

Abstract
Activation of p53 is an important mechanism in apoptosis. However, whether the presence of p53 in mitochondria plays an important role in p53-mediated apoptosis is unclear. Here, we demonstrate that overexpression of NPM (nucleophosmin) significantly suppresses 12-O-tetradecanoylphorbol 13-acetate (TPA)-mediated apoptosis, in part, by blocking the mitochondrial localization of p53. Within 1 h following TPA treatment of skin epithelial (JB6) cells, p53 accumulated in mitochondria. Expression of NPM enhances p53 levels in the nucleus but reduces p53 levels in mitochondria, as detected by immunocytochemistry and Western blot analysis. The suppressive effect of NPM on p53 mitochondrial localization is also observed in TPA-treated primary epithelial cells and in JB6 cells treated with doxorubicin. NPM enhances the expression of p53 target gene p21 and bax. However, the increase in Bax level in the absence of p53 in mitochondria did not lead to an increase in TPA-induced apoptosis, suggesting that the presence of p53 in mitochondria is important. Suppression of NPM by NPM small interfering RNA leads to an increase of p53 levels in mitochondria and apoptosis. Furthermore, suppression of NPM in tumor cells with a high constitutive level of NPM results in p53 translocation to mitochondria and enhances TPA-mediated apoptosis. The results demonstrate the effect of NPM on p53 localization in mitochondria and apoptosis. Together, the data indicate that the presence of p53 in mitochondria plays an important role in stress-induced apoptosis and suggest that NPM may protect cells from apoptosis by reducing the mitochondrial level of p53.
The tumor suppressor functions of p53 are mediated by inhibiting the growth of defective cells through activation of cell cycle arrest and apoptosis (1–3). The apoptotic function of p53 plays a critical role in its tumor suppressor activity (3). p53 level is increased upon exposure to various stimuli, including DNA damage, oncogenic stimuli, hypoxia (4), and oxidative stress (5, 6).
Recently, the linkage between mitochondrial oxidative stress and p53-induced apoptosis has attracted considerable attention for the following reasons. First, p53-mediated apoptosis is preceded by activation of various oxidoreductases and reactive oxygen species generation before mitochondrial perturbation (7). Second, p53 transactivates several mitochondrial proapoptotic proteins, including Bax, a member of the bcl2 family (8); Noxa, a BH3-only member of the bcl2 family (9); and p53AIP1, a p53-regulated apoptosis-inducing protein-1 (10). Third, a fraction of p53 is localized in the mitochondria at the onset of p53-dependent apoptosis preceding changes in mitochondrial membrane potential, cytochrome c release, and caspase activation (11–13). Therefore, it has been suggested that p53 may mediate apoptosis by mechanisms that are both dependent on and independent of its transcriptional activity, thus amplifying its apoptotic function.
p53 is considered to be the guardian of the genome. To enhance the efficiency of p53-dependent transcription of target genes as well as to protect the genome from damage, p53 stabilization is required. It has been reported that NPM (nucleophosmin), a nucleolar phosphoprotein implicated in ribosome biogenesis (14), stabilizes and regulates the transcriptional activity of p53 (15). NPM acts as a molecular chaperone and shuttle between the nucleus and cytoplasm (16). The enhancement of p53 stability and transcriptional ability of NPM may occur through several mechanisms, including inhibition of Mdm2 ubiquitin-ligase activity, competition with p53-Mdm2 bindings, and NPM chaperonage of p53 (15, 17). The stability of p53 proteins is primarily regulated by Mdm2, a ubiquitin E3 ligase, and Arf, a nucleolar protein that binds with p53 and inhibits the ubiquitin ligase activity of Mdm2 for p53 (18). Colombo et al. (19) have reported that lack of NPM expression in NPM−/− embryonic fibroblast cells resulted in widespread apoptosis in wild type p53-expressing cells. However, no significant apoptosis was observed in double knock-out (p53−/−) cells (19), suggesting that NPM suppresses p53-mediated apoptosis. Thus, it remains unclear how the interaction of NPM with p53 affects p53-mediated apoptosis.
NPM is overexpressed in many types of human cancer, including colon (20), ovarian (21), prostate (22), and gastric (23) cancer. It has been proposed that NPM has both antiapoptotic and apoptotic capabilities, depending on its level and subcellular localization (15, 16, 24, 25). Malignant and actively dividing cells expressing large amounts of NPM are resistant to apoptosis (20, 22) induced by UV damage or hypoxia (14, 26). Similar to other proto-oncogenes, NPM overexpression is primarily associated with cell proliferation (27), suggesting that NPM may act as a proto-oncogene by promoting cell survival. It has been demonstrated that NPM may also act as a co-activator for the transcription of survival genes, including bcl2, a member of the Bcl2 protein family identified as an oncogene from human follicular lymphoma of B cell origin (28, 29). It has been well documented that Bcl2 prevents most forms of apoptotic cell death; however, whether the antiapoptotic role of NPM is mediated via bcl2 is not known.
Our study shows that bcl2 does not play a major role in NPM-mediated protection from TPA2 -induced apoptosis and that the antiapoptotic function of NPM is associated with its ability to suppress p53 localization to mitochondria. This is the first study to demonstrate the role of NPM in blocking mitochondrial p53 and preventing p53-mediated apoptosis.
EXPERIMENTAL PROCEDURES
Cell Culture
The mouse skin epithelial cell line (JB6 P+, clone 41, promotable by TPA treatment) was originally provided by Dr. Nancy H. Colburn (NCI-Frederick, National Institutes of Health), maintained as described previously (30), and cultured in modified Eagle's medium supplemented with 4% (v/v) fetal bovine serum (Hyclone Inc., Logan, UT), 200 μm l-glutamine, and 1% penicillin/streptomycin antibiotic (Invitrogen). The primary human mammary epithelial cells were purchased from Clonetics and cultured for three passages in primary culture medium according to the manufacturer's instructions. The human hepatocarcinoma cell line (HepG2) was purchased from the American Type Culture Collection (Manassas, VA) and was cultured in medium consisting of Dulbecco's modified Eagle's/Ham's F-12 (Sigma) supplemented with 10% (v/v) fetal bovine serum (Hyclone Inc., Logan, UT), 200 μm l-glutamine (Invitrogen), and 1% PSN antibiotic (Invitrogen). All cells were grown in 5% CO2 at 37 °C.
Reagents
Unless otherwise stated, all antibodies were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Anti-β-actin monoclonal antibody and wild- type anti-p53 (Ab-11) antibodies were purchased from Sigma and Oncogene (Boston, MA), respectively. Rabbit polyclonal Mn-SOD antibody and glyceraldehyde-3-phosphate dehydrogenase antibody were purchased from Upstate Biotechnology, Inc. (Lake Placid, NY). Nucleophosmin expression vector (pcDNA3.1/ NPM) was purchased from Invitrogen. Both control siRNA and NPM siRNA were purchased from Santa Cruz Biotechnology. Doxorubicin and TPA were purchased from Bedford Laboratories (Bedford, OH) and Sigma, respectively.
Transient Transfection
Cells were grown for 24 h with no antibiotics to obtain 70–80% confluence. The cells were then transfected with plasmids following a Lipofectamine® transfection protocol, as directed by the manufacturer. Cells were transfected with 4 μg of NPM cDNA construct in pcDNA3.1 plasmid vector or pcDNA3.1 (−NPM) vector alone as control. Twenty-four hours after transfection, the cells were washed twice with PBS and incubated in fresh medium.
Cells were grown for another 24 h prior to TPA treatment. One hour post-treatment, the cells were washed with PBS and processed for mitochondrial isolation, nuclear extract preparation, or whole cell lysate preparation. siRNAs (20 nm) were transfected using Transfectin® for 48 h (Santa Cruz Biotechnology) according to the manufacturer's protocol. The siRNA sequence targeting NPM gene silencing is as follows: antisense, 5′-UUCUUAAUGACAACUGGUGtt-3′; sense, 5′-CACCAGUUGUCAUUAAGAAtt-3′. The 2-bp mismatch siRNA (used to verify the specificity of NPM silencing) sequence is as follows: antisense, 5′-GGCUUAAUGACAACUGGUGtt-3′; sense, 5′-CACCAGUUGUCAUUAAGCCtt-3′.
Nuclear Extract Preparation
Nuclei were isolated from JB6 cells. A subconfluent monolayer of cells was collected and centrifuged at 100 × g for 2 min at 4 °C to obtain cell pellets. Cell pellets were then resuspended in buffer A containing 10 mm HEPES (pH 7.9), 1.5 mm MgCl2, 10 mm KCl, 0.5 mm dithiothreitol, and 0.2 mm phenylmethylsulfonyl fluoride with the inclusion of protease inhibitors (pepstatin, aprotinin, and leupeptin) at a concentration of 1 μg/ml. Additionally, the phosphatase inhibitors NaF (5 mm) and Na3VO4 (1 mm) were included. The cell suspension was incubated on ice for 15 min. 12.5 μl of 10% Nonidet P-40 was added, and the mixture was vigorously vortexed for 15 s. The cytoplasmic and nuclear fractions were separated by centrifugation at 17,000 × g at 4 °C for 30 s. Subsequently, the nuclear pellets were resuspended in buffer B containing 20 mm HEPES (pH 7.9), 1.5 mm MgCl2, 420 mm NaCl, 0.2 mm EDTA, 35% glycerol, 0.5 mm dithiothreitol, 0.2 mm phenylmethylsulfonyl fluoride, and protease inhibitor (pepstatin, aprotinin, and leupeptin) at a concentration of 1 μg/ml and incubated on ice for 20 min. Nuclear proteins in the supernatant fraction were collected by centrifugation at 14,000 × g at 4 °C for 2 min.
Isolation of Mitochondria
Approximately 10 million cells were suspended in 1 ml of mitochondrial isolation buffer (225 mm mannitol, 75 mm sucrose, 1 mm EGTA, pH adjusted to 7.4 with a dropwise addition of 500 mm Tris) in a 15-ml plastic tube and homogenized using a Wheaton homogenizer three times with 30-s strokes on ice. The cell debris was removed by centrifugation at 576 × g (2200 rpm) for 5 min in a Sorval SS34 rotor. The supernatant was filtered through a nylon screen cloth (Small Parts Inc., Miami Lakes, FL) and centrifuged at 9000 × g (8700 rpm) for 10 min. The pellet was washed by adding 0.5 ml of mitochondrial isolation buffer and centrifuged at 9000 × g for 5 min. The washing step was repeated to remove cytosolic contamination. The mitochondrial pellet was resuspended in 100 μl of mitochondrial isolation buffer containing 0.1% Triton X-100. Fresh mitochondrial suspension was used for all of the experiments.
Western Analysis
Total cell lysate, mitochondria, and nuclear proteins were analyzed by Western blotting. Samples were boiled with 1× Laemmli buffer and then subjected to 10% SDS-PAGE and transferred to a nitrocellulose membrane. The transfer efficiency was assessed by incubation with 0.1% Ponceau solution. The membrane was washed with distilled water until the dye disappeared completely. The membranes were then blocked by 5% nonfat dried milk in TBS-T (10 mm Tris-HCl, pH 7.8, 150 mm NaCl, and 0.05% (v/v) Tween 20) buffer, pH 7.8, for at least 1 h at room temperature. After a short wash with TBS-T buffer, the membranes were incubated in the primary antibody for at least 2 h at room temperature or overnight at 4 °C. The primary antibodies were diluted in TBS-T buffer containing 5% nonfat dried milk at a dilution range of 1000–5000. The membranes were then washed three times each for 10 min with TBS-T. The membranes were incubated with the secondary antibody at a dilution range of 2000–10,000 for 1–2 h at room temperature. The membranes were washed twice with TBS-T buffer for 10 min and once with PBS for 5 min. Proteins were detected using the ECL® system (Amersham Biosciences). The Quantity One® Image Analyzer software program (Bio-Rad) was used for quantitative densitometric analysis.
Immunoprecipitation
Immunoprecipitation studies were performed on JB6 cells with radioimmune precipitation buffer (9.1 mm Na2HPO4, 1.7 mm NaH2PO4, 150 mm NaCl, pH 7.4, 1% (v/v) Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, 10 μg/ml phenylmethylsulfonyl fluoride, 1 μg/ml aprotinin). The antibodies used for immunoprecipitation were mouse anti-p53 (DO-1). One mg of cellular protein was incubated at 4 °C overnight with 2 μg of corresponding antibodies. After incubation with the antibody, 20 μl of protein A/G (Santa Cruz Biotechnology) was added to the reaction mixture of the antibody and nuclear extract and rotated for 2 h at 4 °C. Immunoprecipitates were collected by centrifugation at 2500 × g for 5 min, followed by washing four times with radioimmune precipitation buffer. Following the final wash, all of the adhering liquids with the protein A/G beads were removed. Samples were then resuspended in the 1× Laemmli buffer, boiled, and subjected to SDS-polyacrylamide gel electrophoresis, and proteins were then detected by Western blotting.
Immunostaining and Fluorescence Microscopy
Mouse epithelial (JB6) cells were transfected with either pcDNA3.1 or pcDNA3.1/NPM expression vector. Twenty-four h post transfection, cells were trypsinized and seeded (104 cells/400 μl/well) into 8-well poly-l-lysine-coated glass chamber slides (BD Biosciences) and grown for 24 h. Cells were then incubated with 200 nmol/liter MitoTracker Red CMXRos (Molecular Probes, Inc., Eugene, OR) in culture medium for 30 min, after which the dye was removed, and cells were treated with TPA (100 nmol/liter) for 1 h. Cells were washed and fixed in 4% paraformaldehyde for 15 min at room temperature. After two washes with cold PBS (pH 7.4), cells were permeabilized with 0.2% Triton X-100 for 15 min at room temperature. Three percent donkey serum was used to block the nonspecific antibody binding. Anti-p53 antibody (Ab-11) or anti-NPM-v5 antibody (catalogue number 46-0705; Invitrogen) was added at a dilution of 1:100 with 3% donkey serum and incubated at 37 °C, followed by incubation with either anti-mouse IgG-FITC or anti-mouse IgG-Cy2 (Jackson ImmunoResearch Laboratories, West Grove, PA) at a dilution of 1:200 for 1 h. Antibodies were then removed and washed three times with 1× PBS (pH 7.4). 100 μl of 1:10,000 diluted Hoechst 33342 dye was added in each well, and after 10 min, cells were washed with 1× PBS (pH 7.4) to visualize the nucleus. Slides were partially dried and mounted with glycerol. Fluorescence was immediately detected using a Leica (Benheim, Germany) laser-scanning confocal microscope with a magnification of ×40.
TUNEL Assay
Cells were plated (1 × 104) in an 8-chambered polystyrene vessel tissue culture-treated glass slide (BD Biosciences) in 400 μl of medium, and the cells were grown for 24 h. Cells were treated with TPA for 24 h and washed with PBS, and apoptotic cells were detected by using terminal deoxynucleotidyltransferase (In Situ Cell Death Detection Kit, fluorescein; Roche Applied Science) assay kit, which catalyzes polymerization of labeled nucleotides to free 3′-OH ends of DNA in a template-dependent manner (TUNEL reaction). Briefly, cells were fixed with 4% paraformaldehyde for 1 h, followed by the permeabilization of cells with 0.1% Triton X and 0.1% sodium citrate solution. After twice washing the cells with 1× PBS (pH 7.4), TUNEL reaction mixture was added, and the slide was then incubated for 60 min in the dark in a humidified atmosphere at 37 °C. TUNEL-positive cells were counted using fluorescence microscopy. The apoptotic cell numbers were scored by counting 3 × 300 cells for each group in a random manner. The experiment was repeated at least three times to ensure reproducibility.
Caspase 3 Activity Assay
Caspase 3 activity assay was performed colorimetrically using a substrate Ac-DEVD-p-nitroanilide in accordance with the protocol supplied by the manufacturer (Sigma). Briefly, JB6 cells were transfected with either pcDNA3.1 or pcDNA3.1/NPM for 48 h. After treatment with TPA for 1 h, the cells were washed with 1× PBS and then lysed with lysis buffer containing 50 nm HEPES, pH 7.4, 5 mm CHAPS, 5 mm dithiothreitol in addition to 1 μg/ml aprotinin and pepstatin. The cell lysates were incubated on ice for 15 min and centrifuged at 16,000 × g for 10 min. The protein concentration was estimated by the Bradford method, and the caspase 3 activity in the supernatant was measured immediately. Fifty-microgram protein samples or the appropriate amount of standard was added to 980 ml of assay buffer. The reaction was initiated by adding 10 μl of 20 mm caspase 3 substrate Ac-DEVD-p-nitroanilide. The tubes were covered and incubated at 37 °C for 2 h. The cleavage of the chemophore from the substrate was detected spectrophotometrically at a wavelength of 405 nm. Caspase 3 activity was calculated by comparing it with the standard caspase 3 activities from the same experiment and was expressed as nmol of p-nitroanilide/μg of protein/min.
Statistical Analysis
Data were analyzed using one-way analysis of variance for comparison. Bonferroni's post-test multiple comparisons procedure was used to determine the statistical significance. Data shown represent mean ± S.D.
RESULTS
p53 Rapidly Accumulates in Mitochondria after TPA Treatment
Previously, we demonstrated that the total cellular level of p53 was increased after TPA treatment. Consistent with the earlier study, we now have found that p53 was increased as early as 30 min with a significant increase at 1 and 3 h and a decline by 24 h after TPA treatment (Fig. 1A). We have also reported that a fraction of p53 translocated to mitochondria during TPA-induced apoptosis (6). The finding that the p53 level in mitochondria was increased after TPA treatment supports our previous findings (Fig. 1B). The purity of isolated mitochondria was monitored by the presence of minimal nucleus-specific protein PCNA after prolonged exposures (Fig. 1B). Mitochondrial marker, VDAC, and Mn-SOD were detected in the mitochondrial fraction (Fig. 1B). These data confirm that p53 is present in mitochondria and is increased after TPA treatment.
FIGURE 1.

TPA enhances p53 levels. A, a time course study of TPA effect (100 nmol/liter) on cellular p53 levels. JB6 cells were treated with TPA for the indicated period of time. Cell lysates were prepared, and p53 protein was detected by Western blotting. Band intensities were quantified by densitometric scanning, and the relative intensity of p53 was calculated by normalizing to corresponding glyceraldehyde-3-phosphate dehydrogenase (GAPDH) band intensity. Each data point represents the mean ± S.D. of four independent samples. *, p < 0.05; **, p < 0.01 compared with the DMSO-treated group. B, detection of p53 in purified mitochondria and following treatment of TPA for 1 h by Western blotting. The purity of mitochondrial fractions was verified by reprobing the membrane with antibodies specific to nuclear protein PCNA.
Overexpression of NPM Suppresses the Level of p53 in Mitochondria
NPM is a nucleolar protein that can stabilize p53; we therefore investigated how NPM affects the level of p53 in the mitochondria by transfecting NPM expression vector in JB6 cells. Cells were then treated with TPA for 1 h, the mitochondrial fraction was isolated, and the presence of p53 was confirmed by Western blotting. The result shows that cells overexpressing NPM lack p53 in mitochondria (Fig. 2A).
FIGURE 2.

NPM suppresses mitochondrial p53 levels. A, suppression of mitochondrial p53 following NPM overexpression in isolated purified mitochondria following DMSO or TPA treatment for 1 h in JB6 cells. B, localization of NPM following NPM overexpression using antibody specific for exogenously expressed NPM (anti-NPM-V5 antibody) staining and confocal microscopy. Green, NPM; red, MitoTracker; blue, nuclear marker 4′,6-diamidino-2-phenylindole (DAPI). C, co-localization of NPM with p53 using NPM and p53 antibody staining and confocal microscopy. Green, p53; red, NPM (anti-NPM-V5 antibody); blue, 4′,6-diamidino-2-phenylindole. D, interaction of p53 and NPM in JB6 cells. E, overexpression of NPM suppresses mitochondrial p53 in isolated purified mitochondria with doxorubicin treatment in JB6 cell mitochondria (a) and total cell lysates (b). GAPDH, glyceraldehyde-3-phosphate dehydrogenase. IP, immunoprecipitation; WB, Western blot.
To monitor whether the absence of p53 in the mitochondria was due to unknown alteration of p53 resulting in the loss of antibody recognition, we used antibodies from two different sources (Ab-11 and DO-1). The results indicate that mitochondrial p53 was detectable in pcDNA3.1-transfected cells but was undetectable in the mitochondria of NPM-transfected cells. In vector only-transfected cells, p53 was present in the mitochondria, and its level was further increased after TPA treatment. The presence of p53 in mitochondria was not due to nuclear contamination, as verified by Western blotting with antibody specific to PCNA (Fig. 2A). To further verify the suppressive effects of NPM on mitochondrial p53 level, we performed immunocytochemistry staining using antibodies to NPM or p53. The overexpressed NPM was localized mainly in the nucleus with and without TPA treatment (Fig. 2B). In vector-transfected control cells, the p53 level was clearly visible in the cytosol. In contrast, the presence of p53 in NPM-overexpressed cells was largely co-localized with NPM in the nucleus (Fig. 2C). To determine whether the co-localization of NPM and p53 is associated with the physical interaction between these two proteins, we performed co-immunoprecipitation studies using antibody to p53 followed by Western blot analysis (Fig. 2D). The results show that p53 antibody is able to immunoprecipitate NPM, which is confirmed by Western blot analysis. Overexpression of NPM increased the immunoprecipitated proteins as compared with vector-transfected control. These results suggest that p53 interacts with both endogenously and ectopically expressed NPM.
To probe whether the effect of NPM on p53 translocation was specific for TPA, we treated JB6 cells with doxorubicin (0.34 μm). Consistent with a more general effect of NPM on mitochondrial p53, treatment with doxorubicin enhanced p53 localization to mitochondria, which was prevented by overexpression of NPM (Fig. 2E).
NPM Enhances the Expression of the p53 Target Gene
Because co-localization and subcellular fractionation experiments suggest that NPM overexpression may retain p53 in the nucleus, we determined p53 levels in isolated nuclear fractions by Western blotting. TPA treatment for 1 h increased nuclear p53 levels both in pcDNA3.1 (−NPM) and pcDNA3.1/NPM (+NPM) expression vector-transfected cells (Fig. 3). Interestingly, overexpression of NPM alone had a positive effect on the nuclear level of p53 (Fig. 3). The increased nuclear level of p53 in NPM-overexpressed cells was supported by the enhanced level of its target protein p21 in nuclear extracts (Fig. 3). This possibility was further supported by the enhanced level of phosphorylated p53 in NPM-overexpressed cells. These results suggest that the enhanced p53 level is trascriptionally active in JB6 cells.
FIGURE 3.
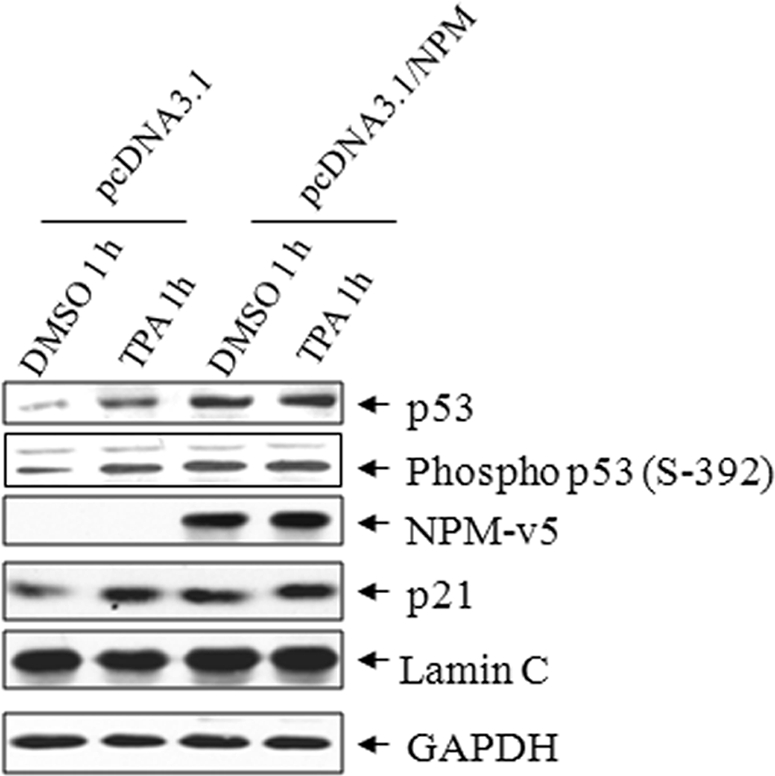
NPM enhances the levels of p53 and its target genes. p53, phospho-p53, and p21 were detected in the isolated purified nuclear extract with or without NPM overexpression in JB6 cells following TPA treatment for 1 h. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
It has been shown that NPM stabilizes p53. To determine whether the lack of mitochondrial p53 in NPM-overexpressed cells is associated with an alteration of p53 stability, we determined the level of p53 in the presence of cycloheximide, an inhibitor of de novo protein synthesis. Transiently transfected cells were treated with TPA for 1 h, and then cycloheximide (20 μg/ml) was added to the medium, and nuclear extracts were collected as a function of time after treatment. The kinetics of p53 degradation in the presence of cycloheximide with or without an NPM-overexpressed condition were monitored by Western analysis (supplemental Fig. 1). The results demonstrate that the rate of decline in p53 levels in NPM-overexpressed cells is slower than the rate of decline of p53 levels in pcDNA3.1 vector-transfected cells. Densitometric quantification of the intensity of p53 bands indicates that the estimated half-life of p53 in NPM-overexpressed cells is longer than the control vector-transfected cells (supplemental Table 1). This result demonstrates that NPM stabilizes p53 in TPA-treated cells.
NPM Protects Cells from TPA-induced Apoptosis
Because overexpression of NPM reduces the level of p53 in the mitochondria, we investigated the effect of NPM in TPA-induced apoptosis. Treatment with TPA significantly increased apoptosis in JB6 cells, as determined by the presence of TUNEL-positive cells. Overexpression of NPM significantly reduced the number of TUNEL-positive cells (Fig. 4A, a). Consistent with the results from the TUNEL assay, overexpression of NPM reduces caspase 3 activity levels (Fig. 4A, b).
FIGURE 4.

NPM suppresses TPA-induced apoptosis. A, JB6 cells were transfected with either pcDNA3.1 or pcDNA3.1/NPM expression vector. a, after treatment of cells with TPA for 24 h, a TUNEL assay was performed, and the TUNEL-positive cells were counted and compared with DMSO control in their randomly selected fields. Data presented are relative -fold changes of TUNEL-positive cells compared with the pcDNA3.1-transfected DMSO control group (10–12% TUNEL-positive cells were observed in the control group). b, caspase 3 activity was determined in cellular extract following treatment with TPA for 1 h. B, HME cells were transfected with either pcDNA3.1 or pcDNA3.1/NPM expression vector and treated with TPA. a, mitochondrial p53 was detected 1 h after TPA treatment by Western blotting. b, -fold changes of TUNEL-positive cells were determined 24 h following TPA treatment. c, overexpression of NPM was confirmed by detecting the ectopic level of NPM. Each data point represents the mean ± S.D. of three experiments. **, p < 0.01 compared with pcDNA3.1-DMSO group; #, p < 0.01 compared with pcDNA3.1-TPA group. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
To determine whether the effect of NPM on mitochondrial p53 localization and apoptosis is specific to JB6 cells, we overexpressed NPM in primary human epithelial (HME) cells. Consistent with the results obtained from JB6 cells, mitochondrial localization of p53 in HME cells was significantly reduced upon overexpression of NPM (Fig. 4B, a). We also monitored the effect of TPA-mediated HME cells apoptosis by a TUNEL assay. Consistent with the role of NPM in JB6 cells, apoptotic cell death of HME cells was increased after the treatment with TPA for 24 h (Fig. 4B, b). A trend of decreased apoptotic cell death was observed in NPM-transfected control cells as compared with the vector alone-transfected control group. Overexpression of NPM significantly suppressed TPA-mediated apoptotic cell death compared with the corresponding vector-transfected cells similarly treated (Fig. 4B, b). Overexpression of NPM was confirmed by Western analysis (Fig. 4B, c).
To verify the effect of NPM in TPA-induced apoptosis, we suppressed endogenous NPM by NPM siRNA. Transfection with NPM siRNA suppressed NPM levels in JB6 cells (Fig. 5A, a). Suppression of NPM also reduced p53 levels in the nucleus (Fig. 5A, b) but enhanced the level of p53 in mitochondria (Fig. 5A, c). The mitochondrial purity was confirmed by the absence of nucleus-specific protein PCNA. Treatment with TPA enhanced p53 level in mitochondria of control siRNA-treated cells, which further increased when the NPM levels were reduced by NPM siRNA. This result confirms that NPM modulates TPA- dependent p53 levels in the mitochondria. The specificity of siRNA was verified by using a 2-bp mismatch NPM siRNA. The endogenous level of NPM did not change in control siRNA- or NPM mismatch siRNA-expressed cells, whereas NPM siRNA clearly suppressed endogenous NPM level (Fig. 5A, d). To further verify the effect of NPM on apoptosis, we examined the effects of NPM siRNA on apoptosis using a TUNEL assay. As shown in Fig. 5A, e, TUNEL-positive cells were increased in NPM siRNA-transfected cells with or without TPA treatment.
FIGURE 5.

NPM siRNA enhances mitochondrial p53 localization and apoptosis. A, JB6 cells. a, NPM siRNA (18.2 pmol or 20 nm final concentration) suppressed NPM protein in total cell lysate. b, NPM siRNA suppressed the nuclear p53 levels in purified nuclear extract. c, p53 was detected in isolated purified mitochondria by Western blotting. Mitochondrial purity was monitored by reprobing the membrane with PCNA, a nucleus-specific protein. Mitochondrial enrichment was confirmed by reprobing the membrane with mitochondrial HSP60. d, specificity of NPM siRNA was determined by comparing with a scrambled or a 2-bp mismatch NPM siRNA on the suppression of NPM. e, cells were transfected with either control or NPM siRNA, and the cells were treated with TPA for 24 h. A TUNEL assay was performed to detect apoptosis. TUNEL-positive cells were counted and compared with DMSO control in their randomly selected fields. Data are presented as -fold changes of TUNEL-positive cells relative to the number of TUNEL-positive cells in the control siRNA-transfected group. Each data point represents the mean ± S.D. of three experiments. **, p < 0.01 compared with the corresponding DMSO group; ##, p < 0.01 compared with the control siRNA DMSO group; #, p < 0.05 compared with the control siRNA-TPA group. B, HME cells. a, the effects of NPM siRNA on NPM protein and p53 in HME cells were monitored by Western blotting in total cell lysate. b, p53 was detected in purified mitochondria following NPM siRNA expression and subsequent TPA treatment. Mitochondrial purity was monitored by reprobing the membrane with PCNA, a nucleus-specific protein, and mitochondrial protein levels were confirmed by reprobing the membrane with mitochondrial HSP60. c, same as in A (e). C, HepG2 cells; a–c, same as in B.
We also verified the effect of NPM on TPA-induced apoptosis in HME cells by suppressing endogenous NPM with NPM siRNA. Transfection of NPM siRNA suppressed NPM levels in HME cells (Fig. 5B, a). Suppression of NPM also reduced the levels of p53 in the total cell lysate upon TPA treatment (Fig. 5B, a) but enhanced the p53 level in mitochondria (Fig. 5B, b). Treatment with TPA increased the p53 level in mitochondria in control siRNA-treated cells, which further increased after the NPM levels were reduced by NPM siRNA. This result confirms that NPM modulates p53 levels in the mitochondria. Under this circumstance, we examined the effects of NPM siRNA on apoptosis of HME cells using a TUNEL assay. As shown in Fig. 5B, c, TUNEL-positive cells were increased in NPM siRNA-transfected cells with or without TPA treatment.
Suppression of NPM in HepG2 Cells Induces Sensitivity to TPA
To further confirm the general effect of NPM on cellular sensitivity to TPA-induced apoptotic cell death, we used malignant human hepatocarcinoma cells, which express a high level of NPM and harbor wild type p53. We reduced the endogenous NPM levels in HepG2 cells using NPM siRNA. The presence of p53 in mitochondria after TPA treatment in control siRNA-transfected cells was undetectable; however, we were able to detect p53 in the mitochondria after suppressing NPM in these cells. Moreover, the mitochondrial level of p53 was further increased after TPA treatment in NPM-suppressed cells compared with NPM siRNA-transfected DMSO-treated control cells (Fig. 5C, b).
Because the suppression of NPM leads to accumulation of p53 in mitochondria, we also investigated the effect of NPM siRNA on TPA-induced apoptosis in HepG2 cells using a TUNEL assay. The results shown in Fig. 5C (c) demonstrate that the number of TUNEL-positive cells was increased in NPM siRNA-transfected cells with or without TPA treatment compared with the corresponding treatment in control siRNA groups.
NPM Overexpression Alters the Levels of Mitochondrial Antiapoptotic and Proapoptotic Proteins
NPM and p53 are known to affect the transcription of proapoptotic and antiapoptotic genes, including bcl2 family members. To probe the effect of NPM and p53 on pro- and antiapoptotic target genes, we measured the protein levels of BCl2 and Bax in the total cell lysate and isolated mitochondria. Consistent with the known transcriptional roles of NPM and p53, cellular levels of Bcl2 and Bax were increased in NPM-transfected cells (supplemental Fig. 2a). Treatment with TPA also increased Bcl2 and Bax in JB6 cells. The increases in Bcl2 and Bax in mitochondria were evident in NPM-transfected cells compared with the pcDNA3.1 (−NPM)-transfected cells (supplemental Fig. 2b). Densitometric analysis of Bcl2/Bax levels demonstrates that the ratio of Bcl2 and Bax in mitochondria is higher in NPM-overexpressed cells. In pcDNA3.1-transfected cells, the mitochondrial Bcl2/Bax ratio significantly decreased after TPA treatment, whereas this ratio did not change significantly in pcDNA3.1/NPM-transfected cells after TPA treatment compared with the corresponding DMSO-treated control (supplemental Fig. 2c). Overall, NPM overexpression increased the Bcl2/Bax ratio, suggesting that overexpression of NPM may protect cells from apoptotic cell death.
Bcl2 Does Not Play a Major Role in the Antiapoptotic Function of NPM
Because overexpression of NPM reduces apoptosis in the presence of higher Bcl2 and Bax levels in mitochondria, we tested whether the antiapoptotic function of NPM is mediated by Bcl2. For this purpose, JB6 cells were co-transfected with NPM expression vector and bcl2 siRNA. Apoptotic cells were detected by a TUNEL assay (supplemental Fig. 2d). The result shows that both bcl2 siRNA- and TPA-mediated apoptosis were significantly reduced by NPM overexpression. Under normal cellular levels of NPM, suppression of Bcl2 by bcl2 siRNA increased apoptotic cell death both in DMSO- and TPA-treated cells (supplemental Fig. 2d). However, overexpression of NPM with control siRNA or bcl2 siRNA significantly reduced the number of apoptotic cells both in the DMSO- and TPA-treated groups. The suppression of Bcl2 expression level by bcl2 siRNA is verified in supplemental Fig. 2e. These results suggest that the presence of Bcl2 does not contribute significantly to the protective role of NPM against TPA-induced apoptosis.
DISCUSSION
We have previously shown that, following TPA treatment in JB6 cells, p53 translocates to both mitochondria and nucleus, as detected by immunofluorescence staining of immunoreactive p53 protein (6). The present study confirms our previous findings and extends them to demonstrate that suppression of mitochondrial p53 plays an important role in preventing stress-induced apoptosis. Importantly, our results demonstrate that suppression of mitochondrial p53 can be accomplished by overexpression of NPM, a nucleolar protein capable of stabilizing p53 by preventing its interaction with MDM2 (17). Our results confirm the role of NPM in stabilizing p53 and in retaining most of the p53 in nucleus. Our results also demonstrate that nuclear p53 is transcriptionally active in NPM-overexpressed cells, as demonstrated by the increased levels of its target genes, such as p21. Our findings that NPM stabilizes p53 and increases its transcription activity are also consistent with an earlier report on murine primary fibroblasts (15).
The increase of p21, a p53 target gene, is implicated in the regulation of the cell cycle. p21 inhibits the activities of cyclin-Cdk2 complexes and is an important regulator of the cell cycle (31). In the present study, we observed that NPM increased p21 expression. The increase of p21 due to the overexpression of NPM suggests that NPM may have an indirect role in cell cycle regulation. Previous studies by us and others have indicated that p53 translocation to mitochondria precedes its nuclear translocation. Thus, it is possible that the effect of p21 on the cell cycle may occur after the direct role of p53 in mitochondria. NPM overexpression also increases Bax, a p53 target gene. However, despite the increase in the proapoptotic Bax, the presence of NPM blocks apoptosis in TPA-treated JB6 cells, confirming an important role of mitochondrial p53 in TPA-induced apoptosis.
Our results indicate that overexpression of NPM suppresses mitochondrial p53 under normal and stress conditions. These data are consistent with the notion that NPM stabilizes p53 in the nucleus, which is consistent with the well documented role of NPM on p53 functions (17) (Figs. 2 and 3). It is well documented that increased phosphorylation of p53 at the Ser392 position stabilizes the tetramer formation of tumor suppressor protein p53 (32). It has been shown that proteasome inhibitor MG132 increases the stability of p53 by enhancing the expression of p53 S-392 phosphorylation (33). It has also been shown that stress-dependent stabilization of p53 in the cytoplasmic pool targets p53 to locate in the mitochondria after monoubiquitylation (34). The stability of p53 is increased by PML (promyolytic leukemia protein), resulting in impairment of p53 ubiquitylation (35, 36). Since we do not know whether NPM-mediated stabilization of p53 inhibits p53 monoubiquitylation, we could not rule out the possible role of NPM in monoubiquitylation of p53. It has been demonstrated that NPM overexpression protects cells from various stress conditions, including DNA damage, hypoxia, and UV exposure through the stabilization and activation of p53 (2, 37, 38). However, the status of mitochondrial p53 in NPM-overexpressed cells remains unknown. Our data provide the first evidence for the role of NPM in suppressing p53 levels in mitochondria. Our data also provide the first evidence to demonstrate that the presence of mitochondrial p53 contributes significantly to TPA-induced apoptosis. Our results are consistent with the results from an elegant study that demonstrated that lack of NPM expression in embryonic fibroblast cells results in widespread apoptosis in wild- type p53-expressing cells (19) and extend them to demonstrate that the lack of NPM may contribute to mitochondria-mediated apoptosis. Our results further demonstrate that although stabilization of p53 in the nucleus results in the activation of its proapoptotic target gene bax, it does not lead to enhanced apoptosis when p53 is absent in mitochondria.
Over a decade ago it was demonstrated that extranuclear p53-dependent apoptosis ensued in the absence of transcriptional activation of p53 target genes (39). Supporting this finding, several lines of studies have provided evidence for a direct transcription-independent apoptotic role of p53 (29, 40, 41). The fact that p53 can have a direct apoptogenic role in mitochondria was first demonstrated by targeting p53 to mitochondria using a mitochondrial target signal link to the N-terminal region of wild-type p53, thereby allowing it to bypass the nucleus (42). It has also been shown that overexpression of p53 lacking nuclear localization signal targets p53 to mitochondria and executes p53-dependent apoptosis in p53 null cells (29). However, the question remains whether the presence of p53 in mitochondria plays an important role in apoptosis despite the presence of p53 in nuclei. Our unique approach to retain p53 in nuclei, by overexpressing NPM, provides an opportunity to directly address this important question. The results demonstrate that the presence of p53 in mitochondria is important for stress-induced apoptosis. Importantly, we show that the proapoptotic function of mitochondrial p53 is needed despite an increase in nuclear p53 with a subsequent increase in Bax level. However, we could not exclude the possibility that increased expression of NPM may also lead to enhancement of other prosurvival genes, leading to the observed protective effect of NPM.
It has been reported that suppression of NPM by siRNA targeting NPM in normal lymphoblast and leukemia cell line UoC-M1 (where NPM levels are very high) increases stress-induced apoptosis (43). Reduced levels of NPM lead to apoptotic cell death of retinoic acid-induced differentiation of leukemia cells (HL-60) (44). Increased expression of NPM leads cells to be resistant to apoptosis by UV exposure in UV-sensitive cells (26, 45). Our finding that overexpression of NPM also leads to an increase in the level of mitochondrial Bcl2 is consistent with this possibility. Bcl2 is a prosurvival member of the Bcl2 family of proteins (46). Bcl2 family proteins act as a critical determinant in the life-or-death decision of cells. Although it is unclear how NPM regulates Bcl2 expression in this instance, our previous study demonstrated that NPM is a NF-κB co-activator (47). bcl2 is a known NF-κB target gene; therefore, it is possible that NPM may transactivate bcl2 gene expression through NF-κB-mediated pathways. Our data show that Bcl2 is also increased after NPM overexpression. However, the suppression of Bcl2 by Bcl2 siRNA with the concomitant overexpression of NPM protects cells from TPA-induced apoptosis, suggesting that the effect of NPM is not dependent on an increase in Bcl2 levels (supplemental Fig. 2).
Although it is well documented that hyperphysiological levels of p53 are associated with the activation of proapoptotic genes (48), our results indicate that NPM can suppress apoptosis despite the increase of the proapoptotic gene bax. This is an interesting observation, which suggests that the proapoptotic function of bax may be dependent on the presence of mitochondrial p53. This possibility is supported by previous studies, which demonstrated that p53 has separate cytoplasmic roles in regulating the Bax-dependent mitochondrial pathway to cell death (49). Thus, an apoptosis-associated specklike protein (ASC) functions as an adapter molecule for Bax and regulates a p53-Bax mitochondrial pathway of apoptosis (50). Overall, these data lead us to suggest a unique mechanism by which NPM prevents apoptosis by reducing the level of mitochondrial p53. Our findings that this effect is operating in other cellular systems, including primary human epithelial cells, with different stress inducers, such as doxorubicin, suggest that the role of mitochondrial p53 in apoptosis is likely to be a general function of p53. It is possible that under normal levels of NPM, stress-activated p53 is localized in mitochondria and executes p53-dependent apoptosis, whereas in NPM-highly expressed cells (which is common in cancer cells), p53 translocation to mitochondria is diminished, enabling the cells to escape from mitochondrial p53-mediated apoptosis, leading to cell survival (Fig. 6). This possibility is supported by our findings that, when NPM was suppressed in high NPM expressing cancer cells (HepG2 cells) by siRNA, p53 localized to mitochondria and increased apoptosis (Fig. 5C).
FIGURE 6.

Schematic depiction of how NPM blocks stress-induced apoptosis. TPA activates NF-κB and p53. The interplay between p53 and NPM leading to apoptosis or survival is dependent on the presence of p53 in mitochondria. NPM enhances p53 stabilization in the nucleus and activates p53 target genes, including bax; NPM as an NF-κB co-activator also induces NF-κB target genes, including bcl2. In normal cells, stress activates p53 and Bax translocation to mitochondria, leading to apoptosis. In cells expressing high levels of NPM, NPM blocks p53 localization to mitochondria, leading to protection from apoptosis despite the increase in Bax level.
Taken together, the present study demonstrates that the presence of p53 in mitochondria plays an important role in stress-mediated apoptosis. This effect can be prevented when cells overexpress NPM. It is possible that pathological elevation of NPM in cancer cells, including skin cancer, is a mechanism for maintaining cell survival and resisting apoptosis. Thus, a NPM-based therapeutic approach could be investigated as a novel mechanism for cancer prevention or therapy.
Supplementary Material
Acknowledgment
We cordially thank Dr. Nancy H. Colburn (NCI-Frederick, National Institutes of Health) for the generous gift of the JB6 cell line.
This work was supported, in whole or in part, by National Institutes of Health Grants CA 49797 and CA 73599 (to D. K. S. C.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1 and 2 and Table 1.
- TPA
- 12-O-tetradecanoylphorbol-13-acetate
- TUNEL
- terminal deoxynucleotidyltransferase dUTP nick end labeling
- HME
- human mammary epithelial
- siRNA
- small interfering RNA
- PBS
- phosphate-buffered saline
- CHAPS
- 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonic acid
- PCNA
- proliferating cell nuclear antigen
- VDAC
- voltage-dependent anion channel.
REFERENCES
- 1.Levine A. J. ( 1997) Cell 88, 323– 331 [DOI] [PubMed] [Google Scholar]
- 2.Prives C., Hall P. A. ( 1999) J. Pathol. 187, 112– 126 [DOI] [PubMed] [Google Scholar]
- 3.Bates S., Vousden K. H. ( 1999) Cell Mol. Life Sci. 55, 28– 37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lohrum M. A., Vousden K. H. ( 1999) Cell Death Differ. 6, 1162– 1168 [DOI] [PubMed] [Google Scholar]
- 5.Dhar S. K., Xu Y., Chen Y., St. Clair D. K. ( 2006) J. Biol. Chem. 281, 21698– 21709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhao Y., Chaiswing L., Velez J. M., Batinic-Haberle I., Colburn N. H., Oberley T. D., St. Clair D. K. ( 2005) Cancer Res. 65, 3745– 3750 [DOI] [PubMed] [Google Scholar]
- 7.Polyak K., Xia Y., Zweier J. L., Kinzler K. W., Vogelstein B. ( 1997) Nature 389, 300– 305 [DOI] [PubMed] [Google Scholar]
- 8.Miyashita T., Reed J. C. ( 1995) Cell 80, 293– 299 [DOI] [PubMed] [Google Scholar]
- 9.Oda E., Ohki R., Murasawa H., Nemoto J., Shibue T., Yamashita T., Tokino T., Taniguchi T., Tanaka N. ( 2000) Science 288, 1053– 1058 [DOI] [PubMed] [Google Scholar]
- 10.Oda K., Arakawa H., Tanaka T., Matsuda K., Tanikawa C., Mori T., Nishimori H., Tamai K., Tokino T., Nakamura Y., Taya Y. ( 2000) Cell 102, 849– 862 [DOI] [PubMed] [Google Scholar]
- 11.Erster S., Mihara M., Kim R. H., Petrenko O., Moll U. M. ( 2004) Mol. Cell. Biol. 24, 6728– 6741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li P. F., Dietz R., Von Harsdorf R. ( 1999) EMBO J. 18, 6027– 6036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marchenko N. D., Zaika A., Moll U. M. ( 2000) J. Biol. Chem. 275, 16202– 16212 [DOI] [PubMed] [Google Scholar]
- 14.Wu M. H., Yung B. Y. ( 2002) J. Biol. Chem. 277, 48234– 48240 [DOI] [PubMed] [Google Scholar]
- 15.Colombo E., Marine J. C., Danovi D., Falini B., Pelicci P. G. ( 2002) Nat. Cell Biol. 4, 529– 533 [DOI] [PubMed] [Google Scholar]
- 16.Bertwistle D., Sugimoto M., Sherr C. J. ( 2004) Mol. Cell. Biol. 24, 985– 996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kurki S., Peltonen K., Latonen L., Kiviharju T. M., Ojala P. M., Meek D., Laiho M. ( 2004) Cancer Cell 5, 465– 475 [DOI] [PubMed] [Google Scholar]
- 18.Ryan K. M., Phillips A. C., Vousden K. H. ( 2001) Curr. Opin. Cell Biol. 13, 332– 337 [DOI] [PubMed] [Google Scholar]
- 19.Colombo E., Bonetti P., Lazzerni E. D., Martinelli P., Zamponi R., Marine J. C., Helin K., Falini B., Pelicci P. G. ( 2005) Mol. Cell. Biol. 25, 8874– 8886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nozawa Y., Van Belzen N., Van der Made A. C., Dinjens W. N., Bosman F. T. ( 1996) J. Pathol. 178, 48– 52 [DOI] [PubMed] [Google Scholar]
- 21.Shields L. B., Gercel-Taylor C., Yashar C. M., Wan T. C., Katsanis W. A., Spinnato J. A., Taylor D. D. ( 1997) J. Soc. Gynecol. Invest. 4, 298– 304 [PubMed] [Google Scholar]
- 22.Subong E. N., Shue M. J., Epstein J. I., Briggman J. V., Chan P. K., Partin A. W. ( 1999) Prostate 39, 298– 304 [DOI] [PubMed] [Google Scholar]
- 23.Tanaka M., Sasaki H., Kino I., Sugimura T., Terada M. ( 1992) Cancer Res. 52, 3372– 3377 [PubMed] [Google Scholar]
- 24.Kondo T., Minamino N., Nagamura-Inoue T., Matsumoto M., Taniguchi T., Tanaka N. ( 1997) Oncogene 15, 1275– 1281 [DOI] [PubMed] [Google Scholar]
- 25.Korgaonkar C., Hagen J., Tompkins V., Frazier A. A., Allamargot C., Quelle F. W., Quelle D. E. ( 2005) Mol. Cell. Biol. 25, 1258– 1271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li J., Zhang X., Sejas D. P., Bagby G. C., Pang Q. ( 2004) J. Biol. Chem. 279, 41275– 41279 [DOI] [PubMed] [Google Scholar]
- 27.Itahana K., Bhat K. P., Jin A., Itahana Y., Hawke D., Kobayashi R., Zhang Y. ( 2003) Mol. Cell 12, 1151– 1164 [DOI] [PubMed] [Google Scholar]
- 28.Bakhshi A., Jensen J. P., Goldman P., Wright J. J., McBride O. W., Epstein A. L., Korsmeyer S. J. ( 1985) Cell 41, 899– 906 [DOI] [PubMed] [Google Scholar]
- 29.Tsujimoto Y., Croce C. M. ( 1986) Proc. Natl. Acad. Sci. U. S. A. 83, 5214– 5218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Colburn N. H., Former B. F., Nelson K. A., Yuspa S. H. ( 1979) Nature 281, 589– 591 [DOI] [PubMed] [Google Scholar]
- 31.Brugarolas J., Moberg K., Boyd S. D., Taya Y., Jacks T., Lees J. A. ( 1999) Proc. Natl. Acad. Sci. U. S. A. 96, 1002– 1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sakaguchi K., Sakamoto H., Lewis M. S., Anderson C. W., Erickson J. W., Appella E., Xie D. ( 1997) Biochemistry 36, 10117– 10124 [DOI] [PubMed] [Google Scholar]
- 33.Chen J. J., Chou C. W., Chang Y. F., Chen C. C. ( 2008) J. Immunol. 180, 8030– 8039 [DOI] [PubMed] [Google Scholar]
- 34.Marchenko N. D., Wolff S., Erster S., Becker K., Moll U. M. ( 2007) EMBO J. 26, 923– 934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bernardi R., Scaglioni P. P., Bergmann S., Horn H. F., Vousden K. H., Pandolfi P. P. ( 2004) Nat. Cell Biol. 6, 665– 672 [DOI] [PubMed] [Google Scholar]
- 36.Louria-Hayon I., Grossman T., Sionov R. V., Alsheich O., Pandolfi P. P., Haupt Y. ( 2003) J. Biol. Chem. 278, 33134– 33141 [DOI] [PubMed] [Google Scholar]
- 37.Schuler M., Green D. R. ( 2001) Biochem. Soc. Trans. 29, 684– 688 [DOI] [PubMed] [Google Scholar]
- 38.Vogelstein B., Lane D., Levine A. J. ( 2000) Nature 408, 307– 310 [DOI] [PubMed] [Google Scholar]
- 39.Caelles C., Helmberg A., Karin M. ( 1994) Nature 370, 220– 223 [DOI] [PubMed] [Google Scholar]
- 40.Chen X., Ko L. J., Jayaraman L., Prives C. ( 1996) Genes Dev. 10, 2438– 2451 [DOI] [PubMed] [Google Scholar]
- 41.Haupt Y., Rowan S., Shaulian E., Vousden K. H., Oren M. ( 1995) Genes Dev. 9, 2170– 2183 [DOI] [PubMed] [Google Scholar]
- 42.Mihara M., Erster S., Zaika A., Petrenko O., Chittenden T., Pancoska P., Moll U. M. ( 2003) Mol. Cell 11, 577– 590 [DOI] [PubMed] [Google Scholar]
- 43.Li J., Zhang X., Sejas D. P., Pang Q. ( 2005) Leuk. Res. 29, 1415– 1423 [DOI] [PubMed] [Google Scholar]
- 44.Hsu C. Y., Yung B. Y. ( 1998) Oncogene 16, 915– 923 [DOI] [PubMed] [Google Scholar]
- 45.Higuchi Y., Kita K., Nakanishi H., Wang X. L., Sugaya S., Tanzawa H., Yamamori H., Sugita K., Yamaura A., Suzuki N. ( 1998) Biochem. Biophys. Res. Commun. 248, 597– 602 [DOI] [PubMed] [Google Scholar]
- 46.Reed J. C., Jurgensmeier J. M., Matsuyama S. ( 1998) Biochim. Biophys. Acta 1366, 127– 137 [DOI] [PubMed] [Google Scholar]
- 47.Dhar S. K., Lynn B. C., Daosukho C., St. Clair D. K. ( 2004) J. Biol. Chem. 279, 28209– 28219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu B., Chen Y., St. Clair D. K. ( 2008) Free Radic. Biol. Med. 15, 1529– 1535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Baptiste N., Prives C. ( 2004) Cell 116, 487– 489 [DOI] [PubMed] [Google Scholar]
- 50.Ohtsuka T., Ryu H., Minamishima Y. A., Macip S., Sagara J., Nakayama K. I., Aaronson S. A., Lee S. W. ( 2004) Nat. Cell Biol. 6, 121– 128 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.