

Abstract
Transition metals are essential enzyme cofactors that are required for a wide range of cellular processes. Paradoxically, whereas metal ions are essential for numerous cellular processes, they are also toxic. Therefore cells must tightly regulate metal accumulation, transport, distribution, and export. Improved tools to interrogate metal ion availability and spatial distribution within living cells would greatly advance our understanding of cellular metal homeostasis. In this work, we present genetically encoded sensors for Zn2+ based on the principle of fluorescence resonance energy transfer. We also develop methodology to calibrate the probes within the cellular environment. To identify both sources of and sinks for Zn2+, these sensors are genetically targeted to specific locations within the cell, including cytosol, plasma membrane, and mitochondria. Localized probes reveal that mitochondria contain an elevated pool of Zn2+ under resting conditions that can be released into the cytosol upon glutamate stimulation of hippocampal neurons. We also observed that Zn2+ is taken up into mitochondria following glutamate/Zn2+ treatment and that there is heterogeneity in both the magnitude and kinetics of the response. Our results suggest that mitochondria serve as a source of and a sink for Zn2+ signals under different cellular conditions.
Although mammalian cells are known to concentrate transition metals, it is now well established that under resting conditions, “free” (e.g. unbound) metals are maintained at extremely low levels. Estimates of the total Zn2+ concentration in mammalian cells typically range from 100 to 500 μm (1); yet free Zn2+ concentrations are tightly buffered by proteins such as metallothionein to maintain cytosolic Zn2+ concentrations in the picomolar to nanomolar range (2–5). However, there is emerging evidence that this static picture is dramatically altered by different cellular conditions, such as redox perturbations caused by oxidative stress (6, 7) and cellular signals such as nitric oxide (8). Consequently, there is a pool of labile Zn2+ that, if mobilized by cellular signals, would result in the generation of transient Zn2+ signals. Recent studies suggest that these Zn2+ signals influence critical biological processes, such as mitochondrial function (7, 9, 10). Elucidation of the sources and dynamics of these Zn2+ signals would greatly advance our understanding of the interplay between metal regulation and cellular function.
There has been a huge effort in the past few years to develop sensitive and selective fluorescent probes to monitor Zn2+ in biological systems. The majority of this work has focused on the generation of small molecule fluorescent indicators (reviewed by Que et al. (11)). Yet there are also examples of sensors based partially on Zn2+-binding proteins, such as carbonic anhydrase (12) and metallothionein (13), and peptide scaffolds (14). Although many of these sensors have begun to provide insight into Zn2+ concentrations within cells, one limitation is that it is challenging to explicitly target them to subdomains within the cell. Localized probes are necessary to generate a complete picture of cellular Zn2+ homeostasis in mammalian cells. For this reason, sensors that are genetically encoded (i.e. generated by translation of a nucleic acid sequence) are attractive platforms for engineering metal-specific sensors. Encoded sensors provide additional benefits such as retention of the sensor over days to weeks permitting long term imaging and the ability to systematically vary the sensor concentration to evaluate the extent to which the sensor perturbs resting Zn2+ concentrations.
Here we present genetically encoded sensors designed with a “Zn2+-sensing domain” sandwiched between two fluorescent proteins. The fluorescent proteins are chosen so that they are capable of undergoing fluorescence resonance energy transfer (FRET).2 Because the mechanism of FRET involves dipole-dipole coupling, it is exquisitely dependent on the distance and orientation of the fluorophores with respect to one another. Therefore, if the binding of Zn2+ induces a conformational change in the sensor, it will alter the energy transfer between the two fluorescent proteins. The advantage of using FRET as the optical readout is that the donor emission will decrease and the acceptor emission will increase upon Zn2+ binding. Hence, by taking the ratio of the acceptor to the donor emission, we can create a ratiometric sensor. These sensors are targeted to the cytosol, mitochondria, and plasma membrane by attachment of signal sequences and fusion to other proteins. These sensors reveal differences in the spatial distribution of Zn2+ and highlight the power and utility of localized probes.
EXPERIMENTAL PROCEDURES
In Vitro Characterization
Details of sensor construction, protein purification, and buffered metal solutions are presented in the supplemental text. Purified sensor protein (0.5 μm) was buffer exchanged into 10 mm MOPS, 100 mm KCl (pH 6.8) and titrated with Zn2+ to obtain the apparent dissociation constant, K′d. We determined the K′d using two approaches. In one approach, the fluorescence intensity at 529 nm (emission maximum of YFP) was plotted as a function of Zn2+, and in the second approach, the change in FRET ratio (R − Rmin) was plotted as a function of Zn2+, where the FRET ratio (R) is defined as the emission maximum of YFP divided by the emission maximum of CFP upon excitation of CFP, and Rmin is the minimum FRET ratio observed under Zn2+-free conditions. These measurements yielded K′d values within experimental error of one another. It should be noted that we measure the apparent Kd because we are monitoring the conformational change that leads to FRET as opposed to directly measuring the Zn2+ binding event. Mixed metal buffer solutions (15) were used to buffer Zn2+ from 9 nm to 1.3 μm (Zn2+/Sr2+/EGTA) and 2–134 μm (Zn2+/Ca2+/EGTA). The Cys2His2 sensor was purified and maintained under anaerobic conditions (see supplemental text for more details). A standard Ellman's assay (16) was used to quantify the percentage of reduced Cys. The observed FRET ratio change (R − Rmin) for the Cys2His2 sensor varied with the percentage of reduced Cys (typically between 50 and 80% of Cys were reduced). The lack of complete Cys reduction likely resulted from our inability to maintain a completely anoxic environment. Normalization to the amount of active, reduced protein yielded the maximal dynamic range for the Cys2His2 sensor of 2.2-fold. Fluorescence measurements were made on a Safire-II fluorescence plate reader (Tecan). FRET ratios (R) were calculated for each titration point by dividing the YFP intensity (λmax = 529 nm) by CFP intensity (λmax = 475 nm) upon CFP excitation at 420 nm. The emission bandwidth was 10 nm.
Cellular Zn2+ Measurements
The His4 sensor was targeted to the extracellular surface of mammalian cells using pDisplay (Invitrogen). The Cys2His2 sensor was targeted to the mitochondrial matrix, referred to as mito-Cys2His2, by attaching four tandem repeats of the cytochrome c oxidase signal sequence (MSVLTPLLLRGLTGSARRLPVPRAKIHSLGDP) to the N terminus of the sensor (17). Sensor constructs were transfected into HeLa cells using TransIt (Mirus) according to the manufacturer's instructions and imaged 48 h post-transfection. The imaging experiments were performed on an Axiovert 200M inverted fluorescence microscope (Zeiss) with a Cascade 512B CCD camera (Roper scientific), and equipped with CFP (430/24 excitation, 455 dichroic, 470/24 emission), YFP (495/10 excitation, 515 dichroic, 535/25 emission), and YFP FRET (430/24 excitation, 455 dichroic, 535/25 emission) filters controlled by a Lambda 10-3 filter changer (Sutter Instruments) and analyzed using Metafluor software (Universal Imaging). Experiments on cytosolically expressed sensors were collected at 40× magnification, whereas mitochondrial experiments were collected at 100× magnification.
The cells were imaged in phosphate-free HEPES-buffered Hanks' balanced salt solution. To determine the in situ K′d values, the cells were treated with 150 μm TPEN to obtain the minimum FRET ratio (Rmin) followed by a wash and the addition of increasing concentrations of a well defined Zn2+ solution (Zn2+/Sr2+/EGTA- or Zn2+/Ca2+/EGTA-buffered solutions). For intracellularly expressed sensors (cytosol and mitochondria), the cells were treated with 150 μm TPEN to obtain the minimum FRET ratio (Rmin) followed by a wash and the addition of 15 μm digitonin (Calbiochem) and a saturating Zn2+ solution (1.5 mm) to establish the maximum FRET ratio (Rmax). The digitonin was used to permeabilize the plasma membrane and facilitate Zn2+ entry into the cell. The Rmax treatment was generally toxic to cells and therefore was always performed at the end of an experiment. For mitochondrial calibrations, the Zn2+ ionophore pyrithione (25 μm) was included along with digitonin to facilitate Zn2+ entry across the mitochondrial membrane. Because for the Cys2His2 sensor, the sensor concentration was similar in magnitude to the K′d, and the FRET ratio versus [Zn2+]total was fit with the following expression (18),
 |
where [sensor]T was taken to be 6 μm. This sensor concentration was determined experimentally as described below. Still, [sensor]T was varied from 0.5 to 10 μm in the above fits with no change in the calculated Kd. For the His4 sensor, the data were fit to a one site binding equation.
Citrine YFP intensity was converted to sensor protein concentration using an established method (19). Briefly, a protein standard curve was generated by measuring the citrine intensities of purified sensor protein of known concentrations in a 50-μm-tall rectangular glass capillary (VitroCom) using identical settings as the imaging experiments.
The FRET ratio (R) was calculated from background corrected FRET and CFP fluorescence images and corrected for photobleaching if necessary. The FRET ratios were converted to Zn2+ concentrations using the following equation (20) along with experimentally derived Rmin, Rmax, and in situ K′d values.
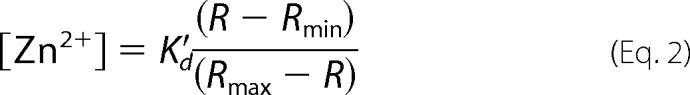 |
It should be noted that Rmin and Rmax are obtained in each experiment and in each individual cell.
Neuronal Culturing and Transfection
Primary hippocampal neurons were prepared from postnatal day 1 Sprague-Dawley rats, with slight modifications (21). Briefly, the hippocampi were dissociated and plated on 35-mm glass-bottomed dishes coated with poly-d-lysine (Sigma) and laminin (Invitrogen) and grown in neurobasal A supplemented with B27 and Glutamax medium (Invitrogen). The neurons were transfected prior to plating by Amaxa nucleofection at 2,000,000 cells/transfection with 2 μg of DNA/manufacturer's recommendations (Amaxa) and grown to 9 days in vitro. The neurons were imaged at 9 days in vitro as above using 100× magnification in a modified Tyrode's salts solution.
RESULTS
Sensor Design
The sensor design consists of two fluorescent proteins and a “sensing domain” that undergoes a conformational change upon Zn2+ binding. A sensor schematic is presented in Fig. 1A. Binding of Zn2+ to the sensing domain changes the distance and orientation between the cyan fluorescent protein (CFP) and the citrine fluorescent protein (YFP), leading to increased FRET from CFP to YFP.
FIGURE 1.

Schematic of sensor function and in vitro characterization. A, mechanism for the sensor response to Zn2+. In the absence of Zn2+, the sensing domain is unstructured, whereas in the presence of Zn2+, it becomes structured leading to a change in the distance and orientation between CFP and YFP. B, emission spectrum of the His4 sensor as a function of Zn2+ upon excitation of CFP at 420 nm. The arrows indicate the decrease in CFP fluorescence intensity and an increase in YFP fluorescence intensity with increasing Zn2+ concentrations. The Zn2+ concentrations were as follows: 0 μm (long dash), 70 μm (short dash), 180 μm (medium dash), solid line (1000 μm). C and D, Zn2+ binding curves for the Cys2His2 (●), His4 (■), and Ala4 (♦) sensors. The FRET ratio change (R − Rmin) is plotted as a function of Zn2+ for each sensor. The lines represent fits to a single site binding equation and yield the following dissociation constants: K′d = 1.7 ± 0.2 μm (●) and 160 ± 4 μm (■). The FRET ratio (R) was calculated as Em529/Em475 upon 420-nm excitation.
The Zn2+-sensing domain is a canonical Cys2His2 zinc finger from the mammalian transcription factor, Zif268. The zinc finger was selected because extensive structural data indicates that these domains are largely unstructured in the absence of metal ion and fold into a compact structure upon Zn2+ binding (22). Three sensor variants were generated: a wild type zinc finger (Cys2His2), a mutant zinc finger containing four histidines (His4), and a mutant zinc finger containing four alanines (Ala4) to abrogate Zn2+ binding.
In Vitro Characterization
To characterize the sensors in vitro, the sensor protein was purified by affinity and size exclusion chromatography. Fig. 1B demonstrates that Zn2+ binding to the His4 sensor causes a decrease in CFP and an increase in YFP emission upon CFP excitation caused by an increase in FRET. The same spectral changes were observed for the Cys2His2 sensor (data not shown). As expected, the sensor is ratiometric, and therefore the sensor response is presented as the FRET ratio (R). The dynamic range of the sensor is defined as Rmax/Rmin and was 4-fold for the His4 sensor and 2.2-fold for the Cys2His2 sensor, making these some of the more sensitive FRET sensors based on conformational change. For comparison, redesigned sensors for Ca2+ exhibit a 5-fold ratio change (17, 23), kinase sensors typically exhibit a 0.2–0.6-fold ratio change (24), and glutamate sensors exhibit a 0.25-fold ratio change (25). We suspect that the difference in the dynamic range between the His4 and Cys2His2 sensors results from slight variation in the overall geometry and hence orientation of the fluorescent proteins with respect to one another. There is some precedence for this because a systematic analysis of analogous Ca2+ sensors revealed large changes in the dynamic range for sensors with different orientations but similar distances between the two fluorescent proteins (23).
To determine the apparent dissociation constant for Zn2+ (K′d), the FRET intensity and FRET ratio for each sensor were measured as a function of Zn2+ concentration. These measurements yielded identical K′d values. The FRET ratio (R) binding curves are presented in Fig. 1 (C and D). The in vitro zinc affinities for the Cys2His2 and His4 sensors were determined to be 1.7 ± 0.2 and 160 ± 4 μm, respectively. The Cys2His2 sensor exhibited a lower Zn2+ affinity than the isolated zinc finger (1.7 μm versus 10 nm). We suspect that this is because attachment of the fluorescent proteins alters the apparent zinc affinity. As expected, mutation of Cys2His2 to His4 led to a reduction in Zn2+ affinity, enabling us to generate both a high and low affinity sensor. Proteolytic removal of the His6 tag did not alter the K′d of either sensor, indicating that the His tag does not participate in Zn2+ binding. Mutation of Cys2His2 to Ala4 abrogates the sensor response, indicating that Zn2+ binding to the Zn2+ finger portion of the sensor is required for the observed FRET ratio change (Fig. 1D).
Although the cellular environment is generally reducing, cellular compartments may be oxidizing, or the cell may experience transient changes in redox potentials. The cysteines of the Cys2His2 sensor are sensitive to oxidization. To test the effects of oxidation on the FRET ratio, we measured the in vitro FRET ratios of the reduced and oxidized Cys2His2 sensor. Rmin was not affected by Cys2His2 sensor oxidization; however, the oxidized form of the sensor was unable to achieve the maximum FRET ratio with the addition of saturating Zn2+ (supplemental Fig. S1). These results indicate that artificial increases in FRET would not be generated by sensor oxidation, but that upon oxidation, the sensor will be less sensitive to Zn2, resulting in a decreased dynamic range.
For sensors to report Zn2+ dynamics accurately within the complex environment of the cell, it is important that they be specific for Zn2+ over other abundant divalent ions (Mg2+ and Ca2+) and biologically relevant transition metals (Cu2+, Mn2+, Fe2+, Fe3+, and Ni2+). To determine the metal specificity, we examined whether other metals could elicit a FRET ratio change in the Cys2His2 and His4 sensors and/or interfere with the Zn2+ response (supplemental Fig. S2). The majority of metals did not elicit a FRET change and did not interfere with the Zn2+ response. The two exceptions were Fe2+ and Cu2+. Fe2+ caused a small perturbation of Zn2+ binding to both sensors but only at the highest concentration tested (250 μm). Fe2+ itself did not cause an artificial FRET change. Cu2+ interfered with Zn2+ binding to the His4 sensor such that the maximum Zn2+ response could only be achieved at the lowest Cu2+ concentration (5 μm). The 5 μm Cu2+ level is still significantly higher than the estimated cytosolic free copper concentration of less than 10−18 m or attomolar (26). Cu2+ only bound to and elicited a FRET change at the highest concentration tested (250 μm), suggesting that Cu2+ signals would be unlikely to give false FRET increases. The metal specificity results suggest that metal cross-reactivity will not be a problem in the cellular environment. However, given that copper ions pose the greatest potential for interference, we further experimentally verified Zn2+ selectivity over copper in cells (see below).
Cellular Expression and in Situ Characterization
Although genetically encoded FRET-based sensors for Zn2+ have been reported previously, they have either not been demonstrated to function in cells (27–29) or were not used explicitly to measure cellular Zn2+ (30). Therefore we sought to express our Cys2His2 and His4 sensors in mammalian cells and characterize the in situ response. We chose to target the low affinity His4 sensor to the extracellular surface of the plasma membrane and the higher affinity Cys2His2 sensor to both the cytosol and the mitochondrial matrix. Fig. 2A presents pseudo color FRET ratio images of HeLa cells expressing the cytosolic Cys2His2 sensor. The addition of TPEN, a Zn2+-specific chelator, results in a decrease in the FRET ratio, whereas an addition of Zn2+ results in an increase. Fig. 2 (B and C) demonstrates sensors targeted to mitochondria and the plasma membrane, respectively. Table 1 summarizes the sensors generated. To measure the apparent dissociation constant in situ, the cells were treated with TPEN to establish the minimum FRET ratio in zero Zn2+ (Rmin) followed by membrane permeabilization with digitonin and the addition of known concentrations of Zn2+. Fig. 2D presents a representative trace illustrating a Zn2+ titration, and Fig. 2E reports the full binding curves. The in situ Zn2+ affinity of each sensor was comparable with that observed in vitro (K′d = 1.5 ± 0.2 μm in situ versus 1.7 ± 0.2 μm in vitro for Cys2His2; and 200 ± 10 μm in situ versus 160 ± 4 μm in vitro for His4). As expected, the cytosolic Ala4 sensor showed no significant FRET ratio change upon addition of Zn2+ to cells. The dynamic range of both sensors was reduced in cells as compared with in vitro (0.25-fold in situ versus 2.2-fold for Cys2His2 and 4-fold for His4 in vitro), even though the affinity for Zn2+ was unchanged. The dynamic range for Cys2His2 was identical in the cytosol and mitochondria, suggesting that possible redox differences in these locations do not affect the sensor functionality and Zn2+ measurements. We speculate that the reduced dynamic range in situ may be due to molecular crowding in the cellular environment, causing the fluorophores of the sensor to be closer together in the absence of Zn2+, resulting in an overall higher Rmin. This is supported by our observation that Rmin is generally higher in cells (∼4) than in purified protein (Rmin = ∼1.5) when measured under the same experimental conditions on the microscope. Although the dynamic range was reduced in cells, the sensors still yielded robust responses to changes in Zn2+ levels and enabled visualization of Zn2+ dynamics in neurons.
FIGURE 2.

Cellular response and sensor calibration in HeLa cells. A, pseudo color FRET ratio images under resting conditions (left panel), following 150 μm TPEN treatment (middle panel), and after treatment with ZnCl2/15 μm digitonin (right panel). B, the Cys2His2 sensor targeted to mitochondria. C, the His4 sensor targeted to the plasma membrane. D, representative microscope trace for a typical calibration experiment of the Cys2His2 sensor in the cytosol. The trace shows the initial treatment of 150 μm TPEN (Rmin) followed by a washout and successive additions of ZnCl2 along with 15 μm digitonin to obtain each titration point. E, in situ calibration for Cys2His2 in the cytosol (●), His4 on the plasma membrane (■) and Ala4 in the cytosol (♦). The FRET ratio change (R − Rmin) is plotted as a function of Zn2+. The data were fit as described in methods yielding the following parameters: K′d = 1.5 ± 0.2 μm (■) and 200 ± 10 μm (●). The scale bar represents 10 μm.
TABLE 1.
Summary of sensors developed in this work
| Sensor | In situ K′d | Targeted locations |
|---|---|---|
| Cys2His2 | 1.5 μm | Cytosol and mitochondria |
| His4 | 200 μm | Cytosol, mitochondria, and plasma membrane |
| Ala4 | Control; does not bind Zn2+ | Cytosol |
Next, we determined whether cellular Cu1+ or Cu2+ ions interfered with Zn2+ measurements in cells. Resting FRET ratios were not affected by the addition of either neocuproine hydrochloride monohydrate (a Cu+1 specific chelator) or bathocuproinedisulfonic acid disodium salt (a Cu+1/+2 chelator) but decreased to a minimum FRET ratio upon addition of TPEN, indicating that the in situ resting ratios were specific to Zn2+ and not influenced by Cu1+ or Cu2+ (supplemental Fig. S3).
The mitochondrially targeted sensor (mito-Cys2His2) exhibited a marked improvement in localization over the small molecule probe RhodZin-3 (Invitrogen; supplemental Fig. S4). Moreover, mito-Cys2His2 stayed localized within mitochondria upon membrane depolarization, whereas RhodZin-3 leaked out into the cytosol under these conditions.
The first step in characterizing cellular Zn2+ homeostasis is to define the levels of Zn2+ in different locations within the cell. There has been some controversy over whether mitochondria contain a labile pool of Zn2+ that is elevated over the cytosol (9, 31). To address this, we measured resting Zn2+ concentrations in the cytosol and mitochondria using our Cys2His2 sensor. Because the addition of a sensor may perturb cellular Zn2+ levels, determinations of the “free” Zn2+ concentrations should be conducted over a range of sensor concentrations, enabling mathematical extrapolation to the “free” [Zn2+] at zero [sensor]. This variation in sensor concentrations is a natural consequence of transient transfection where the numbers of gene copies can vary from cell to cell. Tighter control of sensor expression will likely be possible with the use of inducible promoters, and the establishment of stably transfected cell lines. Fig. 3 (A and B) presents the concentration of Zn2+ as a function of sensor concentration in the cytosol and mitochondria, respectively. For the cytosolic sensor citrine YFP intensity was converted to protein concentration using a protein standard curve (19) (supplemental Fig. S5). In mitochondria, the citrine intensity was not converted to a protein concentration because of the uncertainty in predicting mitochondrial volume.
FIGURE 3.

Comparison of cytosolic and mitochondrial Zn2+. A and B, cytosolic resting Zn2+ concentrations from 46 individual cells (A, ●) and mitochondrial resting Zn2+ concentrations from 10 individual cells (B, ■) as a function of sensor expression. In the cytosol, there is a wide variation in sensor expression from cell to cell, whereas the variation is markedly reduced in mitochondria. C, representative traces of the cytosolic (●), and mitochondrial (■) Cys2His2 sensor. Resting ratios are established at the start, followed by treatment with 150 μm TPEN to obtain Rmin, and 2 mm ZnCl2, 15 μm digitonin (●) or 2 mm Zn2+, 15 μm digitonin, 25 μm pyrithione (■) to obtain Rmax. The number of Rmax data points we can collect is limited by the fact that the Rmax experimental conditions are toxic to the cells.
We observe a direct correlation between sensor protein expression and the calculated Zn2+ values in the cytosol, where higher sensor concentrations give rise to higher estimated Zn2+ levels. A simple saturation model yields the best fit and projects a resting cytosolic Zn2+ concentration of 180 nm when extrapolated to zero sensor concentration. For all of the subsequent experiments, we imaged cells with low (<5 μm) expression levels to minimize perturbation of cellular Zn2+ levels. We did not observe a strong correlation between citrine intensity and Zn2+ levels within mitochondria. This is likely because the sensor expression was naturally low (hundreds of fluorescence counts instead of thousands), and the expression level did not vary significantly. The average Zn2+ level within mitochondria was found to be 680 ± 140 nm. Fig. 3C depicts representative time course experiments from both cytosol and mitochondria and directly illustrates the elevated resting Zn2+ within mitochondria. These data support the hypothesis that mitochondria contain a pool of Zn2+ that serve as a source of Zn2+ signals, as suggested by Sensi et al. (9).
Measurement of Mitochondrial Zn2+ Dynamics in Hippocampal Neurons
A requisite feature of a useful sensor is the ability to observe dynamic fluctuations in living cells. Glutamate and Zn2+ colocalize in presynaptic vesicles in hippocampal neurons, yet the physiological function of this vesicular Zn2+ remains unclear (11). Nolan et al. (32) recently demonstrated that glutamate induces the uptake of exogenous Zn2+ into the cytosol of hippocampal neurons. A natural extension of this work is to track the fate of Zn2+ after entry. Because numerous studies have suggested that mitochondria may sequester Zn2+ (8, 31, 33), we questioned whether transient Zn2+ increases in the cytosol are transmitted to mitochondria in primary rat hippocampal neurons. Upon treatment with glutamate plus Zn2+, we observed an increase of Zn2+ within mitochondria for the high affinity mito-Cys2His2 sensor but not the lower affinity mito-His4 sensor (Fig. 4). Supplemental Fig. S6 presents data traces of seven individual cells from three different experiments. Although there was some variability in the magnitude of the response (percentage of ratio change, i.e. R/Rmin, = 3.2 ± 0.8%, n = 7), 100% of the cells measured showed an increase in mitochondrial Zn2+ upon treatment with glutamate plus Zn2+. Interestingly, the transient mitochondrial Zn2+ signals are dependent on the presence of extracellular Ca2+, because stimulation with glutamate and Zn2+ in Ca2+-free HEPES-buffered Hanks' balanced salt solution lead to no observable increase in mitochondrial Zn2+. The mechanism of Zn2+ uptake is unclear, although the Ca2+ uniporter has been shown to transport Zn2+ (31). We are currently exploring the mechanism of uptake using inhibitors of different transport mechanisms. As depicted in Fig. 4 and supplemental movies, we observe heterogeneity in the magnitude and kinetics of Zn2+ uptake. Fig. 4B depicts the FRET ratio as a function of time for four regions of interest, whereas Fig. 4C shows the actual images. The two cells responded at different rates, with the bottom cell peaking at ∼20 s after stimulation, and the top cell peaking at ∼40 s after stimulation. Likewise there is variability in the response within a given cell. Additionally we often observed transient hot spots (depicted by the white arrows) for one or two frames. These could correspond to individual mitochondria or small clusters of mitochondria. Although we are still exploring the cellular consequences of this heterogeneity, the experiments demonstrate the richness of information possible by subcellular analysis of Zn2+ signals.
FIGURE 4.

Neuronal mitochondria sequester Zn2+ upon treatment with glutamate and exogenous Zn2+. A, treatment of neurons with 100 μm glutamate, 250 μm Zn2+ leads to an increase in mitochondrial Zn2+ as demonstrated by the mito-Cys2His2 response (black circle). The low affinity mito-His4 sensor (blue square) yielded no response as expected given that this probe would be sensitive to Zn2+ concentrations between 20 and 2000 μm. Zn2+ uptake into mitochondria is Ca2+-dependent as mito-Cys2His2 yielded no response when the stimulation was carried out in Ca2+-free/phosphate-free HEPES-buffered Hanks' balanced salt solution (red diamond). The left axis is for circles and squares. The right axis is for diamonds. B, heterogeneity in different regions of interest in response to 100 μm glutamate, 250 μm Zn2+ addition (at time 0). C, pseudo color FRET ratio images showing specific regions of interest. Left panels, zoomed in images under resting conditions (top panel), 10 s following treatment (middle panel), and 25 s following treatment (bottom panel).
Treatment with Zn2+ alone showed no change to the FRET ratio in the mitochondria of hippocampal neurons. However, treatment of hippocampal neurons with glutamate alone resulted in a transient decrease in mitochondrial Zn2+ (Fig. 5). The small molecule Zn2+ indicator FluoZin-3-AM (Invitrogen) indicates that this mitochondrial Zn2+ is released into the cytosol. Importantly, the decrease in mitochondrial Zn2+ was not observed with the low affinity mito-His4 sensor.
FIGURE 5.

Neuronal mitochondria release Zn2+ upon treatment with glutamate alone. Cytosolic FluoZin3 response (black triangle), mito-Cys2His2 response (black circle), mito-His4 response (gray diamond), and mito-Cys2His2 response in the absence of extracellular Ca2+ (light gray square) upon treatment with 100 μm glutamate are shown.
Glutamate-stimulated Zn2+ release was slightly affected by extracellular Ca2+ because glutamate treatment in Ca2+-free buffer altered the kinetics (but not the magnitude) of the response. Zinc release was measured in six individual cells from two separate experiments (supplemental Fig. S6), where the percentage of ratio change (R/Rmin) was 3.5 ± 1.0% (n = 6), and every cell measured showed release of Zn2+ upon treatment with glutamate alone. Glutamate stimulation of neurons leads to acidification followed by alkalinization of the cytosol. Given that fluorescent proteins, particularly the citrine YFP, can be pH-sensitive, we felt it was important to evaluate the pH sensitivity of the mitochondrial targeted sensor and the extent of acidification in mitochondria. As shown in Fig. 6A, the mito-Cys2His2 sensor was unaffected by pH changes between 7.4 and 6.5, but the FRET ratio decreased at pH 6.0. The decrease in FRET ratio is likely caused by H+ quenching of citrine YFP fluorescence as the pKa of citrine is 5.7 (34). To characterize the pH changes within the mitochondrial matrix upon glutamate stimulation in the presence and absence of exogenous Zn2+, we targeted ecliptic pHluorin (35) to mitochondria. Following glutamate treatment in the presence and absence of Zn2+, the pH sensor was calibrated by adding H+ ionophores with buffers of known pH values. As seen in Fig. 6, glutamate stimulation lead to a slight acidification in mitochondria; the pH dropped from >7.4 to ∼7.0. There is no observed effect on the mito-Cys2His2 sensor in this pH range.
FIGURE 6.

Characterization of mitochondrial pH and the effect of pH on the sensor. A, mito-Cys2His2 sensor FRET ratio as a function of pH. B, measurement of mitochondrial pH at rest and upon treatment with 100 μm glutamate stimulation (Glu). C, measurement of mitochondrial pH at rest and upon treatment with 100 μm glutamate, 250 μm Zn2+. For B and C, the pH calibration was performed immediately following stimulation using the same region of interest from three individual cells as described in the supplemental text.
DISCUSSION
Transition metals such as Zn2+ are both essential and toxic. The canonical view of metal homeostasis is that mammalian cells concentrate metal ions from their environment, but the vast majority of these metals are bound to proteins or cellular buffers. An alternate paradigm is emerging in which metals may be mobilized from labile pools, such as metallothionein or organelles. The possibility of transient metal “signals” is forcing us to re-evaluate cellular control of metal availability and its influence on cellular function. To obtain a comprehensive picture of Zn2 regulation, it is necessary to define reservoirs of labile Zn2+ as well as the fate of mobilized Zn2+.
In this work, we present genetically targeted Zn2+ sensors and demonstrate their utility in monitoring the spatial distribution of Zn2+. Our mitochondrially targeted sensor reports an elevated resting Zn2+ level in mitochondria relative to the cytosol. Moreover, treatment of hippocampal neurons with glutamate results in transient release of this zinc into the cytosol, indicating that mitochondria can indeed serve as a source of Zn2+ signals. This is consistent with the finding by Sensi et al. (9) that mitochondrial Zn2+ pools may be mobilized independently of cytosolic pools. We also extended the work of Nolan et al. (32) and demonstrate that upon treatment with glutamate plus Zn2+, Zn2+ is taken up into neurons and is sequestered into mitochondria, thus creating a transient Zn2+ signal in mitochondria. Our results indicate that the role of mitochondria in modulating Zn2+ signals is context-dependent; glutamate treatment alone leads to Zn2+ release, whereas treatment with glutamate plus Zn2+ leads to Zn2+ uptake. This finding may have important biological implications because some hippocampal neurons contain only glutamate in presynaptic vesicles, whereas others contain both glutamate and Zn2+ (4). Our results suggest that perhaps different neurons will elicit different cellular responses, although it is important to note that the cellular consequences of these Zn2+ signals are still unclear.
There is a great challenge in accurately quantifying cellular Zn2+ concentrations. In this work we demonstrate that cellular Zn2+ levels are perturbed, even by expression of the relatively low affinity (Kd = ∼1 μm) Cys2His2 sensor. This perturbation is not surprising given that our sensor concentration is also in the low micromolar range, and hence [sensor] is approximately equal to the Kd. By measuring Zn2+ levels as a function of [sensor], we were able to extrapolate to an estimate of 180 nm Zn2+ in the cytosol. This estimate is higher than reports using other probes in which Zn2+ levels are predicted to be in the picomolar to nanomolar range (2–5). We suspect that all of these estimates, including our own, possess shortcomings that limit their accuracy. As our data indicate, measurements must be made at a range of sensor concentrations. This is particularly relevant for high affinity sensors, which when incorporated into cells at micromolar concentrations result in a situation in which the [sensor] is much greater than the Kd. This is likely to lead to significant perturbation of the cellular free Zn2+ levels. Although we account for this in our measurements, we believe our measurements overestimate Zn2+ because our sensor is at the lower limit of its detection range. Still, we believe the relative estimates of cytosolic and mitochondrial Zn2+ are fairly accurate given that the same sensor is used to monitor Zn2+ in these different locations.
To overcome the shortcomings listed above and acquire accurate measurements of cellular Zn2+, we as a community will require a series of probes with different K′d values, where the [sensor] is kept much lower than the Kd. We are currently working to place our sensors under control of an inducible promoter so that concentrations can be controlled more tightly and varied over a wider range for more accurate extrapolations. One advantage of our sensor design platform is that the fluorescent properties can be readily tuned by changing the fluorescent proteins. We are exploring different fluorescent protein combinations as well as optimizing the brightness of the proteins so that sensors can be incorporated into cells at lower expression levels in an effort to minimize the perturbation of cellular metals. In this regard, genetically encoded probes offer an advantage over small molecule indicators whose concentration is difficult to control because cellular accumulation is a balance of uptake and extrusion (36). Moreover, accurately defining the concentration of small molecule probes whose intensity is modulated by Zn2+ is challenging.
Our localized probe overcomes many of the limitations of RhodZin-3 as a mitochondrial Zn2+ sensor. For example, RhodZin-3 exhibits suboptimal localization; it is lost from mitochondria upon membrane depolarization, and the read-out is intensity-based rather than ratiometric. We attempted to use RhodZin-3 to examine mitochondrial Zn2+ dynamics in neurons, but the localization was so poor that we could not obtain interpretable data (supplemental Fig. S3). Thus, the mitochondrial probe presented here represents a significant advance in our ability to monitor mitochondrial Zn2+.
However, the sensors presented here can still be improved. As illustrated in Fig. 6, they are quenched at low pH (6.0 and below), which would impede measurement of Zn2+ in acidic compartments. The Cys2His2 sensor relies on Cys to bind Zn2+, and thus its use is restricted to reducing environments. Lastly, although we were able to observe reproducible signals, the sensors would benefit from an expanded dynamic range.
In summary, we have developed genetically targeted sensors for Zn2+ and used these to demonstrate that mitochondria contain a labile and releasable pool of Zn2+ under resting conditions. In neurons, mitochondria can serve as a source of Zn2+ signals by releasing Zn2+ into the cytosol, as well as a sink by sequestering elevated cytosolic Zn2+. The observation of a mitochondrial pool of Zn2+ raises the intriguing question of whether other organelles modulate Zn2+ availability by serving as either sources or sinks for Zn2+.
Supplementary Material
Acknowledgment
We thank David Wren for help with setting up the anaerobic protein purification.
This work was supported, in whole or in part, by National Institutes of Health Grants T32 GM-065103 (to P. J. D.) and GM084027 (to A. E. P.). This work was also supported by the University of Colorado and a Whitehall Foundation beginning grant-in-aid.

The on-line version of this article (available at http://www.jbc.org) contains supplemental text, movies, and Figs. S1–S7.
- FRET
- fluorescence resonance energy transfer
- CFP
- cyan fluorescent protein
- YFP
- yellow fluorescent protein
- MOPS
- 4-morpholinepropanesulfonic acid
- TPEN
- N,N,N′,N′-tetrakis(2-pyridylmethyl) ethylenediamine.
REFERENCES
- 1.Eide D. J. ( 2006) Biochim. Biophys. Acta 1763, 711– 722 [DOI] [PubMed] [Google Scholar]
- 2.Maret W., Krezel A. ( 2007) Mol. Med. 13, 371– 375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thompson R. B. ( 2005) Curr. Opin. Chem. Biol. 9, 526– 532 [DOI] [PubMed] [Google Scholar]
- 4.Frederickson C. J., Koh J. Y., Bush A. I. ( 2005) Nat. Rev. Neurosci. 6, 449– 462 [DOI] [PubMed] [Google Scholar]
- 5.Suhy D. A., Simon K. D., Linzer D. I., O'Halloran T. V. ( 1999) J. Biol. Chem. 274, 9183– 9192 [DOI] [PubMed] [Google Scholar]
- 6.Maret W. ( 1994) Proc. Natl. Acad. Sci. U.S.A. 91, 237– 241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maret W. ( 1995) Neurochem. Int. 27, 111– 117 [DOI] [PubMed] [Google Scholar]
- 8.Bossy-Wetzel E., Talantova M. V., Lee W. D., Scholzke M. N., Harrop A., Mathews E., Gotz T., Han J., Ellisman M. H., Perkins G. A., Lipton S. A. ( 2004) Neuron 41, 351– 365 [DOI] [PubMed] [Google Scholar]
- 9.Sensi S. L., Ton-That D., Sullivan P. G., Jonas E. A., Gee K. R., Kaczmarek L. K., Weiss J. H. ( 2003) Proc. Natl. Acad. Sci. U.S.A. 100, 6157– 6162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kroncke K. D., Fehsel K., Schmidt T., Zenke F. T., Dasting I., Wesener J. R., Bettermann H., Breunig K. D., Kolb-Bachofen V. ( 1994) Biochem. Biophys. Res. Commun. 200, 1105– 1110 [DOI] [PubMed] [Google Scholar]
- 11.Que E. L., Domaille D. W., Chang C. J. ( 2008) Chem. Rev. 108, 1517– 1549 [DOI] [PubMed] [Google Scholar]
- 12.Bozym R. A., Thompson R. B., Stoddard A. K., Fierke C. A. ( 2006) ACS Chem. Biol. 1, 103– 111 [DOI] [PubMed] [Google Scholar]
- 13.Hong S. H., Maret W. ( 2003) Proc. Natl. Acad. Sci. 100, 2255– 2260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Walkup G. K., Imperiali B. ( 1996) J. Am. Chem. Soc. 118, 3053– 3054 [Google Scholar]
- 15.Walkup G. K., Burdette S. C., Lippard S. J., Tsien R. Y. ( 2000) J. Am. Chem. Soc. 122, 5644– 5645 [DOI] [PubMed] [Google Scholar]
- 16.Ellman G. L. ( 1959) Arch. Biochem. Biophys. 82, 70– 77 [DOI] [PubMed] [Google Scholar]
- 17.Palmer A. E., Giacomello M., Kortemme T., Hires S. A., Lev-Ram V., Baker D., Tsien R. Y. ( 2006) Chem. Biol. 13, 521– 530 [DOI] [PubMed] [Google Scholar]
- 18.Wilkinson K. D. ( 2004) Methods Mol. Biol. 261, 15– 32 [DOI] [PubMed] [Google Scholar]
- 19.Miyawaki A., Griesbeck O., Heim R., Tsien R. Y. ( 1999) Proc. Natl. Acad. Sci. 96, 2135– 2140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grynkiewicz G., Poenie M., Tsien R. Y. ( 1985) J. Biol. Chem. 260, 3440– 3450 [PubMed] [Google Scholar]
- 21.Banker G., Goslin K. ( 1998) Culturing Nerve Cells, 2nd Ed., MIT Press, Cambridge, MA [Google Scholar]
- 22.Elrod-Erickson M., Rould M. A., Nekludova L., Pabo C. O. ( 1996) Structure 4, 1171– 1180 [DOI] [PubMed] [Google Scholar]
- 23.Nagai T., Yamada S., Tominaga T., Ichikawa M., Miyawaki A. ( 2004) Proc. Natl. Acad. Sci. U.S.A. 101, 10554– 10559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang J., Allen M. D. ( 2007) Mol. Biosyst. 3, 759– 765 [DOI] [PubMed] [Google Scholar]
- 25.Hires S. A., Zhu Y., Tsien R. Y. ( 2008) Proc. Natl. Acad. Sci. 105, 4411– 4416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rae T. D., Schmidt P. J., Pufahl R. A., Culotta V. C., O'halloran T. V. ( 1999) Science 284, 805– 808 [DOI] [PubMed] [Google Scholar]
- 27.vanDongen E. M. W. M., Evers T. H., Dekkers L. M., Meijer E. W., Klomp L. W. J., Merkx M. ( 2007) J. Am. Chem. Soc. 129, 3494– 3495 [DOI] [PubMed] [Google Scholar]
- 28.Evers T. H., Appelhof M. A. M., de Graaf-Heuvelmans P. T. H. M., Meijer E. W., Merkx M. ( 2007) J. Mol. Biol. 374, 411– 425 [DOI] [PubMed] [Google Scholar]
- 29.Miyawaki A., Tsien R. Y. ( 2000) Methods Enzymol. 327, 472– 500 [DOI] [PubMed] [Google Scholar]
- 30.Qiao W., Mooney M., Bird A. J., Winge D. R., Eide D. J. ( 2006) Proc. Natl. Acad. Sci. 103, 8674– 8679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Malaiyandi L. M., Vergun O., Dineley K. E., Reynolds I. J. ( 2005) J. Neurochem. 93, 1242– 1250 [DOI] [PubMed] [Google Scholar]
- 32.Nolan E. M., Ryu J. W., Jaworski J., Feazell R. P., Sheng M., Lippard S. J. ( 2006) J. Am. Chem. Soc. 128, 15517– 15528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sensi S. L., Yin H. Z., Weiss J. H. ( 2000) Eur. J. Neurosci. 12, 3813– 3818 [DOI] [PubMed] [Google Scholar]
- 34.Griesbeck O., Baird G. S., Campbell R. E., Zacharias D. A., Tsien R. Y. ( 2001) J. Biol. Chem. 276, 29188– 29194 [DOI] [PubMed] [Google Scholar]
- 35.Miesenbock G., De Angelis D. A., Rothman J. E. ( 1998) Nature 394, 192– 195 [DOI] [PubMed] [Google Scholar]
- 36.Kao J. P. ( 1994) Methods Cell Biol. 40, 155– 181 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.