

Abstract
The enzymatic activity of the peripheral membrane protein, phosphatidylinositol-specific phospholipase C (PI-PLC), is increased by nonsubstrate phospholipids with the extent of enhancement tuned by the membrane lipid composition. For Bacillus thuringiensis PI-PLC, a small amount of phosphatidylcholine (PC) activates the enzyme toward its substrate PI; above 0.5 mol fraction PC (XPC), enzyme activity decreases substantially. To provide a molecular basis for this PC-dependent behavior, we used fluorescence correlation spectroscopy to explore enzyme binding to multicomponent lipid vesicles composed of PC and anionic phospholipids (that bind to the active site as substrate analogues) and high resolution field cycling 31P NMR methods to estimate internal correlation times (τc) of phospholipid headgroup motions. PI-PLC binds poorly to pure anionic phospholipid vesicles, but 0.1 XPC significantly enhances binding, increases PI-PLC activity, and slows nanosecond rotational/wobbling motions of both phospholipid headgroups, as indicated by increased τc. PI-PLC activity and phospholipid τc are constant between 0.1 and 0.5 XPC. Above this PC content, PI-PLC has little additional effect on the substrate analogue but further slows the PC τc, a motional change that correlates with the onset of reduced enzyme activity. For PC-rich bilayers, these changes, together with the reduced order parameter and enhanced lateral diffusion of the substrate analogue in the presence of PI-PLC, imply that at high XPC, kinetic inhibition of PI-PLC results from intravesicle sequestration of the enzyme from the bulk of the substrate. Both methodologies provide a detailed view of protein-lipid interactions and can be readily adapted for other peripheral membrane proteins.
Bacterial phosphatidylinositol-specific phospholipase C (PI-PLC)2 enzymes aid in organism infectivity (1), whereas the structurally homologous mammalian PLCδ1 and related enzymes are required for phosphoinositide metabolism and are important for intracellular signaling (2, 3). These peripheral membrane proteins often have distinct binding modes for substrates and for other lipids that either anchor the protein to the surface or adjust its conformation to enhance catalysis (4). However, using traditional methods, it has been difficult to determine how lipid composition affects phospholipase binding and, conversely, how protein binding alters the lipid environment, particularly in multicomponent vesicles. Current methods for monitoring peripheral membrane protein binding to mixed component lipid vesicles, including centrifugation, gel filtration, and NMR, can provide information on bulk protein partitioning but do not usually provide insight into how the properties of the individual phospholipids change when protein is bound. Most of these methods also require protein concentrations well above the amounts used in enzyme kinetics. Therefore, even when binding to multicomponent vesicles can be measured, it has been difficult to separate how substrates (or substrate analogues) and activators interact with enzymes, such as PI-PLC, that catalyze interfacial reactions.
Protein binding to lipid vesicles or cells greatly retards protein translational diffusion. For fluorescently labeled proteins, this change can easily be monitored using fluorescence correlation spectroscopy (FCS) (5–7). FCS uses low protein concentrations (<1–50 nm), comparable with what is used in kinetic assays, and experiments can be performed in minutes. Thus, FCS allows us to screen different phospholipid compositions and to identify conditions that can be studied in detail by other techniques aimed at probing how protein binding alters the lipid environment. One such technique is high resolution field cycling 31P NMR spectroscopy (fc-P-NMR) (8, 9).
Bacterial PI-PLCs recognize the headgroups of substrate, substrate analogue, and activator phospholipids, making 31P NMR an obvious choice for studying protein-lipid interactions. Measurements of dipolar relaxation rates could, in principle, determine the effects of protein binding on headgroup orientation with respect to the bilayer normal as well as correlation times for different headgroup motions. However, in a typical modern spectrometer, the slow rotational diffusion time of lipid vesicles, the dominance of chemical shift anisotropy (CSA) relaxation, and a plethora of faster motions at higher fields preclude obtaining information on lipid structure and dynamics from relaxation rates. This limitation is avoided by fc-P-NMR spin lattice relaxation techniques that resolve the dynamics of each phospholipid component in mixed component vesicles by using the high field to separate resonances but allowing nuclei to relax at defined lower fields. To cycle the magnetic field, the sample is mechanically shuttled between the center high field region of the commercial NMR magnet's probe and a substantially lower magnetic field located above the probe before and after the delay times normally used in conventional NMR relaxation sequences (8). Analysis of the field dependence of spin lattice relaxation rates over a wide field range (0.002–11.7 T) provides several correlation times (τ) for each phospholipid molecule on time scales ranging from ps to μs and also allows us to separate dipolar and CSA interactions (8, 9). Of particular interest is the intermediate 5–10 ns τc that appears to be dominated by the wobbling of each phospholipid in the membrane (10). This τc provides a direct probe of lipid dynamics in these multicomponent membranes. A longer correlation time in the μs range, τv, provides information on how vesicle tumbling and lateral diffusion affect the phosphorus nuclei for each type of phospholipid in a mixed vesicle.
We have used FCS and fc-P-NMR to probe the interactions of Bacillus thuringiensis PI-PLC with small unilamellar vesicles (SUVs). This enzyme and related, secreted bacterial PI-PLCs are important for bacterial virulence, and explicit measurements of their interactions with lipids could aid in inhibitor design. Bacterial PI-PLCs are also structurally homologous to the catalytic domain of mammalian PLCδ1 and thus provide a good model for assessing how interactions of this domain with membrane components could modulate enzyme activity. B. thuringiensis PI-PLC has a solvent-accessible active site inserted into a relatively rigid (αβ)-barrel (11). The enzyme exhibits higher specific activity toward small unilamellar vesicles (∼300 Å diameter) over larger vesicles with diameters of ≥1000 Å (12). For soluble substrates, the presence of phosphatidylcholine (PC) in micelles or vesicles activates the enzyme by decreasing the apparent Km and increasing kcat for both the phosphotransferase and phosphodiesterase steps (13). PC activation of PI cleavage, the interfacial reaction, also occurs with vesicle assay systems (12).
Our current model for kinetic activation of B. thuringiensis PI-PLC, based on the crystal structure of an interfacially impaired mutant protein (14) and extensive mutagenesis of surface residues (15–17), is that specific binding to PC in the lipid membrane allows key loop residues to penetrate the interface, promoting an enzyme conformation (suggested to be a transient dimer (14, 17)) with enhanced catalytic activity. However, in all PI vesicle assay systems examined, when the bulk PI is kept constant but the surface concentration decreases, PI-PLC specific activity decreases dramatically above XPC = 0.50 (12, 18). This surface dilution inhibition is often interpreted as the result of the substrate surface concentration decreasing below a two-dimensional Km measured in mole fraction PI (XPI) units (19, 20). The affinity of the diluent (in this case PC) for the enzyme can also complicate a detailed interpretation of this kinetic effect. To truly understand this PI-PLC, where two discrete phospholipids are critical for optimal catalysis, we need a molecular level description of surface dilution inhibition.
Our results clearly indicate a role for lipid dynamics in PI-PLC activation by PC at low XPC and reduced activity of PI-PLC at XPC > 0.5. Specifically, these experiments reveal the following. (i) A relatively small amount of activator PC in the anionic lipid membrane promotes PI-PLC binding to vesicles. (ii) The affinity of the protein for vesicles increases as the amount of PC is increased. Affinities for SUVs reach a maximum for very PC-rich, but not pure PC, bilayers, indicating that both phospholipids contribute to tight vesicle binding (a synergistic effect between activator and active site binding of lipids). (iii) At moderate XPC, 0.1 < XPC < 0.5, the dynamics of both phospholipids are affected similarly, and the restricted, slower motions, which are more or less independent of XPC, parallel the high enzymatic activity. (iv) In the regime of tight binding to PC-rich bilayers, PC dynamics are preferentially slowed, suggesting that this activator lipid sequesters PI-PLC from the bulk of the substrate analogues, leading to significantly lower specific activity than would be expected. This latter observation provides a very specific interpretation of surface dilution inhibition as the result of tight binding of the protein to PC-rich regions that prevents its access to the bulk of the PI.
EXPERIMENTAL PROCEDURES
Vesicle Preparation and Sizing by Dynamic Light Scattering (DLS)
All lipid stock solutions (dioleoyl chains for all of the anionic phospholipids and 1-palmitoyl-2-oleoyl phosphatidylcholine as the PC species) in chloroform were purchased from Avanti Polar Lipids. The chloroform was removed under a stream of nitrogen gas, and the resultant film was lyophilized overnight. The lipid film was rehydrated with phosphate-buffered saline buffer, pH 7.3, for FCS experiments or 50 mm HEPES, pH 7.5, for enzyme assays and for 31P NMR field cycling. Large unilamellar vesicles (LUVs) were prepared by multiple passages of the aqueous lipid solutions through polycarbonate membranes (100-nm pore diameter) using a miniextruder from Avanti. SUVs in phosphate-buffered saline, pH 7.3, were prepared by sonication (Branson Sonifier cell disrupter) of lipid mixtures for 30 min on ice. To introduce a fluorescent lipid into the SUVs, 1:20,000 mol fraction dipalmitoyl-lissamine-rhodamine phosphatidylethanolamine (Avanti) was added into the SUVs, and the mixture was sonicated for another 3 min. Under these conditions, a very small amount of fatty acid may be generated by hydrolysis of the phospholipids. However, no resonances for lyso-phospholipids were observed in a sample of PC/PI (1:1) treated in this fashion, indicating that the amount of lipid hydrolysis in this preparation was less than 0.5 mol %. Previous kinetic experiments have shown that small amounts of fatty acids do not affect enzyme specific activity (13).
The SUVs had a narrow vesicle size distribution as assessed by dynamic light scattering (21) using a Protein Solutions DynaProdynamic light scattering instrument from Wyatt Technology (22). The anion-rich SUVs were slightly smaller, with a narrower size distribution than PC-rich SUVs. All of the SUVs were used within 1 week of preparation.
PI-PLC
Recombinant B. thuringiensis PI-PLC was overexpressed in E. coli and purified as described previously (15). Protein concentrations were assessed by both 280 nm absorption and Lowry assays. Samples for fc-P-NMR were prepared with 10 mm total (dioleoylphosphatidylmethanol (PMe) plus PC) phospholipid and 0.5 or 3 mg/ml PI-PLC (0.014 or 0.086 mm) in 50 mm HEPES, pH 7.5. EDTA (1 mm) was present to scavenge any paramagnetic ions in solution. Fluorescently labeled PI-PLC was prepared from the N168C mutant protein, constructed using the QuikChange methodology (Stratagene). The altered gene sequence was confirmed by DNA sequencing (Genewiz). N168C was fluorescently labeled using Alexa Fluor 488 C5 maleimide (AF488 maleimide; Invitrogen), according to the manufacturer's protocol. AF488 does not bind to vesicles (7), and this was confirmed by performing FCS experiments in the presence of free AF488 and SUVs. A labeling ratio of 100 ± 10% was determined by comparing the absorption of the protein at 280 nm with that of the probe at 495 nm.
PI-PLC Activity toward SUVs
Specific activities of recombinant B. thuringiensis PI-PLC toward PI in SUVs with varying mole fractions of PC were measured by 31P NMR spectroscopy (13, 23). Each sample was in 50 mm HEPES, pH 7.5, with 1 mm EDTA and 0.1 mg/ml bovine serum albumin. In parallel to the fc-P-NMR studies, the total phospholipid concentration of the vesicles was 10 mm, with the mole fraction PC varying from 0.0 to 0.9. The amount of enzyme added, 0.2–5 μg (corresponding to concentrations of 5.8–144 nm), and the incubation time were chosen to allow less than 20% PI cleavage for each sample. Samples were incubated for fixed times at 28 °C, and the reaction was quenched by adding 40 μl of acetic acid to a 1-ml sample. Triton X-100 (100 μl) was added to solubilize the phospholipids for analysis by 31P NMR. In these assays, myoinositol 1,2-(cyclic)-phosphate was the only product, and the specific activity was calculated according to integration of the myoinositol 1,2-(cyclic)-phosphate resonance compared with an internal standard (protons were not decoupled in these experiments).
FCS Measurements
FCS experiments were performed using a previously described (24) home-built confocal setup based on an IX-70 inverted microscope (Olympus). Further details on the apparatus and data treatment are provided in the supplemental material.
FCS experiments were carried out at 22 °C on 300-μl samples in phosphate-buffered saline, pH 7.3, plus 1 mg/ml bovine serum albumin, to stabilize PI-PLC, in chambered coverglass wells (LabTek). Prior to use, the chambers were coated with 10 mg/ml bovine serum albumin and rinsed with phosphate-buffered saline buffer to prevent protein adhesion to the sides of the wells. For PI-PLC vesicle binding experiments, 3.5 nm labeled protein was titrated with unlabeled vesicles. The substrate, PI, was not used for FCS experiments, because PI cleavage by PI-PLC produces diacylglycerol, leading to vesicle fusion (25). Thus, the anionic phospholipids PMe, PG, PA, or PS were used as substrate analogues. PG is a PI-PLC substrate and can be cleaved over a period of days by >1 mg/ml concentrations of PI-PLC (18, 26). However, at the low protein concentrations used in the FCS experiments, no detectable hydrolysis of PG occurred.
The auto- and cross-correlation, Gj,k(τ), were calculated from the time-dependent fluorescence intensities (27, 28). To measure the average diffusion coefficient of the SUVs, FCS experiments were performed on SUVs containing a small amount (0.5 μm) of rhodamine-labeled PC. Diffusion coefficients of free, AF488-labeled PI-PLC (DPI-PLC) and labeled SUVs (DSUV) were 58 ± 5 μm2 s−1 (corresponding to a diffusion time (τD) = 173 μs) and 10.4 ± 1.0 μm2 s−1 (τD = 1.48 ms), respectively.
In the presence of labeled PI-PLC and unlabeled lipid vesicles, FCS auto- and cross-correlations, G(τ), were analyzed using two-component, diffusion-only fits (5, 29),
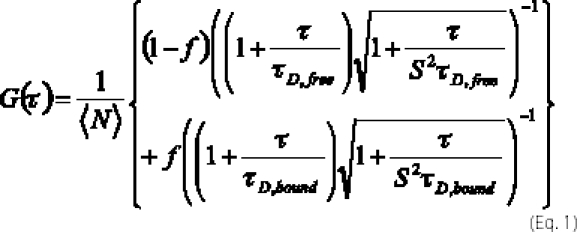 |
where 〈N〉 is the time-averaged number of PI-PLC molecules in the observation volume, f is the fraction of PI-PLC bound to vesicles, and τD,free and τD,bound are the diffusion times for free and vesicle-bound PI-PLC, respectively. For 488-nm excitation, values of S (related to the observation volume) were between 6.8 and 8.
High Resolution 31P Field Cycling
The 31P field cycling spin lattice relaxation rate (R1) experiments were made at 25 °C on a Varian Unityplus 500 spectrometer using a standard 10-mm Varian probe in a custom-built device that moves the sample, sealed in a 10-mm tube, from the sample probe location to a higher position within, or just above, the magnet, where the magnetic field is between 0.06 and 11.7 T (8, 30). Spin lattice relaxation rates at each field strength were measured using 6–8 programmed delay times and analysis of the data with an exponential function to extract R1 = 1/T1. Although the details of the R1 measurement for SUVs and the analysis to obtain and model R1 as a function of field have been described previously (8, 9), a more detailed discussion of the methodology, data analysis, and associated assumptions is presented in the supplemental material.
The field dependence of the 31P relaxation rates was analyzed in two discrete segments. R1 from 0.06 up to 11.7 T was fit with contributions from three terms (8, 30): (i) dipolar relaxation associated with a correlation time, τc, typically in the 5–10 ns range and Rc(0), the maximum relaxation rate for this correlation time, which is directly proportional to τc and inversely proportional to rP-H6 (Rc(0) = τc (μo/4π)2 (h/2π)2γP2γH2 rPH−6), where rP-H indicates the average distance between the phosphorus and its nearest proton neighbors; (ii) CSA relaxation associated with the same slow correlation time (from which we can extract SC2, an order parameter squared (8)); and (iii) CSA relaxation due to a faster motion (it is this term that dominates relaxation at high fields and is distinguished by a square law dependence). R1 in the field range 0.06–11.7 T is then the sum of these three terms,
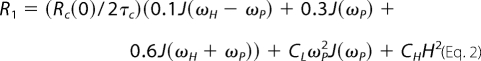 |
where J(ω) = 2τc/(1 + ω2τc2), and the field, H, is the magnetic field measured in tesla; the coefficient CL = (1/15)(1 + η2/3)σ2SC2. In these expressions, γ represents the gyromagnetic ratio (for 1H or 31P), and σ and η are the CSA interaction size and asymmetry for phospholipids (8). The last term is for a faster motion and is not of interest in this work. A protein interacting with a given phospholipid would be expected to slow the motion associated with τc as well as increase SC2.
A second distinct increase in R1 can be measured at very low fields (<0.03 T). The data in this low field regime are fit to a term added to Rc(0) for dipolar relaxation with a submicrosecond correlation time, τv, and a maximum extrapolated relaxation rate of Rv(0) for this slower motion (9).
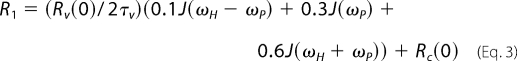 |
The parameter τv primarily reflects vesicle tumbling, although lateral diffusion also contributes to this value. For two phospholipids in the same vesicle, a change in τv in the presence of protein can provide information on segregation of phospholipids (and faster lateral diffusion of one versus the other).
RESULTS AND DISCUSSION
PI-PLC Activity
The specific activity (μmol min−1 mg−1) of PI-PLC toward PI-containing SUVs with a fixed enzyme and total lipid concentration (10 mm) and increasing mole fractions of the activator PC, XPC, was determined. To emphasize the initial activation and eventual inhibition, the PI-PLC relative activity (specific activity normalized to the value obtained for pure PI SUVs, 91.3 ± 9.2 μmol min−1 mg−1) is presented in Fig. 1. Although there is considerable error when averaging different series of experiments with SUVs, the trends are always the same. As little as 0.10 XPC increases the enzyme activity toward PI 2–3-fold, and the activity is more or less constant from 0.10 to 0.50 XPC. The magnitude of this activation depends on the concentration of PI and on the PC species; for example, a 7-fold activation was reported previously using 0.2 mol fraction dimyristoylphosphatidylcholine (12). Apparent Km values for pure PI SUVs (derived from treating the system as if it followed Michaelis-Menten kinetics) have been estimated as 3–5 mm (12), which might suggest that the large kinetic effect of PC is from enhanced protein binding (12, 26).
FIGURE 1.

Relative activity of B. thuringiensis PI-PLC toward SUVs with 10 mm total phospholipids as a function of the mole fraction of PC, XPC. The arrow indicates the XPC value at which there is half-maximal enzyme activity as the amount of substrate is decreased.
Further increases in XPC lead to a drop in PI-PLC-specific activity toward PI. In this case, the total phospholipid is kept constant, whereas the total and surface concentration of PI are decreased. Operationally, one can extract an interfacial “Xm” that represents the mole fraction of substrate in the interface needed for 50% of the maximum activity (analogous to a two-dimensional Km for substrate). For B. thuringiensis PI-PLC, such an Xm is ∼0.3 mol fraction PI (XPI = 1 − XPC). A wide variety of factors, including overall decreased vesicle binding, binding of nonsubstrate lipid in the active site, and/or altered lipid dynamics that make the substrate less accessible to bound protein, could decrease the specific activity as XPC is raised while the total lipid concentration is fixed. To understand such kinetic effects, we need a separate analysis of bulk PI-PLC partitioning to vesicles with different compositions as well as insight into how the dynamics of each of the two lipids are affected when PI-PLC binds to the different interface compositions.
PI-PLC Binding to Binary Component SUVs
Changes in bulk binding of PI-PLC to SUVs can be assessed by FCS as long as a fluorescent probe attached to the protein does not alter the binding or enzyme activity. B. thuringiensis PI-PLC was labeled with AF488 maleimide at a single Cys residue introduced at Asn168 (N168C). Asn168 is on an exterior helix of B. thuringiensis PI-PLC, far from both the proposed protein dimerization interface (14) and the proposed lipid interface (11). N168C labeled with AF488 had a PI cleavage rate of 1600 ± 160 μmol min−1 mg−1 at 28 °C toward 8 mm PI dispersed in 32 mm diC7PC micelles, the same value as for wild-type PI-PLC assayed under the same conditions.
Large unilamellar vesicles (typically ≥1000 Å in diameter) have generally been used for FCS studies of peptide or protein association with vesicles due to their higher thermodynamic stability and relatively uniform size (6, 7), although SUVs have occasionally been used (6, 29). The B. thuringiensis PI-PLC preferentially binds to small vesicles of PC (31), necessitating the use of SUVs for these binding experiments. SUVs composed of different ratios of PC to PMe or other anionic phospholipids, prepared by sonication, exhibited a narrow distribution of vesicle sizes as measured by DLS (Table 1). The average mass-weighted radius of the SUVs was 14.9 ± 2.7 nm, with 85 or more weight % of the lipids in vesicles with radii between 10 and 20 nm for all compositions.
TABLE 1.
Dependence of PC/PMe SUV average radius on vesicle composition measured by dynamic light scattering
| XPC | rDLSa | 10 nm ≤r≤ 20 nmb |
|---|---|---|
| nm | weight % | |
| 0.00 | 10.6 (10.0)c | 98.5 (99.2)c |
| 0.10 | 12.5 | 98.2 |
| 0.20 | 12.3 | 93.5 |
| 0.35 | 11.6 | 96.6 |
| 0.50 | 12.7 | 91.5 |
| 0.70 | 17.7 | 84.6 |
| 0.90 | 15.5 | 94.8 |
| 0.95 | 14.2 | 86.2 |
| 1.00 | 14.4 | 87.7 |
a The average radius for SUVs was calculated from the average diffusion coefficientDavobtained from the light scattering experiment using the Stokes-Einsteinrelation.
b The weight percentage of the small vesicles whose radius fell between 10 and 20 nm as measured by DLS.
c The value in parentheses is for pure PG SUVs prepared by sonication.
For FCS binding experiments, 3.5 nm fluorescently labeled N168C PI-PLC was titrated with unlabeled SUVs. PI-PLC binding to SUVs shifted the correlation curves to longer times (Fig. 2A). The fraction of PI-PLC bound to the vesicles, f, was determined from two-component fits to the cross-correlations using the diffusion coefficients determined for Rho-labeled SUVs as well as for free PI-PLC. An example of how f varies with total phospholipid concentration at two fixed XPC values is shown in Fig. 2B. A single PI-PLC binding site may be formed by multiple lipids, and the apparent dissociation constant Kd, representing the partitioning of the enzyme to the vesicle surface, as well as the coefficient, n, used to account for a sigmoidal binding profile, were determined using Equation 4,
where [PL] is the total phospholipid concentration, not the total vesicle concentration. For pure PC SUVs, the binding curve appears hyperbolic (n = 1).
FIGURE 2.

A, cross-correlation curves for free N168C-AF488-PI-PLC (○), Rho-labeled PC SUVs (□), and Rho-labeled PMe SUVs (▴). The lines through the data represent the best fit to Equation 1, with f = 1 (single species fit). The cross-correlation for 3.5 nm N168C-AF488-PI-PLC binding to 0.5 mm PC/PMe (XPC = 0.50) SUVs (×), corresponding to 48% bound protein. The line through these data is the best fit to Equation 1. B, titration curves for N168C-AF488 PI-PLC binding to pure PC SUVs (●) and to 9:1 PMe/PC SUVs (○). The lines through the data represent the best fits to Equation 4. At high XPC, the binding is not cooperative, although at low XPC, the binding curves are clearly sigmoidal. C, fraction of labeled PI-PLC bound to PC LUVs as a function of PC concentration. The line is the best fit to Equation 4 (n = 1).
The reported preference of B. thuringiensis PI-PLC for interacting with smaller, more curved vesicles (31) was confirmed by examining how the enzyme partitioned to PC LUVs (Fig. 2C). The Kd of PI-PLC binding to these LUVs was 60-fold higher than that for PC SUVs. The average radius for the population of LUVs to which the enzyme bound was determined using the Stokes-Einstein equation and the extracted Dbound. The calculated rFCS was 25 ± 3 nm (corresponding to Dbound = 8 ± 1 μm2 s−1), considerably smaller than the size of Rho-PE-labeled LUVs measured by FCS (rLUV = 40 ± 9 nm, corresponding to D = 5 ± 1 μm2 s−1), or the mass-weighted average size of LUVs measured by light scattering (rDLS = 53 ± 7 nm), indicating that PI-PLC binds primarily to the smaller vesicles in the LUV population.
The composition of the vesicles (i.e. XPC) as well as the identity of the anionic phospholipid affects PI-PLC binding to SUVs. The binding of the enzyme to SUVs as a function of XPC was examined quantitatively using PMe, PG, PA, or PS as the anionic phospholipid. PG, PMe, and PA inhibit the enzyme competitively and serve as substrate analogues (32). Although PA can potentially have a charge of −2, its pKa2 is sufficiently high (8.3 for pure PA SUVs and 7.8 in vesicles with PC/PA = 4:1 (33)) that it is primarily a monoanion, as are the other anionic phospholipids examined. In these mixed vesicles, PS was the poorest PI-PLC binder, consistent with kinetic studies, where it is the weakest of the anionic phospholipid inhibitors (23, 32), and the protein has the highest affinity for mixed PC/PG SUVs (Fig. 3A). With the exception of PS, binding curves for all systems are sigmoidal for low XPC vesicles (Fig. 3B). Although the average XPC is known, individual SUVs can have XPC values that are higher or lower than the average, and at low XPC, PI-PLC may bind more tightly to the small subpopulation of vesicles with higher PC content (the number of vesicles in this subpopulation would of course increase as the total concentration of vesicles increased). Alternatively, several PC lipids may be needed to form the initial PI-PLC binding site, and the probability that PI-PLC will encounter such a transient PC-rich region increases in a cooperative fashion as the number of SUVs increases at low XPC. Regardless of the detailed explanation for the sigmoidal appearance of the curves, it is obvious from our observations that a small amount of PC helps to anchor the enzyme to the mostly anionic phospholipid SUVs.
FIGURE 3.

Dependence of the PI-PLC binding to mixed anionic phospholipid/PC SUVs on XPC. Variation of Kd with XPC (A) and variation of the cooperativity coefficient, n, with XPC (B). The SUVs contained PC along with either PMe (●), PG (▴), PA (□) or PS (×).
For all of the anionic phospholipids mixed with PC, the apparent Kd has a bowl-shaped dependence on XPC (Fig. 3A). There is a more than 10-fold drop in Kd for PG and PA at XPC = 0.1, whereas PMe and PS show smaller, but still significant, decreases in Kd. The value of Kd continues to decrease and reaches a minimum at XPC = 0.9 for all of the two-component vesicle systems. Kd then increases (typically 5–20-fold) for XPC > 0.90. The observation of a minimum Kd as a function of XPC demonstrates that both anionic phospholipid (substrate analogue) and PC help anchor the protein to these small vesicles. Clearly, the enzyme activity (Fig. 1) does not track bulk PI-PLC affinity for SUVs (Fig. 3A). Although activity reaches a plateau between 0.1 and 0.5 XPC and then significantly decreases, the apparent Kd continues to decrease until XPC = 0.9. Although one can think about bulk and two-dimensional binding constants to describe enzyme binding to phospholipids in vesicles, neither way of quantifying binding can explain the kinetic behavior on a molecular level.
Effects of PI-PLC Binding on Lipid Dynamics
Using fc-P-NMR, we demonstrated that PI-PLC binding affects the dynamics of the phospholipid in pure PC (but not pure PMe) SUVs (8). Here, we carry this observation further by studying the field dependence of the 31P spin lattice relaxation rate, R1, for PI-PLC binding to mixed PC/PMe SUVs. Examples of R1 field dependence profiles for each phospholipid in the very low field region (0–0.05 T) and higher field region (0.07–11.74 T) are shown in Fig. 4 for two different XPC values. In the presence of PI-PLC, the field dependence above 0.07 T for PC (Fig. 4B) and PMe (Fig. 4D) are similar at XPC = 0.2 but noticeably different at XPC = 0.8, where Rc(0) is clearly larger for the PC component. The relaxation profile in the absence of PI-PLC is shown as a gray line in each panel in Fig. 4. Without protein, τc for the two phospholipids is the same, 5.6 ± 0.5 ns, and does not vary significantly with XPC. Quantifying the changes induced by protein provides insight into how PI-PLC binding affects the dynamics of each phospholipid.
FIGURE 4.

Field dependence of R1 for PC and PMe in mixed SUVs with XPC = 0.20 (filled symbols) or XPC = 0.80 (open symbols) and 3 mg/ml (86 μm) PI-PLC. A, field dependence of the PC R1 from 0 to 0.05 T. B, PC R1 from 0.07 to 11.7 T. The field dependence of the PMe R1 is shown from 0 to 0.05 T (C) and from 0.07 to 11.7 T (D). The curves through these points represent the best fits to the simple model for R1 dependence on field with two correlation times (τv from A and C and τc from B and D). The profile exhibited by the phospholipid in the absence of protein is shown in each panel by the gray line.
The rise in relaxation rate, R1, below 1 T is caused by dipolar relaxation, principally through dipolar interactions with the glycerol C(3) protons (8). As the field is increased above 1 T, relaxation due to CSA becomes important. As described in detail in the supplemental material, a model-free approach was used to fit the field-dependent profile for R1 from 0.07 up to 11.7 T as a sum of dipolar and CSA terms associated with a slow, τc (ns), and fast, τh (ps), motion and to estimate Rc(0), the maximum relaxation rate caused by τc-associated motions. This τc has been modeled by diffusive and wobbling motions of each phospholipid treated as a relatively rigid cylinder encompassing the phosphorus-glycerol-acyl chains in a hexadecane-like medium (10).
The addition of PI-PLC to pure PMe SUVs had very little effect on τc and Rc(0) (8) under conditions (10 mm PMe) where the FCS Kd (3 ± 1 mm) indicates that most of the protein is associated with the SUVs. Therefore, the average PMe molecular environment is not significantly perturbed, and interactions between the SUVs and PI-PLC are probably electrostatic and nonspecific. In contrast, when PI-PLC was added to pure PC vesicles, it significantly increased τc (from 5 to ∼13 ns) as well as increasing Rc(0) 2–3-fold. The tight binding of PI-PLC to PC SUVs, as measured by FCS (Fig. 3A), clearly involves significant retardation of PC motion in the ns range. An order parameter, Sc2, extrapolated from the CSA portion of relaxation (8), is ∼0.5 for both phospholipids in the absence of enzyme. For PC SUVs with PI-PLC bound, this parameter has a value greater than 1 (8). Given uncertainties in several of the input parameters used to determine Sc2 (e.g. CSA interaction size and asymmetry), the absolute value of this order parameter may not be correct. However, for these two-component SUVs, changes in Sc2 serve as a convenient way to compare the effects of PI-PLC binding on lipid motions as a function of XPC.
For XPC between 0 (pure PMe) and 1 (pure PC), both phospholipids show changes in the field dependence of R1 only when the PI-PLC is present. In these experiments, as in the activity assays, the total concentration of phospholipids was fixed at 10 mm, but the PI-PLC concentration was considerably higher (86 μm), resulting in a lipid/PI-PLC ratio of 116:1. Centrifugation of samples through an Amicon Centricon-100 filter showed that about 20–40% of the protein was associated with the SUVs under these conditions. This suggests that there is adequate enzyme to saturate all vesicle binding sites. Two XPC points, 0.1 and 0.65, used a lipid/PI-PLC ratio of ∼700:1 with only minor effects on τc and Rc(0) compared with the higher protein concentration. All protein was bound to the vesicle at the end of the experiment, consistent with saturation of the vesicles with a total phospholipid/PI-PLC of ∼400–500.
For each XPC, analysis of the field dependence of R1 from 0.06 to 11.7 T in the presence of PI-PLC was used to extract Rc(0) and τc for each of the two phospholipids in the SUVs. As shown in Fig. 5, as little as 0.1 XPC increased Rc(0) and τc for PMe significantly, and both parameters were roughly constant from 0.1 to 0.5 XPC and similar to the values for PC in the same vesicle. The presence of PC in these mostly anionic phospholipid SUVs allows PI-PLC to bind, and as a consequence, the ns motion of both phospholipids is hindered, and τc increases from 5 ns. (This is easily seen in Fig. 4, B and D, where R1 at ∼0.1 T is significantly higher in the presence of PI-PLC than it is in the absence of protein (gray line).) These effects of PI-PLC are also reflected in the SC2 values for PMe, which increase as soon as PC is added (Fig. 5C). The PC resonance in the SUVs is broader than for PMe, so the field dependence of R1 for PC at XPC = 0.10 could not be obtained with any accuracy. Nonetheless, the fc-P-NMR data reveal that for XPC values of 0.5 or less, the phosphorus motions of both PMe and PC are similarly restricted with PI-PLC present. These effects of the protein on phospholipid dynamics could reflect chemical exchange between active site or activator site and bulk phospholipid or else a generalized change in membrane fluidity/viscosity as long as the protein is transiently anchored to the bilayer.
FIGURE 5.

Variation of parameters extracted from high resolution 31P field cycling for PC (●) and PMe (□) in mixed vesicles as a function of XPC. A, Rc(0); B, τc; C, SC2; D, τv. The concentration of PI-PLC was 3 mg/ml (86 μm) for all XPC except 0.65 and 0.90, for which 0.5 mg/ml (14 μm) PI-PLC was present. The dotted lines in A and B show the values for PMe in the absence of protein.
At XPC > 0.5, there is a switch in phospholipid behavior with PI-PLC, resulting in a significantly increased Rc(0), τc, and Sc2 for PC but not for PMe in the same SUV (Fig. 5). For PC, the extracted value of Sc2 is greater than 1, a likely reflection of our not quite correct assumption that τc is the same for CSA and dipolar terms (for a discussion of this, see the supplemental material). However, what is important is that Sc2 increases for PC but not for PMe as this lipid becomes the major component in these vesicles. Clearly, PI-PLC differentially hinders the PC motions at XPC > 0.5. The difference in parameters for the two phospholipids suggests that chemical exchange (between protein-bound and “free” in the bilayer) rather than bulk membrane fluidity is the major determinant of τc for each phospholipid.
So what does PI-PLC actually do to PC in high XPC bilayers? One interpretation is that PC can now interact with the enzyme active site. The motion of the larger (compared with PMe) PC headgroup might be more restricted at this second binding site. However, the FCS results indicate that even at high XPC, PMe and PC behave synergistically when binding to PI-PLC. It has also been shown that PI-PLC bound to pure PC SUVs is activated toward the soluble substrate myoinositol 1,2-(cyclic)-phosphate (13). Thus, the active site is unlikely to be occupied by PC acting as a competitive inhibitor. A different explanation for the observed fc-P-NMR effects is that there is a preferential interaction of PI-PLC with PC, resulting in lipid clustering.
The field dependence of the relaxation rate at very low (<0.06-T) fields provides hints that PI-PLC-induced partial separation of PC and PMe may occur at XPC > 0.5. From the low field rise in relaxation rate (Fig. 4, A for PC and C for PMe), we can determine a correlation time, τv, for each phospholipid, which in the absence of protein is related to the vesicle size and includes contributions from vesicle tumbling, lateral diffusion, and possibly other slower (100-ns) motions of the phospholipids (9). For these sonicated vesicles, the average diameter, as measured by DLS, is relatively constant as XPC is increased from 0 to 0.8 (Table 1). At low XPC, τv is similar for both phospholipids (0.4–0.5 μs). This value is 2–3 times shorter than would be expected for overall rotation of SUVs (9), consistent with some contribution from lateral diffusion. However, as XPC increases above 0.5, τv for PC increases slightly (from 0.51 ± 0.03 μs at XPC = 0.5 to 0.62 ± 0.06 μs at XPC = 0.8), whereas the value for PMe decreases to 0.25 ± 0.07 μs at the highest XPC where relaxation in the low field region was examined (Fig. 5D). That τv for PMe is reduced can be seen in Fig. 4C, where the curves for both XPC values have similar Rv(0) values but the midpoint for this change occurs at a higher field for XPC = 0.8, the hallmark of a shorter correlation time. For comparison, the analysis of the low field dependence of pure PMe SUVs (see Fig. S2) yields a τv of 0.39 ± 0.03 μs. In the PC-rich SUVs with PI-PLC added, τv for PMe is considerably less than that for PC, and the decrease in τv for PMe as XPC increases is statistically significant. The decrease in τv for bulk PMe indicates that the PMe lateral diffusion, unfettered by protein (and perhaps neighboring PC molecules), is now more effective at relaxing the residual magnetization of 31P. This would occur if PC molecules sequester the protein from PMe, which now has a shorter path around the vesicle in areas devoid of PC/protein clusters.
PC Activation and Inhibition of PI-PLC, a Phospholipid-centric View
The combination of FCS and fc-P-NMR provides new insights into the role of PC in B. thuringiensis PI-PLC-catalyzed PI cleavage. In moderate ionic strength buffers, PI-PLC has very weak affinity for SUVs containing only anionic lipids, in this case the substrate PI (or analogue, such as PMe), and relatively small amounts of PC serve to anchor the protein to the mixed component bilayers. The hallmark of that binding is that the rotational/wobbling motion of both substrate analogue (PMe) and activator (PC) molecules is similarly reduced for each phospholipid. However, tight binding does not necessarily correlate with higher enzyme activity. As the relative amount of PC, XPC, is increased above 0.5, specific activity decreases, and there is a change in the dynamics of PC bound to PI-PLC; in contrast, the affinity of the protein for SUVs (as measured by the apparent Kd) continues to increase up to 0.90 XPC.
In kinetic models for surface active enzymes (19, 20), a decrease in activity with increasing mole fraction of diluent is often attributed to the reduction of the substrate surface concentration below a threshold two-dimensional Km. Our data reveal that decreased B. thuringiensis PI-PLC activity at high PC is correlated with changes in substrate (PI) and diluent (PC) lipid-protein interactions and lipid dynamics. Assuming that PMe is a reasonable stand-in for the substrate, at high XPC, PC dynamics, reflected in τv, τc, and Sc2 for the PMe/PC system, would be much slower than PI dynamics in the same vesicle. PI may still be bound at the active site even at high XPC, as reflected by the continued synergy between the two types of phospholipids in vesicle binding, but it would not be rapidly exchanging with the bulk PI pool. By analogy to the PMe/PC system, the tightly bound enzyme is clustered with PC and sequestered from much of the PI substrate in the bilayer. This sequestration probably inhibits PI-PLC diffusion around a given vesicle as well as release from one vesicle and binding to another for cleavage of more PI. We propose that the changes in protein-lipid interactions detected by fc-P-NMR generate the observed decrease in PI-PLC activity at high XPC, either in the kinetic experiment shown here, where total phospholipid is a constant and XPC is varied, or in the more usual surface dilution experiments, where PI is constant and XPC increases. Such behavior, differential interactions of an enzyme with substrate and diluent amphiphiles that affect substrate accessibility to enzyme as well as lipid dynamics in the bilayer, may also be responsible for the decreased activity seen in the high diluent regime for many other phospholipases. The combination of FCS and fc-P-NMR encompassing bulk binding and phospholipid dynamics can be easily generalized to other, less well studied phospholipases or other peripheral membrane proteins where specific phospholipids are thought to have differential binding effects on enzymes.
Biological Relevance of PI-PLC/PC Interactions
The effects of PI-PLC on this simple two-component bilayer provide insights into the biological role of this enzyme secreted by different Bacillus strains. B. thuringiensis is an insect pathogen, and earlier work showed that an avirulent mutant lacking both the broad range and PI-specific PLC was much less potent in killing cells (34). In this insect model, wild type B. thuringiensis disappeared from the circulation hours after injection, while the mutant organism was still circulating. This suggested that one or both phospholipases contributed to the early stages of infection and association of the bacteria with insect and mammal cells, since B. thuringiensis is also an opportunistic human pathogen leading to pulmonary infections (35). Membrane-damaging factors, such as the PI-PLC, apparently allow the bacteria to persist in that environment.
More recent work with Bacillus anthracis has also provided evidence that B. anthracis PI-PLC, a secreted PI-PLC with high sequence similarity to B. thuringiensis PI-PLC, contributes to bacterial pathogenicity. The B. anthracis PI-PLC, like the enzyme from B. thuringiensis, can cleave GPI-anchored proteins in plasma membranes as well as intracellular PI, providing two potential roles for this enzyme in virulence. B. anthracis PI-PLC has also been shown to down-modulate the immune response, and this appears to involve its ability to cleave GPI-anchored proteins (1).
Access to GPI anchors on the external leaflet of a target cell would be facilitated by protein binding to PC (or sphingomyelin, also an activator). GPI anchors may exist in clustered domains (36), indicating that PI-PLC cleavage of GPI anchors may occur at low XPC.
In B. anthracis, PI-PLC is one of three phospholipase C enzymes that contribute to macrophage-associated growth of the bacteria (37). Along with aiding bacterial association with the macrophages, the three enzymes may have overlapping roles in pathogenesis by aiding in release from the phagocyte (38). Since the interior leaflet of the phagocyte resembles the external leaflet of the plasma membrane, PC will be present for PI-PLC binding. Even if PI and GPI-anchored targets are not available, the strong interaction of this PI-PLC with PC in the membrane could cause clustering of the PC, making the phagocyte more susceptible to other membrane hydrolytic factors.
Supplementary Material
Acknowledgments
We thank Professor Irving Epstein for use of the DynaProdynamic light scattering instrument and Dr. Vladimir Vanag for assistance with the light scattering experiments.
This work was supported, in whole or in part, by National Institutes of Health Grants GM60418 (to M. F. R.) and GM77974 (to A. G. R.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1 and S2.
- PI-PLC
- phosphatidylinositol-specific phospholipase C
- PI
- phosphatidylinositol
- PLC
- phospholipase C
- AF488
- Alexa Fluor 488 C5
- CSA
- chemical shift anisotropy
- DLS
- dynamic light scattering
- fc-P-NMR
- high resolution field cycling 31P NMR spectroscopy
- FCS
- fluorescence correlation spectroscopy
- LUV
- large unilamellar vesicles
- PA
- dioleoylphosphatidic acid
- PC
- 1-palmitoyl-2-oleoylphosphatidylcholine
- PG
- dioleoylphosphatidylglycerol
- PMe
- dioleoyl-phosphatidylmethanol
- PS
- dioleoylphosphatidylserine
- SUV
- small unilamellar vesicle
- T
- Tesla
- GPI
- glycosylphosphatidylinositol.
REFERENCES
- 1.Zenewicz L. A., Wei Z., Goldfine H., Shen H. ( 2005) J. Immunol. 174, 8011– 8016 [DOI] [PubMed] [Google Scholar]
- 2.Downes C. P., Gray A., Lucocq J. M. ( 2005) Trends Cell Biol. 15, 259– 268 [DOI] [PubMed] [Google Scholar]
- 3.Rhee S. G. ( 2001) Annu. Rev. Biochem. 70, 281– 312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mulgrew-Nesbitt A., Diraviyam K., Wang J., Singh S., Murray P., Li Z., Rogers L., Mirkovic N., Murray D. ( 2006) Biochim. Biophys. Acta 1761, 812– 826 [DOI] [PubMed] [Google Scholar]
- 5.Magde D., Elson E. L., Webb W. W. ( 1974) Biopolymers 13, 29– 61 [DOI] [PubMed] [Google Scholar]
- 6.Rhoades E., Ramlall T. F., Webb W. W., Eliezer D. ( 2006) Biophys. J. 90, 4692– 4700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rusu L., Gambhir A., McLaughlin S., Rädler J. ( 2004) Biophys. J. 87, 1044– 1053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roberts M. F., Redfield A. G. ( 2004) J. Am. Chem. Soc. 126, 13765– 13777 [DOI] [PubMed] [Google Scholar]
- 9.Roberts M. F., Redfield A. G. ( 2004) Proc. Natl. Acad. Sci. U. S. A. 101, 17066– 17071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Klauda J. B., Roberts M. F., Redfield A. G., Brooks B. R., Pastor R. W. ( 2008) Biophys. J. 94, 3074– 3083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heinz D. W., Ryan M., Bullock T. L., Griffith O. H. ( 1995) EMBO J. 14, 3855– 3863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Qian X., Zhou C., Roberts M. F. ( 1998) Biochemistry 37, 6513– 6522 [DOI] [PubMed] [Google Scholar]
- 13.Zhou C., Wu Y., Roberts M. F. ( 1997) Biochemistry 36, 347– 355 [DOI] [PubMed] [Google Scholar]
- 14.Shao C., Shi X., Wehbi H., Zambonelli C., Head J. F., Seaton B. A., Roberts M. F. ( 2007) J. Biol. Chem. 282, 9228– 9235 [DOI] [PubMed] [Google Scholar]
- 15.Feng J., Wehbi H., Roberts M. F. ( 2002) J. Biol. Chem. 277, 19867– 19875 [DOI] [PubMed] [Google Scholar]
- 16.Feng J., Bradley W. D., Roberts M. F. ( 2003) J. Biol. Chem. 278, 24651– 24657 [DOI] [PubMed] [Google Scholar]
- 17.Guo S., Zhang X., Seaton B. A., Roberts M. F. ( 2008) Biochemistry 47, 4201– 4210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Volwerk J. J., Filthuth E., Griffith O. H., Jain M. K. ( 1994) Biochemistry 33, 3464– 3474 [DOI] [PubMed] [Google Scholar]
- 19.Carman G. M., Deems R. A., Dennis E. A. ( 1995) J. Biol. Chem. 270, 18711– 18714 [DOI] [PubMed] [Google Scholar]
- 20.Berg O. G., Gelb M. H., Tsai M. D., Jain M. K. ( 2001) Chem. Rev. 101, 2613– 2654 [DOI] [PubMed] [Google Scholar]
- 21.Maulucci G., De Spirito M., Arcovito G., Boffi F., Castellano A. C., Briganti G. ( 2005) Biophys. J. 88, 3545– 3550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schuck P. ( 2000) Biophys. J. 78, 1606– 1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Volwerk J. J., Shashidhar M. S., Kuppe A., Griffith O. H. ( 1990) Biochemistry 29, 8056– 8062 [DOI] [PubMed] [Google Scholar]
- 24.Liu L., Mushero N., Hedstrom L., Gershenson A. ( 2006) Biochemistry 45, 10865– 10872 [DOI] [PubMed] [Google Scholar]
- 25.Goñi F. M., Alonso A. ( 2000) Biosci. Rep. 20, 443– 463 [DOI] [PubMed] [Google Scholar]
- 26.Zhou C., Qian X., Roberts M. F. ( 1997) Biochemistry 36, 10089– 10097 [DOI] [PubMed] [Google Scholar]
- 27.Elson E. L., Magde D. ( 1974) Biopolymers 13, 1– 27 [DOI] [PubMed] [Google Scholar]
- 28.Thompson N. L. ( 1991) in Topics in Fluorescence Microscopy ( Lakowicz J. ed) pp. 337– 378, Plenum Press, New York [Google Scholar]
- 29.Takakuwa Y., Pack C. G., An X. L., Manno S., Ito E., Kinjo M. ( 1999) Biophys. Chem. 82, 149– 155 [DOI] [PubMed] [Google Scholar]
- 30.Roberts M. F., Cui Q., Turner C. J., Case D. A., Redfield A. G. ( 2004) Biochemistry 43, 3637– 3650 [DOI] [PubMed] [Google Scholar]
- 31.Wehbi H., Feng J., Kolbeck J., Ananthanarayanan B., Cho W., Roberts M. F. ( 2003) Biochemistry 42, 9374– 9382 [DOI] [PubMed] [Google Scholar]
- 32.Zhou C., Roberts M. F. ( 1998) Biochemistry 37, 16430– 16439 [DOI] [PubMed] [Google Scholar]
- 33.Swairjo M. A., Seaton B. A., Roberts M. F. ( 1994) Biochim. Biophys. Acta 1191, 354– 361 [DOI] [PubMed] [Google Scholar]
- 34.Zhang M. Y., Lövgren A., Low M. G., Landén R. ( 1993) Infect. Immun. 61, 4947– 4954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ghelardi E., Celandroni F., Salvetti S., Fiscarelli E., Senesi S. ( 2007) Microbes Infect. 9, 591– 598 [DOI] [PubMed] [Google Scholar]
- 36.Paulick M. G., Bertozzi C. R. ( 2008) Biochemistry 47, 6991– 7000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Heffernan B. J., Thomason B., Herring-Palmer A., Shaughnessy L., McDonald R., Fisher N., Huffnagle G. B., Hanna P. ( 2006) Infect. Immun. 74, 3756– 3764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Heffernan B. J., Thomason B., Herring-Palmer A., Hanna P. ( 2007) FEMS Microbiol. Lett. 271, 98– 105 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.