled units and potentially functional assembly or systems.38 Typically, such repeating motifs yield elongated and stable fibers, which are difficult to control in polymerization and therefore length. Thus, understanding the molecular determinants for the assembly and thermodynamic stability of fibrous proteins is an important goal in structural biology, which has potential applications in protein engineering and nanostructures. In this study, we sought to expand the characterization of fibrous helical proteins by analyzing bacteriophage proteins related to phage P22 tail needle gp26. In particular, we investigated whether the topology and structure (and therefore function) of the phage P22 tail needles is conserved in other phages related to P22 that infect other Gram-negative bacteria.
Characterization of the gp26 family of tail needles
Initial adsorption of the P22 virions to the Salmonella surface is mediated by binding of the tailspike (gp9) to the O-antigen polysaccharide portion of the surface lipopolysaccharide.39; 40; 41 The tailspike protein cleaves the polysaccharide, and the virion somehow works its way down to the surface of the outer membrane (perhaps by a Brownian ratchet type mechanism).12 The role of gp26 in P22 adsorption and DNA injection is not known, but because its position extending from the virion places its distal tip further from the head than the tailspike, the C-terminus of P22 gp26 has been proposed to make the first contact with and perhaps penetration of the host cell surface.8; 9 During the injection process the gp26 needle is released from the virion to open the channel for DNA release.42 It is not known whether the distal tip of gp26 makes a specific contact with some feature of the cell surface, and although no evidence currently exists for such a contact, the existence of a “secondary” receptor for gp26 remains an attractive possibility. It is therefore not unreasonable to speculate that the distal tail needle domain could be the part of gp26 that makes this contact. This idea seems consistent with the observation that the distal domains of the gp26 needle are present as two different apparently unrelated domains, since different hosts could have different surface features. We used bioinformatic analysis to identify thirty-three sequenced homologues of P22 gp26, and it is interesting to note that C-terminal domain types 1a, 1b and 1c are Salmonella phages while types 1d and 2 are limited to Escherichia and Shigella species (Table I). It is known that the Salmonella phage P22 can infect E. coli that carries the S. enterica serovar Typhimurium O-antigen,43 so if essential P22 gp26 receptors exist they appear not to be restricted to Salmonella.
The N-terminal domain of phage P22 gp26 was previously shown to serve as a specific capsid binding domain, essential to plug the portal vertex channel in the phage P22 virion.13 This event occurs as the last step in P22 morphogenesis and is essential to stabilize the newly packaged DNA.11 We isolated the genes encoding three additional representative tail needles and demonstrated that each of their encoded proteins form trimeric elongated fibers in solution that are characterized by conserved heptad repeats and extreme structural stability. The four gp26-like tail needles we studied define two subfamilies, the P22/HK620 and Sf6/HS1 tail needles. The latter two present a globular C-terminal knob and are predicted to adopt a β-stranded structure. Although the moiety downstream of the helical core appears to be highly divergent, the structure and folding of the α-helical core is almost certainly fundamentally similar in all tail needles. The helical core consists of adjacent, repeated heptads. Only a limited number of heptads, typically at position 3, 6, 12, 14 and 16 have conserved hydrophobic residues at position “a” and “d” (Fig. 2). The other heptads have at least one hydrophobic residue at position either at position “a” or “d”. The repeated nature of the helical core promotes self assembly at very low concentration, suggesting an early assembly of tail needle at the translational level. In vitro all needles are characterized by extreme structural stability. The previously identified phage P22 gp26 is actually the least stable needle fiber, as compared to its homologues in HK620, HS1, and Sf6. Interestingly, HK620, the most similar tail needle to gp26, with nearly 60% sequence identity is also the most stable. Its apparent Tm is ~ 5ºC higher than in P22 (90 versus 85ºC), although the high degree of similarity makes it impossible to conclusively determine what makes this tail needle so stable. In the case of longer HS1 and Sf6, both tail needles present helical cores followed by a highly conserved knob. This prompted us to a second question: how does the length of gp26-like tail needle helical core correlate with its structural stability?
Stability versus length of the α-helical core
In P22 gp26 the symmetric distribution of heptads bearing hydrophobic residues at position “a” and “d” provided a useful framework to identify a “Minimal Coiled-coil Repeat Unit”, or MiCRU. This ~55 residue α-helical module contains three identical gp26 helices, which in the structure make roughly half a revolution around the super-helical axis of the fiber.8 One MiCRU was first inserted inside the wild type P22-tail needle and later inside the gp26-foldon chimera (construct gp26-1M-F), which we previously reported to fold into a stable, self-refolding fiber.17 Our data indicates that insertion of one MiCRU increases the structural stability of gp26 fibers by approximately 5ºC as compared to gp26wt (90 versus 85ºC, Table III), which is comparable to the increased stability measured for the HK620 tail needle. Likewise, all gp26-n(M)-F fibers were morphologically straight when visualized by electron microscopy, ruling out the local misfolding of longer chimera. However, insertion of an additional MiCRU did not further increase the stability of the tail needle. The Tm and Cm for gp26-3M-F, the longest fiber in our study, were identical to that of gp26-2M-F (Table III). Remarkably, all gp26-chimeras fused to a C-terminal foldon domain unfolded reversibly irrespective of the length of the α-helical core. This data supports the idea of a “threshold stability” in P22-gp26. In our study, the stability of the fiber increased linearly with the number of trimerization heptads from 11 to 19, in gp26wt and gp26-2M-F, respectively. Insertion of additional 8 heptads in gp26-3M-F had no measurable effect on stability. The concept of a “threshold stability” in α-helical coiled coils is also supported by theoretical studies.44; 45 Holtzer and Skolnick simulated the stability of coiled coils built by the consensus repeat sequence LEALEGK (abcdefg). They predicted that the stability of a LEALEGK-repeated coil coiled structure increases more dramatically with the number of repeated heptads at shorter chain lengths, and diminishes as the polypeptide chain gets longer. This suggests that the 19 heptads in gp26-2M-F may represent the threshold number of repeats necessary to stabilize P22-gp26 helical core. Although plausible, this is not a general rule, as HK620 tail needle, which has 11 heptads and is 60% identical in primary sequence to P22-gp26 is also the most stable tail needle in our study, with Tm and Cm identical to those of gp26-2M-F. Thus, variation in amino acid sequences must affect the structural stability of gp26 tail needles at least as much as the number of repeated heptads in the α-helical core.
A converse experiment was attempted with the longest tail needle from phage HS1 in this study. We generated a mini-HS1 by removing residues 77–153. This shorter tail needle lacks nearly three quarters of the α-helical core, but still melts irreversibly with Cm/Tm of 6.8M/82°C. This data supports the idea that a small number of repeats (5 in this case) may be sufficient to provide the enthalpic stabilization necessary to overcome the entropy of fixing residues in the helical conformation and fold into a trimeric fiber.45 Interestingly, two of the gp26 homologues identified in this study, from phage APSE-2 and φSG1, have only three predicted heptads in the α-coiled coil core (Table I). Although the stability of these tail needles has not been determined experimentally, it is conceivable that these homologues also fold into trimeric stable fibers.
We conclude that in the gp26-family of tail needles, the structural stability of the trimeric fiber is not solely dependent on the number of repeated heptads conserved in the helical core. Thus, the overall stability of gp26-fibers cannot be considered as an additive property, linearly increasing with the number of inter-molecular contacts between the three gp26-protomers. Other factors contribute and modulate the stability of the fiber, namely the nature of the C-terminal domain downstream of the helical core (e.g. the presence of a knob in HS1/Sf6 versus a coiled-coil repeat in P22/HK620); a minimal set of heptads in the helical core (5 in HS1), as well as variation in amino acid sequences. All these factors synergize to stabilize the fiber, in a way that, at least at this stage, is nonlinearly dependent on the simple length of the helical core and number of trimerization heptads.
Materials and Methods
Plasmids and DNA constructs
Phages Sf6 were the kind gifts of Dr. Renato Morona and Dr. A. John Clark, respectively, and E.coli HS was a gift of Dr. James Nataro. Gp26-homologue genes were amplified by PCR from total virion extracts for phages HK620, HS1 and Sf6. All tail needle genes were ligated between the XbaI and HindIII restriction sites of the vector pMal-c2e (New England Biolabs) (plasmid pMal-gp26), which expresses gp26 fused to an N-terminal maltose binding protein (MBP).17 A PreScission Protease cleavage site was engineered between the MBP and gp26 (plasmid pMal-PP-gp26).17 The DNA sequence encoding the MiCRU domain was obtained by amplifying residues 85–140 from wild type gp26 but changing tyrosine 140 to valine. To introduce MiCRU units inside gp26 helical core, the MiCRU gene was blunt ligated inside the pMal-PP-gp26 plasmid linearized by long PCR between residues V84 and D85. Similarly, mini-HS1 tail needles were generated by splicing the DNA coding region between R76 and I154, generating a shorter construct. All constructs generated in this study were entirely sequenced to ensure the correctness of the DNA sequence.
Protein Expression and Purification
All gp26-homologues were overexpressed in the E. coli strain BL21 (pLysE).46 For expression cells were grown at 37°C to an optical density of A600 = 0.6, and induced for 16h at 22°C with 0.5 mM isopropyl 1 thio-β-D-galactopyranoside (IPTG; Sigma-Aldrich, USA). Cells were lysed by sonication in lysis buffer (250 mM NaCl, 20 mM TrisHCl pH8.0) and MBP-fusion protein fibers were purified by sequential passages over amylose-agarose (New England Biolabs). Fusion proteins were digested with PreScission Protease in 170mM NaCl, 20 mM TrisHCl, pH 8.0 and 5 mM β–mercaptoethanol. Typically one liter of E. coli yielded about 5 mg of pure protein, which was concentrated using an Amicon ultra centrifugal filter device (Millipore) with a 30,000 Da molecular weight exclusion limit. The concentrated protein was applied to a Superdex 200 gel filtration column (GE Healthcare) pre-equilibrated in gel filtration buffer (170 mM NaCl, 20 mM sodium phosphate buffer pH 8.0). Elution fractions were analyzed with SDS-PAGE, confirming a purity of >95%. The concentration of all recombinant engineered proteins used in this study was determined spectrophotometrically at 280 nm using theoretical absorption coefficients calculated from the protein primary sequence. In the SDS-resistance assay, 20 μl of gp26-MiCRU foldon fibers at ~1 mg/ml were incubated either at room temperature (22°C) or 95°C for 5 min. Thereafter, samples were left at room temperature (22°C) for 10 min to allow refolding. Laemmli sample buffer was added to the proteins,47 followed by electrophoretic separation on 12.5% SDS-PAGE.
Analytical ultracentrifugation experiments
Sedimentation velocity analysis of gp26 homologues was performed in 0.02 M Tris, pH 8.0, 0.15 M sodium chloride at an initial concentration of 1 mg/ml. 450μL of each gp26 homologue and 400μL of reference buffers were loaded into separate compartments of a 12mm path-length Epon centerpiece cell. Samples were analyzed at 10 °C and 50,000 rpm in a Beckman Optima XL-A Analytical Ultracentrifuge (AUC). The sedimentation coefficient distribution was calculated with a continuous c(s) distribution model with the program SEDFIT (Peter Schuck, NIH http://www.analyticalultracentrifugation.com/download.htm). The fitted distribution of the sedimentation coefficient calculated for gp26 homologues corresponds well with an estimated molecular weight of trimers (Table II).
Negative Stain Transmission Electron Microscopy (TEM)
For negative stain electron microscopy, homologues of gp26 and gp26-MiCRU-Foldon fibers at a concentration of ~ 1 μg/ml were applied to glow discharged carbon coated copper grids and stained with 1 % uranyl formate. Grids were examined in a Joel JEM-2100 transmission electron microscope operated at 200 kV.17 Images were recorded on a charge coupled device (CCD, TVIPS F415MP) at an electron optical magnification of 100,000× and a defocus of −1.5 μm, placing the first zero of the contrast transfer function (CTF) at ~ 0.05 Å−1. The pixel size on the specimen scale was 1.14 Å and was calibrated with catalase two dimensional crystals. Data sets of ~100 images for each specimen were manually selected using the EMAN program boxer. Images were CTF corrected using ctfit, band pass filtered to remove low and high spatial frequencies and a circular mask was applied. Reference-free image alignment and classification was performed using IMAGIC-5 software (Image Science GmbH, Berlin, Germany).
Spectroscopic Techniques
Relative denaturant and thermal stabilities of homologues and engineered fibers were analyzed using equilibrium unfolding studies. Equilibrium unfolding studies for gp26 homologues and gp26-1M, gp26-1M-F, gp26-2M, gp26-2M-F and gp26-3M-F were carried out by monitoring variations in ellipticity at 222 or 220 nm as a function of guanidine hydrochloride (GdnHCl) or temperature as previously described.13 All spectroscopic measurements were performed with a final protein concentration of 6μM in 20mM sodium phosphate (pH 8.0) and 170mM NaCl. Circular dichroism (CD) spectra in the far-UV region (197–260 nm) were recorded using an AVIV 62A DS spectropolarimeter equipped with a Neslab CFT-33 refrigerated recirculator. A rectangular quartz cuvette with a pathlength of 0.1 cm was used to perform the CD measurements. Low-noise CD spectra were obtained by averaging three scans. Reversibility of unfolding by CD was monitored by recording variations in ellipticity at 220nm as a function of temperature in 1°C increments. After each increment, samples were equilibrated for 60s and CD spectra recorded with an integration time of 15s. Slow cooling to 25°C followed by a second run was carried out to check the reversibility of unfolding. The apparent midpoint of the unfolding transition temperature, Tm, and half denaturation concentration, Cm, were determined by fitting the experimental thermal profile to standard two-state unfolding equations.48 We used an in-house linear least squares fitting routine programmed in ORIGIN 6.1 (OriginLab Corporation, USA) to determine the best-fit curve and to calculate the resulting parameters. We defined Tm as the temperature or Cm as GdnHCl concentration at which 50% of the protein is unfolded. Although this thermodynamic analysis is strictly derived for two-state, reversible folding processes, it is also used to determine relative stability temperatures in irreversible processes,48 and we apply it in this manner.
Acknowledgments
We are indebted to Dr. Stephan Wilkens for assistance in collecting and analyzing electron microscopy data. We thank Dr. Adam S. Olia for stimulating discussions. We thank Dr. James Nataro for E. coli HS and Dr. A. John Clark for phage HK620. This work was supported by NIH grants 1R56AI076509-01A1 and RO1 AI074825 to GC and SRC, respectively.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Parry DA, Fraser RD, Squire JM. Fifty years of coiled-coils and alpha-helical bundles: a close relationship between sequence and structure. J Struct Biol. 2008;163:258–69. doi: 10.1016/j.jsb.2008.01.016. [DOI] [PubMed] [Google Scholar]
- 2.Scheibel T. Protein fibers as performance proteins: new technologies and applications. Curr Opin Biotechnol. 2005;16:427–33. doi: 10.1016/j.copbio.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 3.Shoulders MD, Raines RT. Collagen Structure and Stability. Annu Rev Biochem. 2009 doi: 10.1146/annurev.biochem.77.032207.120833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van Raaij MJ, Mitraki A, Lavigne G, Cusack S. A triple beta-spiral in the adenovirus fibre shaft reveals a new structural motif for a fibrous protein. Nature. 1999;401:935–8. doi: 10.1038/44880. [DOI] [PubMed] [Google Scholar]
- 5.Tao Y, Strelkov SV, Mesyanzhinov VV, Rossmann MG. Structure of bacteriophage T4 fibritin: a segmented coiled coil and the role of the C-terminal domain. Structure. 1997;5:789–98. doi: 10.1016/s0969-2126(97)00233-5. [DOI] [PubMed] [Google Scholar]
- 6.Conley MP, Wood WB. Bacteriophage T4 whiskers: a rudimentary environment-sensing device. Proc Natl Acad Sci U S A. 1975;72:3701–5. doi: 10.1073/pnas.72.9.3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berget PB, Poteete AR. Structure and functions of the bacteriophage P22 tail protein. J Virol. 1980;34:234–43. doi: 10.1128/jvi.34.1.234-243.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Olia AS, Casjens S, Cingolani G. Structure of phage P22 cell envelope-penetrating needle. Nat Struct Mol Biol. 2007 doi: 10.1038/nsmb1317. [DOI] [PubMed] [Google Scholar]
- 9.Lander GC, Tang L, Casjens SR, Gilcrease EB, Prevelige P, Poliakov A, Potter CS, Carragher B, Johnson JE. The Structure of an Infectious p22 Virion Shows the Signal for Headful DNA Packaging. Science. 2006 doi: 10.1126/science.1127981. [DOI] [PubMed] [Google Scholar]
- 10.Andrews D, Butler JS, Al-Bassam J, Joss L, Winn-Stapley DA, Casjens S, Cingolani G. Bacteriophage P22 tail accessory factor GP26 is a long triple- stranded coiled-coil. J Biol Chem. 2005;280:5929–33. doi: 10.1074/jbc.C400513200. [DOI] [PubMed] [Google Scholar]
- 11.Strauss H, King J. Steps in the stabilization of newly packaged DNA during phage P22 morphogenesis. J Mol Biol. 1984;172:523–43. doi: 10.1016/s0022-2836(84)80021-2. [DOI] [PubMed] [Google Scholar]
- 12.Israel V. A model for the adsorption of phage P22 to Salmonella typhimurium. J Gen Virol. 1978;40:669–73. doi: 10.1099/0022-1317-40-3-669. [DOI] [PubMed] [Google Scholar]
- 13.Bhardwaj A, Olia AS, Walker-Kopp N, Cingolani G. Domain organization and polarity of tail needle GP26 in the portal vertex structure of bacteriophage P22. J Mol Biol. 2007;371:374–87. doi: 10.1016/j.jmb.2007.05.051. [DOI] [PubMed] [Google Scholar]
- 14.Olia AS, Casjens S, Cingolani G. Structural plasticity of the phage P22 tail needle gp26 probed with xenon gas. Protein Sci. 2009;18:537–548. doi: 10.1002/pro.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Crick FHC. The packing of -helices: simple coiled-coils. Acta Crystallogr. 1953;6:689–697. [Google Scholar]
- 16.Lupas AN, Gruber M. The structure of alpha-helical coiled coils. Adv Protein Chem. 2005;70:37–78. doi: 10.1016/S0065-3233(05)70003-6. [DOI] [PubMed] [Google Scholar]
- 17.Bhardwaj A, Walker-Kopp N, Wilkens S, Cingolani G. Foldon-guided self- assembly of ultra-stable protein fibers. Protein Sci. 2008;17:1475–85. doi: 10.1110/ps.036111.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pedulla ML, Ford ME, Karthikeyan T, Houtz JM, Hendrix RW, Hatfull GF, Poteete AR, Gilcrease EB, Winn-Stapley DA, Casjens SR. Corrected sequence of the bacteriophage p22 genome. J Bacteriol. 2003;185:1475–7. doi: 10.1128/JB.185.4.1475-1477.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Casjens S, Winn-Stapley DA, Gilcrease EB, Morona R, Kuhlewein C, Chua JE, Manning PA, Inwood W, Clark AJ. The chromosome of Shigella flexneri bacteriophage Sf6: complete nucleotide sequence, genetic mosaicism, and DNA packaging. J Mol Biol. 2004;339:379–94. doi: 10.1016/j.jmb.2004.03.068. [DOI] [PubMed] [Google Scholar]
- 20.King MR, Vimr RP, Steenbergen SM, Spanjaard L, Plunkett G, 3rd, Blattner FR, Vimr ER. Escherichia coli K1-specific bacteriophage CUS-3 distribution and function in phase-variable capsular polysialic acid O acetylation. J Bacteriol. 2007;189:6447–56. doi: 10.1128/JB.00657-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Clark AJ, Pontes M, Jones T, Dale C. A possible heterodimeric prophage- like element in the genome of the insect endosymbiont Sodalis glossinidius. J Bacteriol. 2007;189:2949–51. doi: 10.1128/JB.00913-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Toh H, Weiss BL, Perkin SA, Yamashita A, Oshima K, Hattori M, Aksoy S. Massive genome erosion and functional adaptations provide insights into the symbiotic lifestyle of Sodalis glossinidius in the tsetse host. Genome Res. 2006;16:149–56. doi: 10.1101/gr.4106106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van der Wilk F, Dullemans AM, Verbeek M, van den Heuvel JF. Isolation and characterization of APSE-1, a bacteriophage infecting the secondary endosymbiont of Acyrthosiphon pisum. Virology. 1999;262:104–13. doi: 10.1006/viro.1999.9902. [DOI] [PubMed] [Google Scholar]
- 24.Douglas SE. DNA Strider. An inexpensive sequence analysis package for the Macintosh. Mol Biotechnol. 1995;3:37–45. doi: 10.1007/BF02821333. [DOI] [PubMed] [Google Scholar]
- 25.Olia AS, Bhardwaj A, Joss L, Casjens S, Cingolani G. Role of gene 10 protein in the hierarchical assembly of the bacteriophage P22 portal vertex structure. Biochemistry. 2007;46:8776–84. doi: 10.1021/bi700186e. [DOI] [PubMed] [Google Scholar]
- 26.Lupas A, Van Dyke M, Stock J. Predicting coiled coils from protein sequences. Science. 1991;252:1162–1164. doi: 10.1126/science.252.5009.1162. [DOI] [PubMed] [Google Scholar]
- 27.Rasko DA, Rosovitz MJ, Myers GS, Mongodin EF, Fricke WF, Gajer P, Crabtree J, Sebaihia M, Thomson NR, Chaudhuri R, Henderson IR, Sperandio V, Ravel J. The pangenome structure of Escherichia coli: comparative genomic analysis of E. coli commensal and pathogenic isolates. J Bacteriol. 2008;190:6881–93. doi: 10.1128/JB.00619-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schuck P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys J. 2000;78:1606–19. doi: 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rost B, Yachdav G, Liu J. The PredictProtein server. Nucleic Acids Res. 2004;32:W321–6. doi: 10.1093/nar/gkh377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wolf E, Kim PS, Berger B. MultiCoil: a program for predicting two- and three-stranded coiled coils. Protein Sci. 1997;6:1179–89. doi: 10.1002/pro.5560060606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Herrmann H, Aebi U. Intermediate filaments: molecular structure, assembly mechanism, and integration into functionally distinct intracellular Scaffolds. Annu Rev Biochem. 2004;73:749–89. doi: 10.1146/annurev.biochem.73.011303.073823. [DOI] [PubMed] [Google Scholar]
- 32.Herrmann H, Bar H, Kreplak L, Strelkov SV, Aebi U. Intermediate filaments: from cell architecture to nanomechanics. Nat Rev Mol Cell Biol. 2007;8:562–73. doi: 10.1038/nrm2197. [DOI] [PubMed] [Google Scholar]
- 33.Skehel JJ, Wiley DC. Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu Rev Biochem. 2000;69:531–69. doi: 10.1146/annurev.biochem.69.1.531. [DOI] [PubMed] [Google Scholar]
- 34.Smith AE, Helenius A. How viruses enter animal cells. Science. 2004;304:237–42. doi: 10.1126/science.1094823. [DOI] [PubMed] [Google Scholar]
- 35.Thoennes S, Li ZN, Lee BJ, Langley WA, Skehel JJ, Russell RJ, Steinhauer DA. Analysis of residues near the fusion peptide in the influenza hemagglutinin structure for roles in triggering membrane fusion. Virology. 2008;370:403–14. doi: 10.1016/j.virol.2007.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cohen C, Parry DA. Alpha-helical coiled coils: more facts and better predictions. Science. 1994;263:488–9. doi: 10.1126/science.8290957. [DOI] [PubMed] [Google Scholar]
- 37.Engel J, Kammerer RA. What are oligomerization domains good for? Matrix Biol. 2000;19:283–8. doi: 10.1016/s0945-053x(00)00075-5. [DOI] [PubMed] [Google Scholar]
- 38.Bromley EH, Channon K, Moutevelis E, Woolfson DN. Peptide and protein building blocks for synthetic biology: from programming biomolecules to self-organized biomolecular systems. ACS Chem Biol. 2008;3:38–50. doi: 10.1021/cb700249v. [DOI] [PubMed] [Google Scholar]
- 39.Steinbacher S, Baxa U, Miller S, Weintraub A, Seckler R, Huber R. Crystal structure of phage P22 tailspike protein complexed with Salmonella sp. O-antigen receptors. Proc Natl Acad Sci U S A. 1996;93:10584–8. doi: 10.1073/pnas.93.20.10584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Steinbacher S, Miller S, Baxa U, Weintraub A, Seckler R. Interaction of Salmonella phage P22 with its O-antigen receptor studied by X-ray crystallography. Biol Chem. 1997;378:337–43. doi: 10.1515/bchm.1997.378.3-4.337. [DOI] [PubMed] [Google Scholar]
- 41.Steinbacher S, Seckler R, Miller S, Steipe B, Huber R, Reinemer P. Crystal structure of P22 tailspike protein: interdigitated subunits in a thermostable trimer. Science. 1994;265:383–6. doi: 10.1126/science.8023158. [DOI] [PubMed] [Google Scholar]
- 42.Israel V. E proteins of bacteriophage P22. I. Identification and ejection from wild-type and defective particles. J Virol. 1977;23:91–7. doi: 10.1128/jvi.23.1.91-97.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Neal BL, Brown PK, Reeves PR. Use of Salmonella phage P22 for transduction in Escherichia coli. J Bacteriol. 1993;175:7115–8. doi: 10.1128/jb.175.21.7115-7118.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Holtzer A, Skolnick J. Application of the augmented theory of alpha-helix-to-random-coil transitions of two-chain, coiled coils to extant data on synthetic, tropomyosin-analog peptides. Biopolymers. 1988;27:87–96. doi: 10.1002/bip.360270107. [DOI] [PubMed] [Google Scholar]
- 45.Scholtz JM, Baldwin RL. The mechanism of alpha-helix formation by peptides. Annu Rev Biophys Biomol Struct. 1992;21:95–118. doi: 10.1146/annurev.bb.21.060192.000523. [DOI] [PubMed] [Google Scholar]
- 46.Studier FW, Rosenberg AH, Dunn JJ, Dubendorff JW. Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol. 1990;185:60–89. doi: 10.1016/0076-6879(90)85008-c. [DOI] [PubMed] [Google Scholar]
- 47.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–5. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 48.Greenfield NJ. Using circular dichroism collected as a function of temperature to determine the thermodynamics of protein unfolding and binding interactions. Nat Protoc. 2006;1:2527–35. doi: 10.1038/nprot.2006.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–8. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]