

Abstract
FTY720, a sphingosine analog, is in clinical trials as an immunomodulator. The biological effects of FTY720 are believed to occur after its metabolism to FTY720 phosphate. However, very little is known about whether FTY720 can interact with and modulate the activity of other enzymes of sphingolipid metabolism. We examined the ability of FTY720 to modulate de novo ceramide synthesis. In mammals, ceramide is synthesized by a family of six ceramide synthases, each of which utilizes a restricted subset of acyl-CoAs. We show that FTY720 inhibits ceramide synthase activity in vitro by noncompetitive inhibition toward acyl-CoA and uncompetitive inhibition toward sphinganine; surprisingly, the efficacy of inhibition depends on the acyl-CoA chain length. In cultured cells, FTY720 has a more complex effect, with ceramide synthesis inhibited at high (500 nm to 5 μm) but not low (<200 nm) sphinganine concentrations, consistent with FTY720 acting as an uncompetitive inhibitor toward sphinganine. Finally, electrospray ionization-tandem mass spectrometry demonstrated, unexpectedly, elevated levels of ceramide, sphingomyelin, and hexosylceramides after incubation with FTY720. Our data suggest a novel mechanism by which FTY720 might mediate some of its biological effects, which may be of mechanistic significance for understanding its mode of action.
FTY720 (2-amino-(2-2-[4-octylphenyl]ethyl)propane 1,3-diol hydrochloride), also known as Fingolimod, is an immunosuppressant drug currently being tested in clinical trials for organ transplantation and autoimmune diseases such as multiple sclerosis (1). FTY720 is a structural analog of sphingosine, a key biosynthetic intermediate in sphingolipid (SL)2 metabolism (see Fig. 1). In vivo, FTY720 is rapidly phosphorylated by sphingosine kinase 2 (2, 3) to form FTY720 phosphate (FTY720-P), an analog of sphingosine 1-phosphate (S1P) (see Fig. 1A). FTY720-P binds to S1P receptors (S1PRs) (4, 5) and thereby induces a variety of phenomena such as T-lymphocyte migration from lymphoid organs (6–9); accordingly, FTY720 treatment results in lymphopenia as lymphocytes (especially T-cells) become sequestered inside lymphoid organs (10–12). The ability of FTY720 to sequester lymphocytes has stimulated its use in treatment of allograft rejection and autoimmune diseases (13), and FTY720 is currently under phase III clinical trials for treatment of relapsing-remitting multiple sclerosis (14).
FIGURE 1.

SL structure and metabolism. A, structures of SLs and SL analogs used in this study. B, metabolic inter-relationships between SLs and the metabolism of FTY720. The enzymes are denoted in italics. LPP3, lipid phosphate phosphatase 3; LPP1α, lipid phosphate phosphatase 1α.
Apart from the binding of FTY720-P to S1PRs, the ability of FTY720 to inhibit S1P lyase (15) (see Fig. 1B), and its inhibitory effect on cytosolic phospholipase A2 (16), whose activity can be modulated by ceramide 1-phosphate (17), little is known about whether FTY720 or FTY720-P can modulate the activity of other enzymes of SL metabolism. Because FTY720 is an analog of sphingosine, one of the two substrates of ceramide synthase (CerS) (see Fig. 1), we now examine whether FTY720 can modulate CerS activity. CerS utilizes fatty acyl-CoAs to N-acylate sphingoid long chain bases. Six CerS exist in mammals, each of which uses a restricted subset of acyl-CoAs (18–23). We demonstrate that FTY720 inhibits CerS activity and that the extent of inhibition varies according to the acyl chain length of the acyl-CoA substrate. Surprisingly, FTY720 inhibits CerS activity toward acyl-CoA via noncompetitive inhibition and toward sphinganine via uncompetitive inhibition. Finally, the mode of interaction of FTY720 with CerS in cultured cells depends on the amount of available sphinganine. Together, we show that FTY720 modulates ceramide synthesis, which may be of relevance for understanding its biological effects in vivo and its role in immunomodulation.
EXPERIMENTAL PROCEDURES
Materials
d-Erythro-[4,5-3H]Sphinganine (80 Ci/mmol), FTY720, (R)-FTY720-P and (R)-FTY-phosphonate were synthesized as described (24, 25). Fatty acyl-CoAs, ceramides, dimethylsphingosine, lysophosphatidic acid, and the internal standards for liquid chromatography electrospray ionization-tandem mass spectrometry (ESI MS/MS) were from Avanti Polar Lipids (Alabaster, AL). Sphinganine was from Matreya (Pleasant Gap, PA). Silica gel 60 TLC plates were from Merck. Defatted bovine serum albumin (BSA), suramin, S1P, and a protease inhibitor mixture were from Sigma-Aldrich. Dulbecco's modified Eagle's medium was from Invitrogen or Sigma-Aldrich. All of the solvents were of analytical grade and were purchased from Biolab (Jerusalem, Israel).
Cell Culture and Transfection
Human embryonic kidney (HEK) 293T cells, RBL-2H3 mast cells, and hepatocellular liver carcinoma (HEPG2) cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum, 100 IU/ml penicillin, and 100 μg/ml streptomycin. HEK cells were transfected with human CerS genes (26) using the calcium phosphate method (0.25 μg of plasmid/cm2 of culture dish), which gave ∼90% transfection efficiency.
CerS Assay
The cells were removed from culture dishes using trypsin (0.25%, w/v) followed by centrifugation (4 min, 150 × gav). The cell pellets were washed with phosphate-buffered saline and homogenized in 20 mm HEPES-KOH, pH 7.4, 25 mm KCl, 250 mm sucrose, and 2 mm MgCl2 containing a protease inhibitor mixture. Protein was estimated using the Bradford protein assay (Bio-Rad). CerS activity was assayed essentially as described (26). Briefly, cell homogenates were incubated with [4,5-3H]sphinganine/sphinganine/20 μm defatted BSA and fatty acyl-CoA for 20 min at 37 °C. Stearoyl-CoA (C18-CoA) was used for CerS1 and 4 (18, 19), lignoceroyl-CoA (C24-CoA) or behenoyl-CoA (C22-CoA) was used for CerS2 (22), and palmitoyl-CoA (C16-CoA) was used for CerS5 (19). The amount of homogenate used for each CerS was taken so that the reaction rate was linear with respect to the amount of protein (26). The reactions were terminated using chloroform/methanol (1/2; v/v), and the lipids were extracted (27). Lipids were separated by TLC using chloroform/methanol/2 m ammonium hydroxide (40/10/1; v/v/v) as developing solvent. [3H]Lipids were identified using authentic standards, visualized using a phosphorimaging screen (Fuji, Tokyo, Japan), and recovered from the TLC plates by scraping the silica directly into scintillation vials containing 1 ml of methanol and 5 ml of Optima Gold scintillation fluid (Packard, Downers Groove, IL). Radioactivity was determined using a Packard 2100 beta radiospectrometer equipped with the Transformed Spectral Index of the External Standard/Automatic Efficiency Control program for quench correction.
Metabolic Labeling
HEK and RBL-2H3 cells were incubated with 25 μm FTY720 for 1.5 h followed by incubation with [4,5-3H]sphinganine or [4,5-3H]sphinganine/sphinganine during the last 30 min of the incubation period; when transfected HEK cells were used, they were incubated with FTY720 and sphinganine 24 h after transfection. The cells were removed from the culture dishes by scraping into methanol, and the lipids were extracted (28). The lipids were separated by TLC using chloroform/methanol (50:3.5; v/v) as developing solvent, identified, and quantified as above.
ESI-MS/MS
SL analyses by ESI-MS/MS were conducted using a PE-Sciex API 3000 triple quadrupole mass spectrometer and an ABI 4000 quadrupole linear ion trap mass spectrometer as described (18, 19, 29, 30). HEK cells were harvested by trypsinization, collected by centrifugation, washed twice with ice-cold phosphate-buffered saline, and lyophilized. The samples were spiked with a SL internal standard mixture (Avanti Polar Lipids), then extracted, and analyzed by liquid chromatography ESI-MS/MS.
RESULTS
Inhibition of CerS Activity
To determine whether FTY720 inhibits CerS activity in vitro, homogenates from HEK cells overexpressing CerS2 were incubated with increasing concentrations of FTY720. Significant inhibition of CerS2 activity was observed (see Fig. 2A). In contrast, another sphingosine analog, dimethylsphingosine (Fig. 1), had no effect on CerS2 (Fig. 2A) or CerS4 and 5 activity (not shown). The lack of effect of dimethylsphingosine also acts as an important control, demonstrating that the inhibition of CerS2 by FTY720 does not occur via a nonspecific mechanism. For instance, it is conceivable that FTY720 could displace sphinganine from the complex it forms with BSA (which acts as a lipid carrier in the reaction mixture; see Refs 24 and 26), thus rendering sphinganine inaccessible for the reaction. Based on these results, we conclude that FTY720 specifically inhibits CerS2.
FIGURE 2.

Effect of FTY720 on CerS2 activity. CerS2 activity was assayed in homogenates (100 μg of protein) from CerS2-overexpressing cells using 0.25 μCi of [4,5-3H]sphinganine, 15 μm sphinganine, 20 μm defatted BSA and 50 μm C22-acyl-CoA, with FTY or dimethylsphingosine (A) or FTY720-P, lysophosphatidic acid, or S1P (B). The specific activity of CerS2 in the absence of inhibitors was 84 ± 4.6 pmol ceramide formed per min per mg of protein. The data in A is a typical experiment for HEK cells overexpressing CerS2, and similar results were obtained for cells overexpressing CerS4 and 5. In B, the results are the means ± S.D. for FTY720-P and S1P (n = 4) and the means (n = 2) for lysophosphatidic acid.
We have recently shown that CerS2 contains an S1PR-like motif via which S1P inhibits CerS2 activity (22). To determine whether FTY720-P, an analog of S1P (Fig. 1A), can also inhibit CerS2 via this motif, homogenates from CerS2-overexpressing cells were incubated with FTY720-P. No inhibition was observed, whereas S1P significantly inhibited CerS2 (Fig. 2B), as reported previously (22). This result also demonstrates that FTY720 itself (Fig. 2A) and not FTY720-P, is responsible for CerS2 inhibition. Similarly, FTY720-P had no effect on CerS5 activity (data not shown). Likewise, lysophosphatidic acid did not inhibit CerS2 (Fig. 2B), confirming the specificity of the CerS2 S1P receptor-like motif for S1P.
We next examined the level of inhibition of FTY720 using different acyl-CoAs for ceramide synthesis (Fig. 3). In three different cell lines, namely HEK, RBL-2H3 mast cells, and hepatocellular liver carcinoma (HEPG2) cells, the EC50 of inhibition was lowest using C18-CoA, which gave an EC50 value of between 35 and 47 μm (Fig. 3). In contrast, the EC50 values using either C16-CoA or C24-CoA were considerably higher (Fig. 3). The dependence on acyl-CoA chain length was further examined by comparing the EC50 value using HEK cells overexpressing either CerS5 or CerS1, which use C16- and C18-CoA, respectively (18, 19). FTY720 inhibited CerS1 activity to a greater extent than CerS5 (Fig. 4A). Finally, we analyzed the activity of CerS4 in HEK cells using C18- or C20-CoA; CerS4 can utilize either of these acyl-CoAs for ceramide synthesis (19). There was a significant difference in the EC50, with values of 32 and 65 μm for C18- and C20-CoA, respectively (Fig. 4B). Together, these results suggest that the efficacy of inhibition of CerS by FTY720 depends on the acyl-CoA chain length. This result is unexpected because FTY720 is a sphingosine analog (Fig. 1A), and therefore we assumed that the chain length of the acyl-CoA would not be relevant for CerS inhibition by FTY720.
FIGURE 3.

Inhibition of CerS activity by FTY720 using various acyl-CoAs. CerS activity was assayed in homogenates (100 μg of protein) from HEK, RBL-2H3, and HEPG2 cells using 0.25 μCi of [4,5-3H]sphinganine, 15 μm sphinganine, 20 μm defatted BSA, and 50 μm of acyl-CoA, as indicated. The results are the means ± S.D. (n = 3) for HEK cells and the means ± S.D. of two independent experiments for RBL and HEPG2 cells.
FIGURE 4.

Dependence on a cyl-CoA chain length of inhibition of CerS activity by FTY720. A, CerS activity was assayed in homogenates (100 μg of protein) of HEK cells using 0.25 μCi of [4,5-3H]sphinganine/15 μm sphinganine, 20 μm defatted BSA and 50 μm of either C18- (for CerS1) or C16-acyl-CoA (for CerS5). B, CerS4 activity was assayed in homogenates (100 μg of protein) from CerS4-overexpressing cells using 0.25 μCi of [4,5-3H]sphinganine, 15 μm sphinganine, 20 μm defatted BSA, and 50 μm of either C18- or C20-acyl-CoA. The specific activity of CerS4 in the absence of FTY720 toward C18-CoA was 444 pmol of ceramide formed per min per mg of protein and 389 pmol of ceramide formed per min per mg of protein toward C20-CoA in a typical experiment. The results are the means ± S.D. of two independent experiments, each of which was repeated at least three times with similar results.
We also examined whether FTY720 could be acylated by CerS. HEK cells overexpressing CerS4 or CerS5 were incubated with 1-[14C]stearoyl-CoA or 1-[14C]palmitoyl-CoA, respectively. Although [14C]stearoyl-sphinganine (C18-ceramide) and [14C]palmitoyl-sphinganine (C16-ceramide) were both synthesized and their formation significantly inhibited in the presence of FTY720, no additional radioactive bands could be detected by TLC after incubation with FTY720 (data not shown).
Kinetic Analysis
We next examined the inhibition kinetics of FTY720 in homogenates from HEK cells overexpressing CerS4. Because FTY720 is a structural analog of the sphingoid long chain base (Fig. 1A), we assumed that FTY720 would act as a competitive inhibitor toward sphinganine. Surprisingly, however, FTY720 was an uncompetitive inhibitor toward sphinganine (Fig. 5A).
FIGURE 5.

Kinetic analysis of CerS4 inhibition by FTY 720. Homogenates (100 μg of protein) were prepared from HEK cells overexpressing CerS4 and incubated with (squares) or without (circles) 25 μm FTY720. A, CerS4 activity was assayed using 50 μm C18-CoA and increasing amounts of [4,5-3H]sphinganine/sphinganine, 20 μm defatted bovine serum albumin (such that the ratio of [4,5-3H]sphinganine to sphinganine was the same at each sphinganine concentration). The data were plotted according to Lineweaver-Burk. The slopes of the lines were parallel, with values of 0.0169 ± 0.0019 and 0.0167 ± 0.0019 with and without FTY720, respectively, demonstrating that FTY inhibits CerS4 activity toward sphinganine via uncompetitive inhibition. The results are shown for a typical experiment repeated three times. B, CerS4 activity was assayed using 0.25 μCi of [4,5-3H]sphinganine, 15 μm sphinganine, 20 μm defatted bovine serum albumin, and increasing amounts of C18-acyl-CoA. The data were plotted according to the best fit of a sigmoid dose-response curve, which gave r2 values of 0.99 for both curves, and demonstrate that FTY720 inhibits CerS4 via noncompetitive inhibition toward C18-acyl-CoA. The inset shows a Lineweaver-Burk plot from a different experiment in which defatted bovine serum albumin was not included in the reaction buffer. Noncompetitive inhibition toward C18-acyl-CoA is also observed. The results are the means ± S.D. from two independent experiments repeated five or six times.
In contrast, FTY720 acted as a noncompetitive inhibitor toward C18-CoA (Fig. 5B). The inhibition of FTY720 toward C18-CoA was allosteric (Fig. 5B) under the normal reaction conditions used to assay CerS (i.e. using defatted BSA as lipid carrier, see above). However, when the assay was performed in the absence of defatted BSA, classical noncompetitive inhibition was obtained using the Lineweaver-Burk plot (Fig. 5B, inset). These data indicate that FTY720 has a complex mode of inhibition on CerS in vitro, acting as an uncompetitive inhibitor toward sphinganine and a noncompetitive inhibitor toward acyl-CoA.
Modulation of Ceramide Synthesis in Cultured Cells
To determine whether FTY720 also inhibits ceramide synthesis in cultured cells, mock-transfected HEK cells were incubated with 25 μm FTY720 and then metabolically labeled with 1 μCi of [4,5-3H]sphinganine/2 μm sphinganine. Significant inhibition of ceramide synthesis was observed under these conditions (Fig. 6A). Similarly, ceramide synthesis was inhibited by FTY720 in CerS1- and CerS4-overexpressing HEK cells (Fig. 6A). FTY720 also inhibited ceramide synthesis in RBL-2H3 cells (Fig. 6A), demonstrating that the inhibition of ceramide synthesis by FTY720 also occurs in cells of immune origin.
FIGURE 6.

Effect of FTY720 on ceramide synthesis in cultured cells. A, HEK cells overexpressing CerS1 and CerS4 or mock-transfected HEK and RBL-2H3 cells were incubated with 25 μm FTY720 for 90 min. The cells were incubated with 1 μCi of [4,5-3H]sphinganine, 2 μm sphinganine (for HEK cells) or with 10 μm sphinganine (for RBL-2H3 cells) during the last 30 min of the incubation. The results are the means ± S.D. of two independent experiments for HEK cells (repeated at least three times) and the means ± S.D. for RBL cells (n = 3). B, HEK cells were incubated with 25 μm FTY720 for 90 min. The cells were incubated with increasing amounts of [4,5-3H]sphinganine/sphinganine (such that the ratio of [4,5-3H]sphinganine to sphinganine was the same at each sphinganine concentration) during the last 30 min of the incubation period. The results are the means ± S.D. of three independent experiments.
The amount of [4,5-3H]sphinganine/sphinganine used in these metabolic labeling experiments (∼12 nmol/mg protein) was significantly higher than endogenous sphinganine levels in HEK cells (∼6 pmol/mg protein) (19). Because FTY720 has a complex mode of inhibition of CerS activity (see above), we examined the concentration dependence of the effect of FTY720 versus sphinganine concentration. At high sphinganine concentrations, i.e. 500 nm to 5 μm, FTY720 (25 μm) significantly inhibited ceramide synthesis (Fig. 6B; cf. Fig. 6A). However, at lower sphinganine concentrations (<200 nm), no inhibition of ceramide synthesis was observed. Indeed, although not statistically significant, a tendency toward an increase in ceramide synthesis was detected at the lowest (<1 nm) sphinganine concentrations.3 Thus, the modulation of ceramide synthesis by FTY720 in cultured cells depends on the ratio of the concentrations of sphinganine to FTY720.
This unexpected result was supported by data obtained using ESI-MS/MS to determine total ceramide, sphingomyelin, and hexosylceramide levels in HEK cells and the fatty acid composition of these SLs (Fig. 7). Upon incubation of HEK cells with FTY720 for 90 min (the same time of incubation as used for the metabolic labeling experiments) (Fig. 6A), an increase in ceramide levels was observed, with no change in endogenous sphinganine levels (Fig. 7); similar increases were observed for hexosylceramide and sphingomyelin. Moreover, levels of C18-C22-ceramide were significantly increased, as were levels of C18-C22-hexosylceramide and C18-C22-sphingomyelin. This result is consistent with a complex mode of interaction of FTY720 with CerS, perhaps involving an allosteric element that is not preserved in vitro, whereby ceramide synthesis is stimulated in cells despite its inhibition by FTY720 in vitro.
FIGURE 7.

SL composition of HEK cells treated with FTY720. The fatty acid composition of Cer, hexosylceramide, and sphingomyelin was measured after 90 min of incubation with 25 μm FTY720. The inset in the top panel shows the sphinganine levels and total ceramide levels. The results are the means ± S.D. (n = 3). *, p < 0.05, comparing cells treated with FTY720 versus the respective untreated control. No S.D. value is given for total ceramide levels (in the inset) because this value is the sum of ceramide species with different acyl chain lengths.
Finally, to determine that the changes in ceramide synthesis in cultured cells were not due to the conversion of FTY720 to FTY720-P (9), which could bind to an S1PR at the cell surface and thus influence ceramide synthesis, we examined the effect of FTY720 in the presence of suramin, an inhibitor of G-protein-coupled receptors (31). No changes were observed in ceramide synthesis after incubation with FTY720 in the absence or presence of suramin (Fig. 8A). We next incubated cells with (R)-FTY-phosphonate, a nonhydrolyzable analog of FTY-P (Fig. 1), at concentrations (1 μm) that would saturate binding to an S1PR. There was no difference in the extent of inhibition of ceramide synthesis by FTY720 in the absence or presence of (R)-FTY-phosphonate (Fig. 8B). Thus, we can exclude the possibility that the modulation of ceramide synthesis by FTY720 is related to its metabolism to FTY720-P and binding to S1PRs.
FIGURE 8.

Effect of suramin and FTY720-phosphonate on inhibition of CerS activity by FTY720. HEK cells were incubated with suramin (50 μm, 10 min) (A) or with FTY-phosphonate (FTY-P) (1 μm, 30 min) prior to incubation with FTY720 (25 μm, 90 min). The cells were incubated with 1 μCi of [4,5-3H]sphinganine/2 μm sphinganine during the last 30 min of the incubation. The results are the means ± S.D. (n = 4) for A and the means for B (n = 2).
DISCUSSION
In the current study we demonstrate that FTY720 can interfere with SL metabolism via modulation of ceramide synthesis. Previously, FTY720 was thought to largely mediate its biological effects after its conversion to FTY720-P and subsequent binding to S1PRs. We now propose an additional mode of action, namely via modulation of ceramide synthesis.
The mechanism by which FTY720 modulates ceramide synthesis is surprisingly complicated. FTY720 inhibits CerS activity in vitro but under certain conditions activates ceramide synthesis in cultured cells. The reasons for these differences are not known, but FTY720 is not the first compound that inhibits CerS activity in vitro and has an opposite effect in cells. For instance, in cells overexpressing CerS, fumonisin B1, a well characterized CerS inhibitor in vitro (32), has little or no inhibitory effect and in some cases results in elevated ceramide levels (18, 19). The acyl chain length profile of the ceramides whose synthesis is elevated in cultured cells by fumonisin B1 is the same as the profile of the ceramides whose synthesis is inhibited in vitro (18, 19), demonstrating a direct connection between the apparently disparate effects of fumonisin B1. In contrast to the dual effects of fumonisin B1, which was only observed in cells overexpressing CerS, FTY720 elevates ceramide levels in untransfected cells (Fig. 7). Recently, another sphingosine analog, spisulosine (33), was shown to induce de novo ceramide synthesis in prostate tumor cell lines (33), whereas it inhibits CerS activity in vitro.4
A clue concerning the differences observed between the in vitro analysis and experiments in cultured cells might be found in the mode of inhibition of CerS activity by FTY720. Because FTY720 is a sphingosine analog, we predicted that FTY720 would act as a competitive inhibitor toward sphinganine, having little if any effect toward acyl-CoA. This prediction proved to be wrong, because we found that FTY720 was an uncompetitive inhibitor toward sphinganine and a noncompetitive inhibitor toward acyl-CoA (Fig. 5). Uncompetitive inhibitors bind to enzymes only after formation of the E-S complex (34). This type of inhibition is most commonly encountered in multi-substrate reactions where the inhibitor is competitive with respect to one substrate but uncompetitive with respect to another. CerS are bi-substrate enzymes, requiring the binding of both sphinganine and acyl-CoA for N-acylation of the former. Nothing is known about the active site of any CerS or about the order of binding of the substrates in the reaction pathway. Our data imply that sphinganine first binds to CerS to form an E-S (CerS-sphinganine) complex, and only after formation of this complex can FTY720 bind (Fig. 9). Alternatively, there may be two sphinganine-binding sites that act allosterically with respect to one another, or CerS themselves may form dimers that interact allosterically. The possibility of allosteric interactions is also supported by the noncompetitive mode of inhibition of acyl-CoA binding by FTY720. Noncompetitive inhibitors bind at an allosteric site on the enzyme and leave the active site unblocked (34). Thus, the binding sites of FTY720 and acyl-CoA appear to be distinct, but the interaction between sphinganine binding and FTY720 binding nevertheless impacts the rate of the reaction with respect to acyl-CoA. Finally, the experiment showing that FTY720 inhibits ceramide synthesis at high sphinganine concentrations in vivo, but not at low concentrations (Fig. 6), supports a complex, possibly allosteric mode of interaction between sphinganine and FTY720 and is consistent with uncompetitive inhibitors being most effective at high substrate concentrations (35); this explanation might also be relevant for the mode of interaction between sphinganine and other sphinganine analogs, such as spisulosine and fumonisin B1.
FIGURE 9.
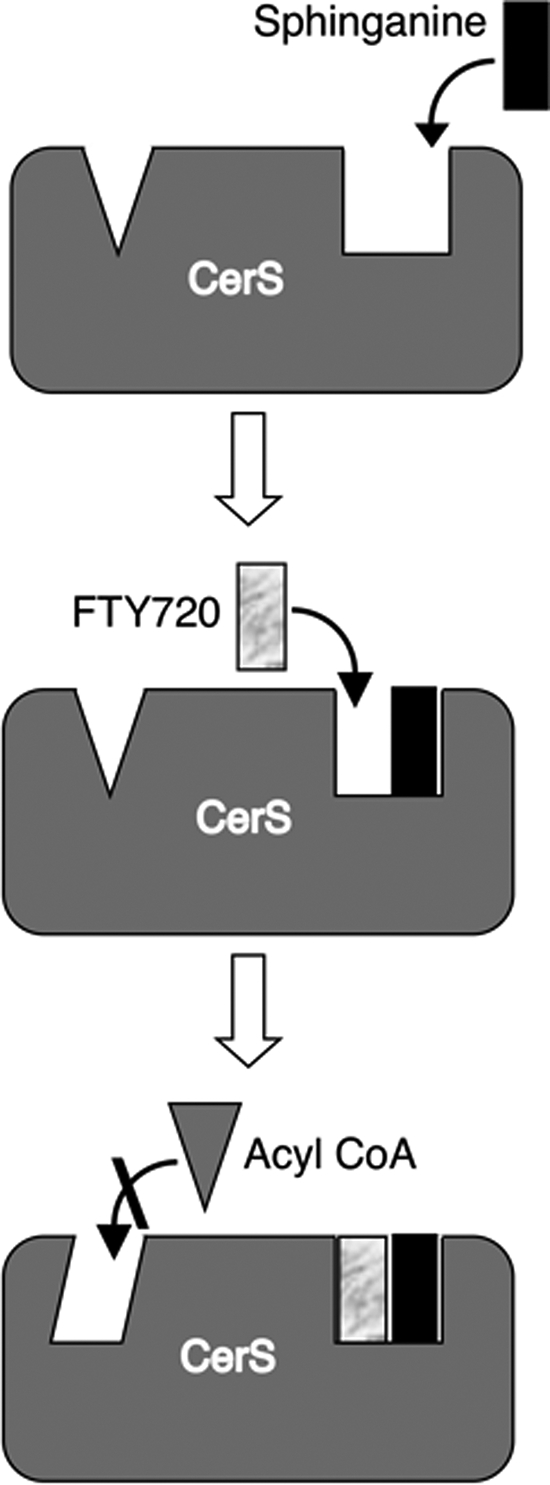
Possible mode of interaction of FTY720 with CerS. The data presented in this study are consistent with the idea that sphinganine first binds to CerS and that subsequent binding of FTY720 leads to an allosteric change that prevents binding of acyl-CoA. See “Discussion” for further details.
The range of concentrations (low micromolar) needed to inhibit CerS activity by FTY720 is considerably higher than that needed to bind and activate (mid nanomolar) S1P receptors after phosphorylation of FTY720 to FTY720-P. However, this cannot exclude the possibility that some of the biological effects of FTY720 may be mediated by FTY720 itself, prior to its metabolism to FTY720-P, or after its dephosphorylation by either lipid phosphate phosphatase 3 or lipid phosphate phosphatase 1α (Fig. 1B). A number of studies have examined the biological effects of FTY720 using micromolar concentrations (4, 15, 36–42), and these studies are consistent with the notion that the effects of FTY720 that are mediated via binding to S1PRs occur in the range expected of receptor-mediated signaling pathways, whereas additional and novel effects of FTY720, such as the pathway described herein, which is independent of S1PR binding, occur at much higher concentrations.
Whether FTY720 ever reaches the concentrations used in the current study (and in the study of Berdyshev et al. (42); see below) after its administration to either animals or to human patients is not known. In at least one study, FTY720 was shown to accumulate at levels of ∼1 μm in various rat and dog tissues soon after its injection (43). However, because FTY720 is hydrophobic, its concentration within intracellular membranes might be much higher. The precise concentrations to which CerS enzymes may be exposed after FTY720 treatment is therefore not known, and the clinical relevance of our data to understanding the mode of FTY720 action would be strengthened by determination of the local membrane concentration of FTY720 in, for instance, the endoplasmic reticulum, where the CerS enzymes are located.
While this manuscript was under review, a study was published by Berdyshev et al. (42) demonstrating inhibition of CerS activity by FTY720. In contrast to our study, Berdyshev et al. (42) reported competitive inhibition of CerS with respect to sphinganine and did not include any information on the mode of inhibition toward acyl-CoA, because the competitive mode of inhibition toward sphinganine would imply direct competition between sphinganine and FTY720, alleviating the need to examine inhibition toward acyl-CoA. In contrast, our study suggests a much more complex mode of inhibition, as discussed above. The conclusion by Berdyshev et al. (42) is based on CerS assays that do not appear to have been performed under optimal conditions. For example, concentrations of acyl-CoA were only 5 μm, which is significantly below the amount required to elicit a maximal rate of the reaction (24). Likewise, a sphinganine concentration of 100 nm was used, which is significantly lower than the Km of the reaction (which is in the range of 2–5 μm) (26). Moreover, the effect of FTY720 reported by Berdyshev et al. (42) might be related to the 100-fold molar excess of inhibitor (i.e. 10 μm FTY720) versus substrate (100 nm sphinganine), which could lead to displacement of sphinganine from the BSA complex, a possibility that we excluded in our study (Fig. 2), and thus lead to apparent competitive inhibition. Clearly, conclusions concerning modes of kinetic inhibition cannot be accurately derived from reactions performed under suboptimal assay conditions. Thus, the data in our study suggest a completely different mode of action of FTY720, as illustrated in Fig. 9.
One other major difference between the two studies is our analyses of the dependence of the inhibition by FTY720 on acyl-CoA chain length. Whereas Berdyshev et al. (42) did not observe any significant dependence on acyl-CoA chain length, perhaps due to some of the kinetic considerations discussed above, we observed that FTY720 inhibits ceramide synthesis using C18-CoA to a greater extent than other acyl-CoAs. This may be of relevance for understanding the mode of action of FTY720 as a modulator of ceramide synthesis in tissues that express different CerS enzymes and thus contain ceramides with different acyl chain lengths. Finally, our study examined the effect of FTY720 on CerS activity in three different cell lines, including RBL-2H3 mast cells, lending mechanistic significance to our study with relation to the possible effect of FTY720 in immunomodulation.
In summary, we have shown a multifaceted mode of interaction between FTY720 and CerS. This may be of importance for understanding the mode of action of FTY720, particularly because FTY720 is currently under investigation in a number of clinical trials, which apparently all assume that conversion to FTY720-P is necessary for its clinical efficacy. Our current study suggests a new mechanistic interpretation for some of the observed effects of FTY720, namely via modulation of ceramide synthesis.
Acknowledgments
We thank Dr. Adi Mesika for help in the initial stages of this study and Dr. Al Merrill for the ESI-MS/MS analyses.
This work was supported, in whole or in part, by National Institutes of Health Grant GM076217 (to A. M. and A. H. F.), and HL-083187 (to R. B.). This work was also supported by Israel Science Foundation Grant 1404/07 and by funds from the Minerva Foundation (to A. H. F.).
At the lowest [4,5-3H]sphinganine concentrations, the amount of [3H]ceramide formed (5,000 dpm) is extremely low, even though the specific activity of [4,5-3H]sphinganine is high (80 Ci/mmol). Thus, these experiments are difficult to perform to a high degree of experimental accuracy.
The EC50 of spisulosine toward CerS4 in vitro was ∼25 μm (data not shown).
- SL
- sphingolipid
- BSA
- bovine serum albumin
- CerS
- ceramide synthase
- ESI-MS/MS
- electrospray ionization-tandem mass spectrometry
- FTY720-P
- FTY720 phosphate
- HEK
- human embryonic kidney
- HEPG2
- hepatocellular liver carcinoma
- RBL
- rat basophilic leukemia
- S1P
- sphingosine 1-phosphate
- S1PR
- S1P receptor.
REFERENCES
- 1.Mansoor M., Melendez A. J. ( 2008) Rev. Recent Clin. Trials 3, 62– 69 [DOI] [PubMed] [Google Scholar]
- 2.Kharel Y., Lee S., Snyder A. H., Sheasley-O'Neill S. L., Morris M. A., Setiady Y., Zhu R., Zigler M. A., Burcin T. L., Ley K., Tung K. S., Engelhard V. H., Macdonald T. L., Pearson-White S., Lynch K. R. ( 2005) J. Biol. Chem. 280, 36865– 36872 [DOI] [PubMed] [Google Scholar]
- 3.Billich A., Bornancin F., Dévay P., Mechtcheriakova D., Urtz N., Baumruker T. ( 2003) J. Biol. Chem. 278, 47408– 47415 [DOI] [PubMed] [Google Scholar]
- 4.Brinkmann V., Davis M. D., Heise C. E., Albert R., Cottens S., Hof R., Bruns C., Prieschl E., Baumruker T., Hiestand P., Foster C. A., Zollinger M., Lynch K. R. ( 2002) J. Biol. Chem. 277, 21453– 21457 [DOI] [PubMed] [Google Scholar]
- 5.Mandala S., Hajdu R., Bergstrom J., Quackenbush E., Xie J., Milligan J., Thornton R., Shei G. J., Card D., Keohane C., Rosenbach M., Hale J., Lynch C. L., Rupprecht K., Parsons W., Rosen H. ( 2002) Science 296, 346– 349 [DOI] [PubMed] [Google Scholar]
- 6.Yopp A. C., Ledgerwood L. G., Ochando J. C., Bromberg J. S. ( 2006) Clin. Transplant. 20, 788– 795 [DOI] [PubMed] [Google Scholar]
- 7.Zemann B., Kinzel B., Müller M., Reuschel R., Mechtcheriakova D., Urtz N., Bornancin F., Baumruker T., Billich A. ( 2006) Blood 107, 1454– 1458 [DOI] [PubMed] [Google Scholar]
- 8.Zhang Z., Schluesener H. J. ( 2007) Mini Rev. Med. Chem. 7, 845– 850 [DOI] [PubMed] [Google Scholar]
- 9.Kihara A., Igarashi Y. ( 2008) Biochim. Biophys. Acta 1781, 496– 502 [DOI] [PubMed] [Google Scholar]
- 10.Yagi H., Kamba R., Chiba K., Soga H., Yaguchi K., Nakamura M., Itoh T. ( 2000) Eur. J. Immunol. 30, 1435– 1444 [DOI] [PubMed] [Google Scholar]
- 11.Chiba K., Matsuyuki H., Maeda Y., Sugahara K. ( 2006) Cell Mol. Immunol. 3, 11– 19 [PubMed] [Google Scholar]
- 12.Pinschewer D. D., Ochsenbein A. F., Odermatt B., Brinkmann V., Hengartner H., Zinkernagel R. M. ( 2000) J. Immunol. 164, 5761– 5770 [DOI] [PubMed] [Google Scholar]
- 13.Brown B. A., Kantesaria P. P., McDevitt L. M. ( 2007) Ann. Pharmacother. 41, 1660– 1668 [DOI] [PubMed] [Google Scholar]
- 14.Horga A., Montalban X. ( 2008) Expert Rev. Neurother. 8, 699– 714 [DOI] [PubMed] [Google Scholar]
- 15.Bandhuvula P., Tam Y. Y., Oskouian B., Saba J. D. ( 2005) J. Biol. Chem. 280, 33697– 33700 [DOI] [PubMed] [Google Scholar]
- 16.Payne S. G., Oskeritzian C. A., Griffiths R., Subramanian P., Barbour S. E., Chalfant C. E., Milstien S., Spiegel S. ( 2007) Blood 109, 1077– 1085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pettus B. J., Bielawska A., Subramanian P., Wijesinghe D. S., Maceyka M., Leslie C. C., Evans J. H., Freiberg J., Roddy P., Hannun Y. A., Chalfant C. E. ( 2004) J. Biol. Chem. 279, 11320– 11326 [DOI] [PubMed] [Google Scholar]
- 18.Venkataraman K., Riebeling C., Bodennec J., Riezman H., Allegood J. C., Sullards M. C., Merrill A. H., Jr., Futerman A. H. ( 2002) J. Biol. Chem. 277, 35642– 35649 [DOI] [PubMed] [Google Scholar]
- 19.Riebeling C., Allegood J. C., Wang E., Merrill A. H., Jr., Futerman A. H. ( 2003) J. Biol. Chem. 278, 43452– 43459 [DOI] [PubMed] [Google Scholar]
- 20.Koybasi S., Senkal C. E., Sundararaj K., Spassieva S., Bielawski J., Osta W., Day T. A., Jiang J. C., Jazwinski S. M., Hannun Y. A., Obeid L. M., Ogretmen B. ( 2004) J. Biol. Chem. 279, 44311– 44319 [DOI] [PubMed] [Google Scholar]
- 21.Lahiri S., Futerman A. H. ( 2005) J. Biol. Chem. 280, 33735– 33738 [DOI] [PubMed] [Google Scholar]
- 22.Laviad E. L., Albee L., Pankova-Kholmyansky I., Epstein S., Park H., Merrill A. H., Jr., Futerman A. H. ( 2008) J. Biol. Chem. 283, 5677– 5684 [DOI] [PubMed] [Google Scholar]
- 23.Mizutani Y., Kihara A., Igarashi Y. ( 2006) Biochem. J. 398, 531– 538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hirschberg K., Rodger J., Futerman A. H. ( 1993) Biochem. J. 290, 751– 757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lu X., Sun C., Valentine W. J., E S., Liu J., Tigyi G., Bittman R. ( 2009) J. Org. Chem. 74, 3192– 3195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lahiri S., Lee H., Mesicek J., Fuks Z., Haimovitz-Friedman A., Kolesnick R. N., Futerman A. H. ( 2007) FEBS Lett. 581, 5289– 5294 [DOI] [PubMed] [Google Scholar]
- 27.Bligh E. G., Dyer W. J. ( 1959) Can. J. Biochem. Physiol. 37, 911– 917 [DOI] [PubMed] [Google Scholar]
- 28.Folch J., Lees M., Sloane-Stanley G. H. ( 1957) J. Biol. Chem. 226, 496– 509 [PubMed] [Google Scholar]
- 29.Sullards M. C., Merrill A. H., Jr. ( 2001) Science's STKE 67, pl1. [DOI] [PubMed] [Google Scholar]
- 30.Merrill A. H., Jr., Sullards M. C., Allegood J. C., Kelly S., Wang E. ( 2005) Methods 36, 207– 224 [DOI] [PubMed] [Google Scholar]
- 31.Alderton F., Sambi B., Tate R., Pyne N. J., Pyne S. ( 2001) Br. J. Pharmacol. 134, 6– 9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Merrill A. H., Jr., Liotta D. C., Riley R. T. ( 1996) Trends Cell Biol. 6, 218– 223 [DOI] [PubMed] [Google Scholar]
- 33.Sánchez A. M., Malagarie-Cazenave S., Olea N., Vara D., Cuevas C., Díaz-Laviada I. ( 2008) Eur. J. Pharmacol. 584, 237– 245 [DOI] [PubMed] [Google Scholar]
- 34.Segel I. H. ( 1993) Enzyme Kinetics: Behavior and Analysis of Rapid Equilibrium and Steady-State Enzyme Systems, Wiley-Interscience, New York [Google Scholar]
- 35.Cornish-Bowden A. ( 1995) Fundamentals of Enzyme Kinetics, Portland Press, London [Google Scholar]
- 36.Neviani P., Santhanam R., Oaks J. J., Eiring A. M., Notari M., Blaser B. W., Liu S., Trotta R., Muthusamy N., Gambacorti-Passerini C., Druker B. J., Cortes J., Marcucci G., Chen C. S., Verrills N. M., Roy D. C., Caligiuri M. A., Bloomfield C. D., Byrd J. C., Perrotti D. ( 2007) J. Clin. Investig. 117, 2408– 2421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nagaoka Y., Otsuki K., Fujita T., Uesato S. ( 2008) Biol. Pharm. Bull. 31, 1177– 1181 [DOI] [PubMed] [Google Scholar]
- 38.Miron V. E., Hall J. A., Kennedy T. E., Soliven B., Antel J. P. ( 2008) Am. J. Pathol. 173, 1143– 1152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Azuma H., Takahara S., Ichimaru N., Wang J. D., Itoh Y., Otsuki Y., Morimoto J., Fukui R., Hoshiga M., Ishihara T., Nonomura N., Suzuki S., Okuyama A., Katsuoka Y. ( 2002) Cancer Res. 62, 1410– 1419 [PubMed] [Google Scholar]
- 40.Sanchez T., Estrada-Hernandez T., Paik J. H., Wu M. T., Venkataraman K., Brinkmann V., Claffey K., Hla T. ( 2003) J. Biol. Chem. 278, 47281– 47290 [DOI] [PubMed] [Google Scholar]
- 41.Matsuoka Y., Nagahara Y., Ikekita M., Shinomiya T. ( 2003) Br. J. Pharmacol. 138, 1303– 1312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Berdyshev E. V., Gorshkova I., Skobeleva A., Bittman R., Lu X., Dudek S. M., Mirzapoiazova T., Garcia J. G., Natarajan V. ( 2009) J. Biol. Chem. 284, 5467– 5477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meno-Tetang G. M., Li H., Mis S., Pyszczynski N., Heining P., Lowe P., Jusko W. J. ( 2006) Drug. Metab. Dispos. 34, 1480– 1487 [DOI] [PubMed] [Google Scholar]