

Abstract
Colicin Ia is a soluble, harpoon-shaped bacteriocin which translocates across the periplasmic space of sensitive Escherichia coli cell by parasitizing an outer membrane receptor and forms voltage-gated ion channels in the inner membrane. This process leads to cell death, which has been thought to be caused by a single colicin Ia molecule. To directly visualize the three-dimensional structure of the channel, we generated two-dimensional crystals of colicin Ia inserted in lipid-bilayer membranes and determined a ∼17 three-dimensional model by electron crystallography. Supported by velocity sedimentation, chemical cross-linking and single-particle image analysis, the three-dimensional structure is a crown-shaped oligomer enclosing a ∼35 Å-wide extrabilayer vestibule. Our study suggests that lipid insertion instigates a global conformational change in colicin Ia and that more than one molecule participates in the channel architecture with the vestibule, possibly facilitating the known large scale peptide translocation upon channel opening.
Colicin Ia is a pore-forming water-soluble bacterial toxin produced by some strains of Escherichia coli to kill other competing bacteria (1, 2). It belongs to a functionally and structurally similar group of proteins that also includes colicins A (3), E1 (4), and N (5). Each of these proteins consist of three domains with distinct properties; the receptor domain (R), which binds a specific outer membrane receptor on the target cell, and the translocation domain (T) at the N terminus, responsible for traversing the outer membrane and the periplasmic space to deliver the channel-forming domain (C) at the C terminus to the bacterial inner membrane. The bundle of 10 α-helices that compose the C domain changes its conformation to form a voltage-gated ion channel in the plasma membrane. Opening of the channel produces an efflux of ions that depletes the cellular energy resources and ultimately leads to cell death.
The x-ray structure of full-length, soluble colicin Ia (69 kDa) has been determined (6). The monomeric molecule is mostly α-helical, with the R domain separated from the T and C domains by a pair of unusually long (∼160 Å) α-helices thought possibly to span the periplasmic space during channel formation (6). The C domain is characterized by two hydrophobic helices (VIII and IX; residues Ala-580—Ile-612) that is surrounded by the remaining eight largely amphipathic α-helices. The same structural motif for the C domain is conserved in other members of the colicin family and is also present in the channel-forming domains of diphtheria toxin, exotoxin A, and the Bcl family of pro- and anti-apoptotic proteins (7). This pair of helices, termed the hydrophobic hairpin, is instrumental in driving the initial membrane insertion event (8) that is followed by a series of large scale pH and voltage-dependent conformational changes in the C domain, resulting in the opening of the ion channel in the plasma membrane (9, 10). In the absence of a high resolution membrane-inserted structure of a channel-forming colicin, solid-state NMR (11, 12), streptavidin binding (8) and cross-linking of site-directed cysteine mutants (9) have suggested that the initial membrane-bound intermediate exists as a two-dimensional helical array of the eight amphipathic helices (I-VII and X) spread across the membrane surface, with the hydrophobic helices (VIII and IX) embedded in the bilayer. A recent electron paramagnetic resonance study using preparations of spin-labeled ColA proteoliposomes has supported a similar umbrella model where the eight amphipathic helices reside at the air-water interface for the closed-channel state (13). Biotin-labeled cysteine mutants have also been used to determine how much of the C domain (aside from the hydrophobic hairpin) crosses the plasma membrane (14, 15) for colicin Ia. A large region of the amphipathic sequence (helices II-V; residues Leu-474—Tyr-541) has been found to cross from the cis to the trans side of the membrane in planar lipid bilayer experiments, resulting in a four-transmembrane segment molecule that is thought to form the ion channel.
Because the 12–13 residue α-helices of the C domain are well short of the ∼20 residues required to span the plasma membrane, it has been proposed that conformational changes causing helix extension take place during the channel formation process. 13C spin diffusion NMR has indicated that whereas the overall secondary structure of the C domain is preserved, most of the helices undergo “opening,” and modulation of the tertiary structure allows for the required extension of the helices to cross the plasma membrane and form the channel (16). The internal structure of the colicin Ia channel has been investigated by examining the effect of different nonelectrolyte molecules on the single-channel conductance in planar lipid bilayer membranes (17). It was determined that the diameter at the cis entrance (equivalent to the outside of the cell) is 18 Å, and the diameter at the trans entrance (inside the membrane) is 10 Å, with a 7 Å diameter constriction located in close proximity to the trans entrance of the channel. More recent studies (18) employing the substituted cysteine accessibility method to determine what residues line the open colicin Ia channel suggest an hourglass-shaped pore with the most constricted part near the cis rather than the trans side, as opposed to the conclusion of Krasilnikov et al. (17). Both studies point to a pore constriction inside the membrane, and as pointed out by Kienker et al. 18), there exist plausible explanations to reconcile some of the differing results. The large diameter of the colicin Ia channel coupled with the studies which indicate that each colicin Ia molecule contributes four transmembrane segments in the membrane integrated state (14) suggests that the ion channel is formed by a multimer of colicin Ia molecules. However, all of the past studies directed at determining the oligomeric state of any of the colicin channels indicate a monomeric structure. The question as to how a four-transmembrane monomeric protein can form an ion channel of sufficient diameter to allow the passage of ions as large as tetraethyl ammonium (19) has remained unanswered.
In this work we have subjected colicin Ia incorporated into lipid bilayer membranes to structural and biochemical investigations. We show, based on cross-linking and velocity sedimentation experiments, single-particle analysis of electron micrographs and results from electron crystallographic analysis of two-dimensional crystals of colicin Ia that the protein forms oligomers upon insertion into the bilayer. The suggested architecture of this oligomer based on the ∼17 Å resolution three-dimensional model and the biological implications, are discussed.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification
Recombinant full-length colicin Ia was expressed in pKSJ311-containing E. coli TG1 cells and purified (supplemental text S1). Colicin Ia channel-forming domain (CFD)3 (residues Glu-438—Ile-626) was expressed from pKSJ155 (called CT-M in Kienker et al. (20)) in BL21(DE3) cells and purified (supplemental Text S1). Purified protein was either freshly used for subsequent study or stored in aliquots at −80 °C until required in sodium citrate buffer (full-length colicin Ia) or Tris buffer (colicin Ia CFD). Samples of the purified proteins were shown to possess normal channel-forming properties in planar lipid bilayer experiments, as described in Kienker et al. (20).
Preparation and Incorporation of Colicin Ia into Large Unilamellar Vesicles
1,2-Dimyristoyl-sn-glycero-3-[phospho- rac-(1-glycerol)] (DMPG) (Avanti) and 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) (Avanti) or 1-palmitoyl-2-oleoyl-sn-glycero-3-[phospho-rac-(1-glycerol)] (POPG) and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) (Avanti) in chloroform were mixed 1:1 and dried to a thin film initially under N2 and then by vacuum evaporation by centrifugation. The lipid film was resuspended at a final concentration of 10 mg/ml in 50 mm sodium acetate at pH 4.5, 150 mm NaCl and hydrated for 1 h at 37 °C. Large unilamellar vesicles (LUVs) were prepared by extrusion through a 200-nm polycarbonate filter using a Mini-Extruder (Avanti). To incorporate colicin Ia into the preformed liposomes, purified protein was added to the LUV suspension at lipid-protein ratio of 6:1 (w/w) in sodium acetate buffer, and the mixture was incubated for 2–3 h in a water bath at 37 °C.
The integration of colicin Ia CFD into lipid bilayer membranes was tested by subjecting the proteoliposomes to high salt/high pH as follows. A suspension of LUVs loaded with the colicin Ia CFD (lipid-protein ratio 8:1) was centrifuged through a 10% (w/v) sucrose cushion at 100,000 × g for 1 h at 4 °C. The pellet was resuspended in 50 mm sodium phosphate buffer at pH 7 for 5 min using a H2O bath sonicator (Fisher). The suspension was treated with 4.5 m urea and 2.5 m NaCl for 1 h at room temperature as described (21) and then titrated to pH 11–12 with 1 m NaOH stock and further incubated for 30 min at room temperature. Next, the reaction mixture was titrated back to pH 7 with 1 m HCl stock solution and then dialyzed against 50 mm sodium phosphate at pH 7 for 1 h at room temperature to remove excess urea and NaCl. Finally, the so-treated liposomes were recovered by centrifugation as above through a 10% (w/v) sucrose cushion. Pellets and corresponding supernatants were analyzed by SDS-PAGE.
Cross-linking of Full-length Colicin Ia Incorporated into Proteoliposomes
Bis[sulfosuccinimidyl]suberate (BS3) (Thermo Scientific) was used for cross-linking experiments and prepared fresh for each cross-linking reaction. The suspension of colicin Ia-incorporated LUVs prepared as described above in acetate buffer was dialyzed against 20 mm HEPES at pH 7.5, 150 mm NaCl for 2 h at 25 °C to change the pH to within the recommended range for the cross-linking reaction (i.e. pH 7–9). The dialyzed proteoliposome suspension was then centrifuged in an Airfuge (Beckman) at 20 p.s.i. (∼100,000 × g) for 10 min to remove any remaining protein that was not incorporated into the liposomes. The proteoliposome pellet was resuspended in the above buffer and repelleted as before to ensure complete removal of unincorporated colicin Ia. BS3 in 20 mm HEPES at pH 7.5, 150 mm NaCl was added to the proteoliposome suspension at a final concentration of 0.5 mm. The reaction mixture was incubated at 25 °C for 30 min, and the cross-linking was quenched by the addition of 1 m Tris-HCl at pH 7.5 stock solution to a final concentration of 50 mm followed by a further incubation at 25 °C for 15 min. Samples were analyzed by electrophoresis on NuPAGE 3–8% Tris acetate gels (Invitrogen).
Size-exclusion Chromatography (SEC) to Assay Membrane-inserted Cross-linked Colicin Ia
Proteoliposomes containing cross-linked colicin Ia were solubilized by the addition of 2% (w/v) n-dodecyl-β-d-maltopyranoside (DDM) (Anatrace) and incubation at 25 °C overnight with gentle stirring. Insoluble material was removed by centrifugation in an Airfuge (Beckman) at 20 p.s.i. for 10 min. The supernatant (100 μl) was applied to a Superose 6 column (30 × 10 mm) (GE Healthcare) equilibrated in 20 mm HEPES at pH 7.5, 150 mm NaCl, 0.05% (w/v) DDM and controlled by an AKTA chromatography system (GE Healthcare). The column was calibrated with molecular weight standards (of known Stokes radii) thyroglobulin (8.5 nm), apoferritin (6.1 nm), catalase (5.22 nm), aldolase (4.8 nm), and bovine serum albumin (3.55 nm). The Stokes radii for colicin Ia peaks were estimated by comparison of the elution volume against the elution volumes for the calibration standards.
Rate Zonal Sedimentation Analysis of Solubilized Cross-linked Colicin Ia by Sucrose Gradient
100 μl of solubilized cross-linked colicin Ia was layered onto a preformed linear 5–20% (w/v) sucrose gradient containing 20 mm HEPES at pH 7.5, 150 mm NaCl, 0.05% (w/v) DDM and prepared using a SG15 gradient maker (Hoefer Scientific Instruments). Additional gradients were prepared and layered with 100 μl of 2 mg/ml each of the following molecular mass standards: thyroglobulin (669 kDa; 19 S), apoferritin (440 kDa; 17 S), catalase (232 kDa; 11.4 S), aldolase (158 kDa; 7.88 S), and bovine serum albumin (67 kDa; 4.4 S). The gradients were centrifuged for 15 h at 5 °C in an AH-650 rotor (Sorvall) at 30,400 rpm (87,400 × g). After centrifugation, 350-μl fractions were removed from the bottom of the tube using a peristaltic pump, and aliquots of the standards were analyzed for absorbance at 280 nm. The fractions of the gradient containing solubilized, cross-linked colicin Ia were precipitated by trichloroacetic acid then acetone and analyzed by electrophoresis on NuPAGE 3–8% Tris acetate gels (Invitrogen).
Using the estimated sedimentation coefficient (s20,w) and Stokes radius (RS) values, the experimental molecular weight range of multimeric colicin Ia was calculated (22) using the equation, molecular weight = s20,wN6πn RS/(1 − υρ), where Avogadro's number (N) = 6.023 × 1023, viscosity coefficient (n) = 1 × 10−2 g·s−1·cm−1, solution density (ρ) = 1 g·cm−3, and partial specific volume (υ) = 0.72 cm3·g−1 (assumed to be that of an average soluble protein).
Two-dimensional Crystallization of Colicin Ia in Lipid Bilayer Membranes
For two-dimensional crystallization trials, purified colicin Ia in 20 mm sodium citrate at pH 5.2, 50 mm NaCl, at a final concentration of 1 mg/ml, was mixed with a 70:30 mixture of DMPG and DMPC at lipid-protein ratio of 1:2–1:4 (w/w). Lipid dissolved in chloroform was dried under N2 and then solubilized for 1–2 h into a 2% (w/v) aqueous solution of n-octyl-β-d-glucopyranoside (Anatrace) at 25 °C with gentle stirring. After incubation of the lipid/protein/detergent mixture at 25 °C for 2–3 h, 50-μl aliquots were dialyzed against 50 mm sodium acetate at pH 4.5, 150 mm NaCl, 0.02% (w/v) NaN3 (SPECTRAPOR 10-kDa cut-off) at 37 °C until the detergent was deemed to have been completely removed (indicated by inability to break the surface tension of a water droplet).
Specimen Preparation, Electron Microscopy, and Image Processing
Typically 5-μl samples of two-dimensional crystallization trials and samples from colicin Ia/lipid reconstitution assays were applied to glow-discharged (hydrophilic) 300-mesh copper electron microscopy grids coated with plastic-supported continuous carbon film. The sample was allowed to adsorb for 90 s before being washed three times with filtered, distilled H2O and stained with 1.5% (w/v) uranyl acetate. Excess stain was removed by blotting with filter paper (Whatman No. 1), and the grid was allowed to air dry.
For screening two-dimensional crystallization trials, a Philips CM12 operated at 120 kV and equipped with a BIOSCAN CCD camera (GATAN Inc) was used. For data collection, samples were examined using a Philips Tecnai 12 electron microscope operated at 120 kV and equipped with a LaB6 filament. Images were recorded at a nominal magnification of 52,000 at 2.0–3.0 μm under-focus (for two-dimensional crystals) or 1.5–3.0 μm under-focus (for single particle images). Electron microscopy image film (Kodak SO-163) was used, which was developed for 10 min in D19 (diluted 1:1 with H2O). Images of tilted crystals (up to ±50°) were recorded at ∼10° intervals in a tilt series starting with the nominally untilted view. Micrographs with minimal astigmatism and drift, as judged by optical diffraction, were selected for digitization using a Super CoolScan 9000 ED (Nikon). For images of two-dimensional crystals, typically areas of ∼0.4 × 0.4 μm (on the specimen) were digitized at 4,000 dpi, corresponding to sampling at ∼1.2 Å on the specimen. For images used for single particle analysis, digitization was carried out at 2442 dpi, corresponding to ∼2 Å on the specimen.
The processing of the images of two-dimensional crystals was carried out using the MRC suite of programs (23, 24) with usually two rounds of unbending for each image, as described e.g. in Hyun et al. (25) (supplemental Text S2). For single-particle image analysis, 4305 tilted pairs (0° and 45°) of isolated, membrane-inserted Colicin Ia particles were reconstructed by using the Xmipp program (26) suite and the random-conical tilt approach (27). Reference models for simultaneous three-dimensional maximum-likelihood refinement (28) were constructed after separating the dataset into three classes using maximum-likelihood classification (29, 30). Approximately 1250 particles were assigned to the final reconstruction after 25 cycles of iterative refinement (supplemental Text S2).
RESULTS
Incorporation of the Channel-forming Domain of Colicin Ia into Preformed Liposomes
Colicin Ia CFD incorporated into proteoliposomes (POPG:POPC = 1:1) at pH 4.5 were treated with high concentrations of urea and NaCl as well as extreme alkaline pH and then centrifuged at 100,000 × g. SDS-PAGE analysis of this sample showed that ∼80% of the protein was retained in the proteoliposome pellet (Fig. 1A). This demonstrates that under the protocol of using acidic pH and negatively charged lipids that we have followed throughout this work for inducing insertion into lipid bilayers, a major fraction of the protein is in a stable, membrane-integrated state rather than peripherally attached to the lipid bilayer.
FIGURE 1.

Incorporation and cross-linking of colicin Ia in large unilamellar vesicles. A, lanes 1 and 3 correspond to the supernatants and pellets of 100,000 × g centrifugation of DMPG/DMPC LUVs incubated with recombinant full-length colicin Ia at pH 4.5 (lipid-protein ratio of 6:1 (w/w)), whereas lanes 2 and 4 correspond to those for equivalent centrifugation runs after dialysis of the proteoliposomes to pH 7.5 buffer. The samples are analyzed on a 10% SDS-PAGE. B, lipid-incorporated full-length colicin Ia cross-linked by BS3 resolved on a 4–10% SDS-PAGE. Lanes 1, 2, and 3 are for 0, 0.25, and 0.5 mm BS3. Species with large molecular mass can be seen to be retained in the stacking portion of the gel in lanes 2 and 3.
Chemical Cross-linking of Full-length Colicin Ia in the Lipid Bilayer
After cation exchange chromatography of cell lysate overexpressing recombinant colicin Ia, SEC using Superdex 200 in 20 mm sodium citrate at pH 5.2, 50 mm NaCl produced a large peak and several small peaks. The SDS-PAGE of the major colicin Ia peak, which runs anomalously at ∼60 kDa (supplemental Fig. S1), shows that the protein used in all subsequent experiments has been purified to homogeneity.
To use the cross-linking reagent BS3, unilamellar vesicles composed of DMPC and DMPG (1:1) loaded with full-length colicin Ia at pH 4.5 was dialyzed against HEPES buffer at pH 7.5. About 50% of the protein was retained in the lipid-associated state, as judged by subsequent centrifugation and SDS-PAGE analysis (Fig. 1B). This stably inserted colicin Ia was then subjected to cross-linking experiments. The SDS-PAGE band for colicin Ia monomer at ∼60 kDa disappears after cross-linking, and a faint smear of ∼150-kDa appears in the range where we expect to see a dimer or a trimer of colicin Ia (estimated molecular mass of 138 or 208 kDa) (Fig. 1B). However, the majority of the cross-linked protein sample migrates only a short distance into the separating portion of the SDS-PAGE and, therefore, represents an assembly possessing a much larger molecular mass. These observations taken together suggest that (a) colicin Ia is not monomeric but oligomeric in the lipid bilayer, and (b) whereas it may initially organize as a dimer (or trimer) after insertion into the lipid bilayer, ultimately a much larger multimeric state of the protein is formed, which conventional SDS-PAGE fails to resolve.
Characterization of Colicin Ia Oligomer by SEC and Sucrose Gradient Sedimentation
For accurate estimation of the mass of oligomeric colicin Ia in the lipid bilayer, cross-linked protein from proteoliposomes was solubilized with 2% (w/v) DDM, then analyzed by SEC on a Superose 6 10/300 column calibrated with protein standards of known molecular weight and Stokes radii (RS) and sedimentation coefficients (s20,w). When soluble, monomeric colicin Ia was subjected to SEC under the same buffer conditions, the retention volume corresponded to a Stokes radius of 3.35 nm (data not shown). SEC of cross-linked solubilized colicin Ia sample demonstrated that it eluted across a broad range of retention volumes (Fig. 2A) with RS ranging from 6.09 to 7.91 nm (Fig. 2B). This shift in elution peak from 3.35 nm indicates that colicin Ia likely exists in a multimeric form(s) upon insertion into the lipid bilayer. Eluted fractions from the Superose 6 column were resolved by electrophoresis on a 4–8% Tris acetate gel and showed the presence of multiple protein species of molecular mass corresponding potentially to monomer, dimer, trimer, up to hexamer (∼460 kDa) (Fig. 2C). Examination of these fractions by transmission electron microscopy after staining with 1.5% (w/v) uranyl acetate revealed a protein population of large heterogeneous multimers (data not shown). Peaks that eluted at higher retention volumes (13.8 and 14.24 ml), corresponding to RS of 5.55 and 5.29 nm, respectively, displayed a relatively more homogeneous population of irregularly shaped objects (data not shown). When analyzed by SDS-PAGE, these fractions were found to contain high molecular mass species (larger than 460 kDa), which may represent colicin Ia oligomers with tightly bound lipid/detergent molecules displaying an anomalous elution profile.
FIGURE 2.

SEC of detergent-solubilized cross-linked colicin Ia. A, elution profiles of cross-linked colicin Ia in proteoliposomes after solubilization with 2% DDM and using Superose 6 column. B, calibration curve using the retention volumes of standard proteins with known Stokes radii. C, 0.5-ml fractions for the elution profiles in A precipitated with trichloroacetic acid and resolved on a 3–8% Tris acetate polyacrylamide gel; lanes 1–7 correspond to the range 9.5–13 ml; lanes 8 and 9 correspond to 13.5–14.5 ml. Species with a large molecular mass are retained in the stacking portion of the gel particularly in lanes 8 and 9. BSA, bovine serum albumin; AU, absorbance units.
Rate zonal sedimentation analysis was used to estimate the sedimentation coefficient for cross-linked colicin Ia after solubilization from proteoliposomes by 2% (w/v) DDM. Sucrose gradients were run, and the migration distance for molecular mass standards was used to plot a calibration curve (Fig. 3A). Migration of cross-linked colicin Ia was compared with those for the protein standards of known sedimentation coefficients (Fig. 3B). Results of this experiment agreed with the results from the SEC experiment described above. Once again, there appear to be several protein species of large molecular mass with estimated s20,w ranging from 9.0 to 16.75 S (where S = Svedberg unit = 1 × 10−13 s). When compared with the migration of monomeric colicin Ia, these enhanced sedimentation coefficient values again indicate the presence of colicin Ia multimer that is generated upon protein insertion into the lipid bilayer. Quantitatively, using the dependence of molecular mass on sedimentation coefficient and Stokes radius (see “Experimental Procedures”) and the quoted S values above, the sedimentation of these colicin Ia multimers translate to species of molecular mass in the range of ∼222 to ∼537 kDa. These values would correspond to apparent masses of hydrodynamic complexes consisting of approximately three to eight colicin Ia monomers (monomer = 69 kDa) and including masses of any associated lipids and detergent molecules.
FIGURE 3.

Rate zonal sedimentation analysis of cross-linked colicin Ia solubilized in DDM by sucrose gradient. A, calibration curve using the migration distances for proteins with known sedimentation coefficients upon centrifugation on 5–20% (w/v) sucrose gradients (see “Experimental Procedures”). B, cross-linked solubilized colicin Ia run on the same sucrose gradient analyzed by a 3–8% Tris acetate gel. Lanes 1–15 correspond to fractions from the bottom to the top of the gradient. Lanes 4–8 correspond to sedimentation coefficients of 16.75 S to 9.0 S based on the curve shown in A. BSA, bovine serum albumin.
Generation and Analysis of Two-dimensional Crystals of Membrane-integrated Colicin Ia
The successful two-dimensional crystallization trials produced small (∼0.5 × 0.5 μm), well ordered crystalline patches of ring-shaped particles on vesicles or sheets arranged in a close-packed hexagonal array (Fig. 4, A and B). This represents the first instance that a crystalline array of colicin Ia in lipid bilayer membranes has been observed.
FIGURE 4.

Incorporation of colicin Ia in lipid bilayers in the form of isolated oligomers and two-dimensional crystals. A, an electron micrograph of colicin Ia incorporated into lipid bilayers in a crystallization experiment and visualized in a negative stain. Individual ring-shaped particles assemble in-plane to produce two-dimensional crystal lattices. B and C are close-ups of the two-dimensional lattice and isolated ring-shaped particles, respectively. Scale bar = 200 nm (A) and 50 nm (B and C).
With the constraint that in vitro, acidic pH and negatively charged lipids promote membrane insertion of colicin Ia, many different reconstitution regimens were explored (31, 32). Supplemental Table S1 provides a summary of the various reconstitution/crystallization conditions that were explored. A dispersed accumulation of ring-shaped particles, such as in the crystals, was often seen adjacent to the crystalline lattice (Fig. 4C). A high concentration of such particles distinguished by areas of sequestered uranyl acetate stain was often indicative of the existence of two-dimensional crystals in the vicinity when crystallization trials were screened.
The diffraction spots in the calculated Fourier transform of a nominally untilted crystal extended to ∼18 Å resolution after correcting for lattice distortions (Fig. 5A). The phase residual for symmetry-related reflections calculated using the program ALLSPACE indicated that the plane group of symmetry p3 was the most likely choice (Table 1). The averaged projection map is shown in Fig. 5B with the unit cell dimensions a = b = 94 Å, γ = 120°. The 3-fold symmetry is apparent in the unsymmetrized projection map (supplemental Fig. S2). The variations in amplitude and phase along lattice rods (supplemental Fig. S3) were fitted using the program LATLINE with p3 plane group of symmetry. These profiles were uniformly sampled at 1/300 Å−1 and then reverse Fourier-transformed to generate a three-dimensional reconstruction that used data up to ∼16 Å resolution.
FIGURE 5.

Image processing of nominally untilted colicin Ia two-dimensional crystals. A, the computed power spectrum of a negatively stained, colicin Ia two-dimensional crystal (shown in Fig. 4B) after applying corrections for lattice distortions. The hexagonal symmetry is apparent. The 4, −2 reflection (encircled) corresponds to ∼18 Å resolution. B, averaged projection density map calculated by using the h, k, 0 reflections from the merged three-dimensional data set generated using p3 symmetry. One unit cell is demarcated. Positive and negative densities representing stain-excluding protein and stain-permeated regions are indicated by continuous and dashed contour lines, respectively.
TABLE 1.
Phase residuals for the different choices of a plane group of symmetry of the colicin Ia two-dimensional crystal in lipid bilayer membrane
| Plane group of symmetry | Phase residualvs. other spotsa(number of comparisons) | Phase residualvs. theoretical spotsb(number of comparisons) |
|---|---|---|
| p3c | 7.7 (60) | |
| p312c | 16.0 (138) | 17.7 (12) |
| p321d | 18.9 (144) | 22.7 (24) |
| p6e | 23.4 (150) | 18.1 (60) |
| p622e | 24.1 (312) | 18.1 (60) |
a90° random.
b45° random.
c Acceptable.
d Should be considered.
e Possible.
Three-dimensional Model of Membrane-inserted Colicin Ia Determined from Tilted Negatively Stained Two-dimensional Crystals
Fig. 6 displays views of the three-dimensional density map that was shown in projection in Fig. 5B. The density volume with a nominal in-plane resolution of ∼17 Å has a crown shape and is characterized by a maximum outer diameter of ∼81 Å, an inner diameter of ∼34 Å, and an averaged height of ∼43 Å. Notwithstanding the uncertainty in the threshold level used for the display, the size of this stain-excluding protein density volume is clearly more than that expected for a full-length colicin Ia monomer. In the full density map, a few additional density peaks located at the symmetry axes and disconnected to the main density volume were observed, which may have arisen due to errors in the phases. The p3 symmetry translates to an assembly of at least three colicin molecules. At the current resolution the boundaries between the monomers cannot be discerned, and the asymmetric volume, chosen arbitrarily, is indicated in Fig. 6, B and C. This multimerization is consistent with the hydrodynamic results described above, reflecting the oligomeric organization of colicin Ia in the lipid bilayer.
FIGURE 6.

Three-dimensional density map calculated using data from tilted images of negatively stained two-dimensional crystals of colicin Ia. Solid views of the reconstructed three-dimensional density map calculated using p3 symmetry and rendered at 1.35× S.D. in the density. The view in A is normal to the bilayer, whereas those in B and C are parallel to the bilayer, with the relatively featureless region at the lower half of the figure delineating the bilayer. The ellipse in A and B indicates an arbitrarily demarcated asymmetric unit. The relatively flatter surface displayed in D is attributed to the bilayer-proximal face of the colicin Ia oligomer. Because of the lack of permeation by the hydrophilic stain, very little density is seen in the bilayer region. The figure was created using PyMOL. The scale bar is 20 Å.
Three-dimensional Model of Membrane-integrated Colicin Ia from Single Particle Analysis
Example class averages produced from untilted views showed an apparent 3-fold symmetry for the isolated, ring-shaped colicin Ia oligomer (supplemental Fig. S4). After the random-conical tilt reconstruction and refinement using the maximum-likelihood procedure, a three-dimensional model for membrane-reconstituted colicin Ia oligomer was generated. The reconstructed ring-shaped model shown in Fig. 7 has an outer diameter of ∼82 Å and an inner opening of ∼40 Å and formally appears similar to the reconstruction produced from analysis of two-dimensional crystals. The resolution of the structure based on Fourier shell correlation criterion of 0.5 was determined to be ∼29 Å.
FIGURE 7.
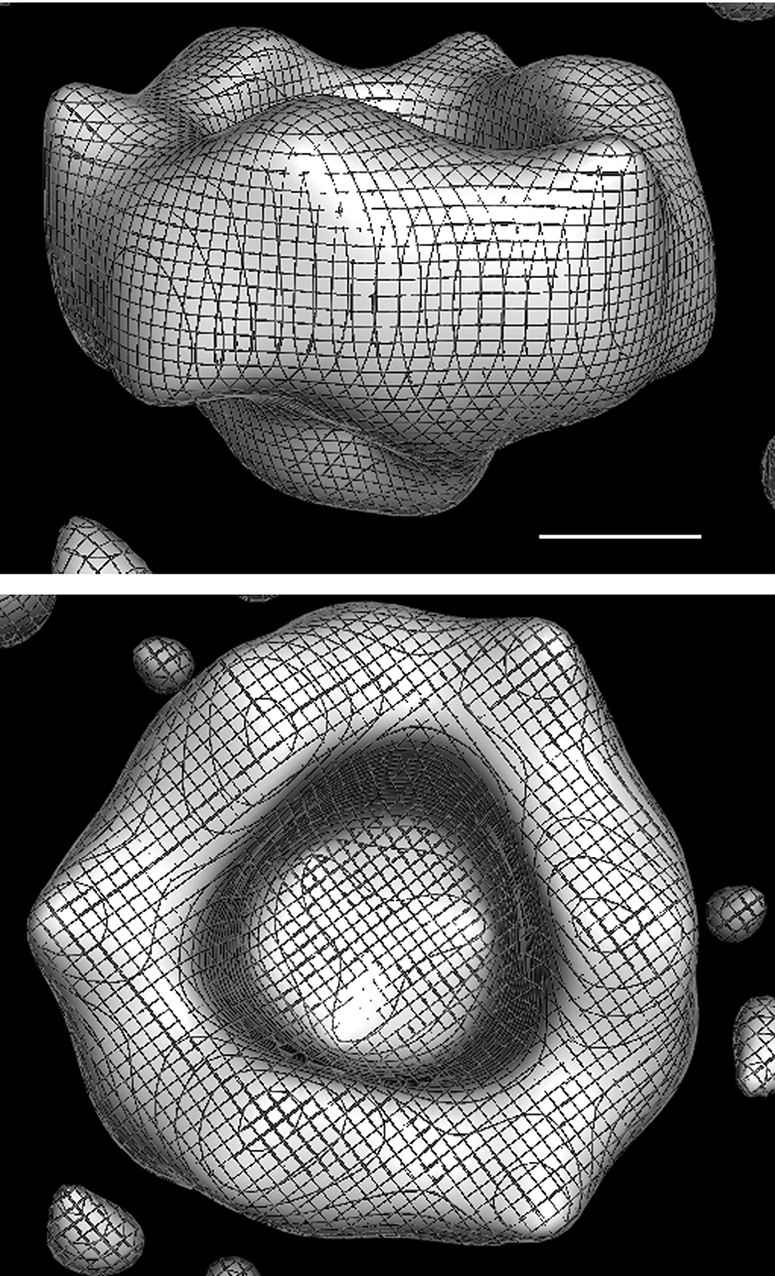
Three-dimensional reconstruction of membrane-integrated colicin Ia oligomers by single particle analysis. Two views of the reconstructed density generated by the application of random-conical tilt analysis followed by refinement using the maximum-likelihood based approach. C3 symmetry was imposed, and the maps rendered at 2.0× S.D. of the density were created using PyMOL. The scale bar is 20 Å.
DISCUSSION
This is the first study in which colicin Ia has been directly imaged in the membrane-inserted conformation. We have examined the biophysical and structural properties of this membrane-integrated state to investigate the structural changes accompanying the transition from the soluble conformation.
Our initial protein incorporation assays agree with previous findings that under acidic pH, soluble colicin Ia spontaneously inserts into lipid bilayers containing negatively charged lipids (33). Treatment of the resultant proteoliposomes with high salt, urea, and alkali conditions confirmed that the majority (∼80%) of the colicin Ia CFD, incorporated at pH 4.5, is stably integrated into the lipid bilayer. This fraction is somewhat higher compared with that in the case of full-length colicin Ia, which could very likely be because of the differences in the channel-forming characteristics between the two peptides (34).
Cross-linking Studies Reveal Multimeric Organization of Colicin Ia in the Lipid Bilayer
Cross-linking of lipid-integrated colicin Ia and analysis by SEC and sedimentation velocity gradients revealed that the protein has undergone a process of oligomerization involving several species of molecular mass up to ∼540 kDa, which translates to multimers involving potentially up to 6 or 8 monomers including any associated lipid and/or detergent molecules. Similar to these results, previous studies investigating the oligomeric state of a membrane protein after chemical cross-linking using BS3 have observed a “ladder” of high molecular mass species, e.g. in the case of MP20 (35) and CaiT (36). Further confirmation of multimeric organization of colicin Ia in the lipid bilayer was provided by electron micrographs of proteoliposomes produced via detergent-mediated incorporation at low pH. These allowed for the first time direct visualization of a membrane-bound state of colicin Ia, which was seen as ring-shaped oligomers (∼80 Å in diameter), either isolated or in the form of hexagonally packed ordered arrays. Although BS3 (with a spacer arm of 11.4 Å) has higher specificity than glutaraldehyde, the possible presence of some aggregation artifacts during cross-linking cannot be discounted. However, the biophysical data taken together with the visualization by electron microscopy and image processing makes a strong case for the existence of colicin Ia in a multimeric state in the lipid bilayer.
In this context we had also observed that at low pH, SEC of the colicin E1 CFD suggested the presence of oligomeric species with an apparent molecular mass of approximately four times the expected size for the monomer (supplemental Fig. S5). Bax, a member of the Bcl-2 family of apoptotic regulators which has a putative CFD showing structural homology to that of colicin Ia and E1, forms channels in the lipid membrane of mitochondria in eukaryotic cells (37). Dimerization of Bax and the formation of higher molecular weight structures have been shown to be essential for its function (38). This behavior is also similar, for example, to that seen for the bacterial toxin aerolysin, which oligomerizes before insertion into membranes and subsequent formation of membrane channels (39).
Two-dimensional Crystallization of Colicin Ia in the Lipid Bilayer
Examination of the crystallization trials revealed that there was a progressive formation of two-dimensional crystals in the lipid bilayer by the ring-shaped colicin Ia oligomers, presumably through a process of annealing. We suspect that the close packing of the extrabilayer portion drives crystallization, as experiments with the recombinant colicin Ia CFD produced only small, poorly ordered crystals. In general, the colicin Ia two-dimensional crystals were difficult to identify under the low magnification search conditions used for traditional low dose electron microscopy. The paucity of crystalline areas necessitated examination at high magnification, which compromised image acquisition under strict low dose conditions and the possible use of cryo-electron microscopy of frozen-hydrated specimens. We were able to collect a handful of tilt series of negatively stained crystals, of which data from two tilt series were finally utilized for three-dimensional reconstruction. On the other hand, images of the dispersed ring-shaped colicin Ia oligomers on vesicles were collected under low dose conditions, as regions potentially containing incorporated colicin Ia could be distinguished from those devoid of protein by the higher contrast due to sequestration of uranyl acetate stain.
It is possible that two-dimensional crystallization was challenging because upon membrane insertion the colicin Ia molecules are not all in the same conformation. Treatment of colicin Ia proteoliposomes reconstituted at pH 4.5 with pH 7.5 buffer released ∼50% of the protein, suggesting that different conformers with possibly different degrees of membrane-integration were generated. Earlier studies have suggested a number of models for the intermediates of colicin Ia that exist before channel opening upon application of voltage (14, 16). The fact that the soluble structure of colicin Ia is predicted to undergo radical conformational changes to form the resultant ion channel (40) suggests that these changes may happen through a series of intermediate steps.
Three-dimensional Reconstruction from Negatively Stained Specimens and Structural Insight into the Membrane-integrated State of Colicin Ia
The visualized stain-embedded oligomeric structure is expected to correspond to the “closed state” of colicin Ia channel. Overall, the three-dimensional model derived from the analysis of two-dimensional crystals (Fig. 6) correlates well with the model obtained from the independent single-particle analysis using the random-conical tilt method (Fig. 7), demonstrating the fidelity of the visualized oligomeric structure and the gleaned structural interpretation. There are less pronounced features in the single-particle reconstruction and, therefore, also a somewhat higher inner diameter of the oligomer. These differences could be primarily attributed to lower resolution (∼29 Å against ∼17 Å) and possibly to effects of close packing in the periodic array.
The density volume (Fig. 6), described approximately as a hollow cylinder of ∼43 Å average height and ∼77 Å and ∼32 Å average outer and inner diameters, corresponds to a molecular mass of ∼130 kDa. Because the hydrophilic stain is expected to poorly contrast the hydrophobic lipid bilayer, only the stain-excluded protein volume, largely representing the extrabilayer portion of the molecule, is seen with very little density displayed for the bilayer region (Fig. 6, B and C). This crown-shaped density is relatively flatter on one side (bottom of Fig. 6, B and C, and the top of D), which we attribute to the face of the oligomer proximal to the lipid bilayer.
Based on the projection data from two-dimensional crystals (Table 1), a higher plane group of symmetry p321 or p312 i.e. with an in-plane 2-fold axis, could conceivably have been chosen as opposed to p3 symmetry. We found that the average phase residual per image using, for instance p312 during merging of three-dimensional data, was 11.9°, close to the value of 7.9° when p3 was used. This observation could point to the presence at least locally of an in-plane 2-fold symmetry characterizing the three-dimensional density leading to a hexamer. However, this conclusion must await higher resolution investigation. An exact in-plane 2-fold symmetry can be ruled out as no significant density is seen “below” the expected featureless extent of the bilayer (Fig. 6, B or C). The asymmetric disposition of colicin Ia could be because of relatively differing concentration of DMPG in the two leaflets driving preferential association of colicin Ia molecules to the more negatively charged leaflet. Under the conditions used to achieve membrane insertion, the absence of any significant density on the other face of the bilayer could be because of two reasons; (a) no part or a relatively small portion of the polypeptide chain has translocated across the bilayer, as is indicated from movement of tagged full-length colicin Ia (20, 34) and/or that (b) the translocated peptide is heterogeneous in extent or in conformation or both, leading to markedly reduced signal in the averaged map. An exercise in density modification by excising the isolated noise peaks at the 3-fold symmetry locations in between the oligomers mentioned above and iterative phase-combination4 produced an improved map. This map displayed density in the putative bilayer region (supplemental Fig. S6), similar to that seen also in the independently generated map from single particle analysis (Fig. 7).
Large scale conformational changes in the colicin Ia CFD are known to accompany the opening and formation of the voltage-gated ion channel (40). In the closed state, helices VIII and IX insert in the membrane in the absence of an applied voltage (8). In the open state, two additional segments (parts of helices VI and VII; residues Tyr-541—Gly-577 and part of helix I; residues Ile-454—Leu-474) are inserted in the membrane. The entire region Leu-474—Tyr-541 (inclusive) is on the trans side of the membrane (helices II–V of the crystal structure). This equates to two additional transmembrane segments of 20–30 amino acids each and at least 68 residues (474–541) translocated across the membrane (40). Kienker et al. (20) have shown that the long helix I (∼160 Å) and all of the R domain are translocated in the case of experiments with recombinant colicin Ia fragments but not the whole colicin Ia. The space created by the large ∼32 Å-wide and ∼48 Å-long vestibular entrance (Fig. 6) in principle could facilitate accommodation of the peptide fragments en route to translocation across the lipid bilayer and eventual formation of the open channel. Although as mentioned above, the full extent of the colicin Ia molecule has not been visualized in the current study, it appears very likely that the dimension perpendicular to the bilayer will be substantially smaller than the ∼210 Å length of the harpoon-like molecule revealed in the x-ray structure. Fig. 8 shows a schematic model linking the x-ray structure with the observed three-dimensional model for the oligomeric membrane-inserted state. This figure reiterates the necessity of substantial conformational change accompanying the membrane integration, which in the in vitro experiment, is likely facilitated by acid induced unfolding at the low pH used.
FIGURE 8.

Conformational change of colicin Ia upon membrane integration. The three-dimensional model for the colicin Ia oligomer integrated into the lipid bilayer is the reconstruction generated from negatively stained two-dimensional crystal after refinement (supplemental Fig. S6). The extent of density in the bilayer is compromised by the inability of the hydrophilic stain to penetrate the membrane. The figure for the outer membrane Cir receptor complexed with recombinant R domain of colicin Ia is based on Buchanan et al. (47), and the x-ray model for colicin Ia is from Wiener et al. (6). All structural components are drawn approximately to scale.
Although our results strongly indicate that colicin Ia channel exists as a multimer, they do not implicate that an oligomer is required to form the functional channel. Electrophysiological studies on both colicin E1 and colicin Ia have supported the hypothesis that the protein inserts into lipid bilayers as a monomer and that the functional channel is a monomer (18, 41, 42). However, consistent with this, modeling the colicin Ia channel as composed of four transmembrane helices leads to an asymmetric pore of only a 5 Å diameter at the narrowest constriction (43), which is insufficient to allow the passage of tetraethyl ammonium ion (19). On the other hand, a pentameric bundle of helices involving e.g. more than one monomer can enclose a pore with sufficient diameter of ∼7.5 Å (43). Many oligomeric channel/transporter membrane proteins harbor multiple functional channels, e.g. OmpF (44), and aquaporins (45), ClC channel/transporter family (46). This may be the case for colicin Ia channel also, pending high resolution detail of the channel architecture that could be revealed using our two-dimensional crystals.
Supplementary Material
Acknowledgments
We thank P. Kienker for testing the channel properties of the recombinant peptides in lipid bilayers and K. Jakes for initially providing the purified protein samples, expression vectors, and purification protocols and helpful advice throughout the course of this work. We thank C. Wright for help with protein expression and purification.Wethank P. Kienker and K. Jakes for critical reading and comments on the manuscript and D. Christie and J. Kistler for useful comments. We thank V. Ward for help with Fig. 8, A. Turner for maintaining the microscopes, Y. Halytskyy for support with the BeSTGRID and CERES computer cluster at University of Auckland, and J.-K. Hyun for help with the preparation of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grant R21 DK060827 (NIDDK). This work was also supported by a Marsden Fund (New Zealand) grant (to A. K. M.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental text, Figs. S1–S6, and Table S1.
A. K. Mitra, manuscript in preparation.
- CFD
- channel-forming domain
- DMPG
- 1,2-dimyristoyl-sn-glycero-3-[phospho-rac-(1-glycerol)]
- DMPC
- 1,2-dimyristoyl-sn-glycero-3-phosphocholine
- POPG
- 1-palmitoyl-2-oleoyl-sn-glycero-3-[phospho-rac-(1-glycerol)]
- POPC
- 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- LUV
- large unilamellar vesicles
- BS3
- bis[sulfosuccinimidyl]suberate
- DDM
- n-dodecyl-β-d-maltopyranoside
- SEC
- size-exclusion chromatography
- RS
- Stokes radius.
REFERENCES
- 1.Cramer W. A., Heymann J. B., Schendel S. L., Deriy B. N., Cohen F. S., Elkins P. A., Stauffacher C. V. ( 1995) Annu. Rev. Biophys. Biomol. Struct. 24, 611– 641 [DOI] [PubMed] [Google Scholar]
- 2.Slatin S., Kienker P. K. ( 2003) in Pore Forming Peptides and Protein Toxins ( Menestrina G., Dalla Serra M., Lazarovici P. eds) pp. 102– 131, Taylor and Francis, London [Google Scholar]
- 3.Parker M. W., Postma J. P., Pattus F., Tucker A. D., Tsernoglou D. ( 1992) J. Mol. Biol. 224, 639– 657 [DOI] [PubMed] [Google Scholar]
- 4.Elkins P., Bunker A., Cramer W. A., Stauffacher C. V. ( 1997) Structure 5, 443– 458 [DOI] [PubMed] [Google Scholar]
- 5.Vetter I. R., Parker M. W., Tucker A. D., Lakey J. H., Pattus F., Tsernoglou D. ( 1998) Structure 6, 863– 874 [DOI] [PubMed] [Google Scholar]
- 6.Wiener M., Freymann D., Ghosh P., Stroud R. M. ( 1997) Nature 385, 461– 464 [DOI] [PubMed] [Google Scholar]
- 7.Parker M. W., Pattus F. ( 1993) Trends Biochem. Sci. 18, 391– 395 [DOI] [PubMed] [Google Scholar]
- 8.Kienker P. K., Qiu X., Slatin S. L., Finkelstein A., Jakes K. S. ( 1997) J. Membr. Biol. 157, 27– 37 [DOI] [PubMed] [Google Scholar]
- 9.Duché D., Parker M. W., González-Mañas J. M., Pattus F., Baty D. ( 1994) J. Biol. Chem. 269, 6332– 6339 [PubMed] [Google Scholar]
- 10.Lindeberg M., Zakharov S. D., Cramer W. A. ( 2000) J. Mol. Biol. 295, 679– 692 [DOI] [PubMed] [Google Scholar]
- 11.Yao X. L., Hong M. ( 2006) Biochemistry 45, 289– 295 [DOI] [PubMed] [Google Scholar]
- 12.Wei Z., White D., Wang J., Musse A. A., Merrill A. R. ( 2007) Biochemistry 46, 6074– 6085 [DOI] [PubMed] [Google Scholar]
- 13.Padmavathi P. V., Steinhoff H. J. ( 2008) J. Mol. Biol. 378, 204– 214 [DOI] [PubMed] [Google Scholar]
- 14.Slatin S. L., Qiu X. Q., Jakes K. S., Finkelstein A. ( 1994) Nature 371, 158– 161 [DOI] [PubMed] [Google Scholar]
- 15.Qiu X. Q., Jakes K. S., Kienker P. K., Finkelstein A., Slatin S. L. ( 1996) J. Gen. Physiol. 107, 313– 328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Luo W., Yao X., Hong M. ( 2005) J. Am. Chem. Soc. 127, 6402– 6408 [DOI] [PubMed] [Google Scholar]
- 17.Krasilnikov O. V., Da Cruz J. B., Yuldasheva L. N., Varanda W. A., Nogueira R. A. ( 1998) J. Membr. Biol. 161, 83– 92 [DOI] [PubMed] [Google Scholar]
- 18.Kienker P. K., Jakes K. S., Finkelstein A. ( 2008) J. Gen. Physiol. 132, 693– 707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cascales E., Buchanan S. K., Duché D., Kleanthous C., Lloubès R., Postle K., Riley M., Slatin S., Cavard D. ( 2007) Microbiol. Mol. Biol. Rev. 71, 158– 229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kienker P. K., Jakes K. S., Finkelstein A. ( 2000) J. Gen. Physiol. 116, 587– 598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miercke L. J., Betlach M. C., Mitra A. K., Shand R. F., Fong S. K., Stroud R. M. ( 1991) Biochemistry 30, 3088– 3098 [DOI] [PubMed] [Google Scholar]
- 22.Post P. L., Tyska M. J., O'Connell C. B., Johung K., Hayward A., Mooseker M. S. ( 2002) J. Biol. Chem. 277, 11679– 11683 [DOI] [PubMed] [Google Scholar]
- 23.Henderson R., Baldwin J. M., Ceska T. A., Zemlin F., Beckmann E., Downing K. H. ( 1990) J. Mol. Biol. 213, 899– 929 [DOI] [PubMed] [Google Scholar]
- 24.Crowther R. A., Henderson R., Smith J. M. ( 1996) J. Struct. Biol. 116, 9– 16 [DOI] [PubMed] [Google Scholar]
- 25.Hyun J. K., Coulibaly F., Turner A. P., Baker E. N., Mercer A. A., Mitra A. K. ( 2007) J. Virol. 81, 11075– 11083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sorzano C. O., Marabini R., Velázquez-Muriel J., Bilbao-Castro J. R., Scheres S. H., Carazo J. M., Pascual-Montano A. ( 2004) J. Struct. Biol. 148, 194– 204 [DOI] [PubMed] [Google Scholar]
- 27.Radermacher M., Wagenknecht T., Verschoor A., Frank J. ( 1987) J. Microsc. 146, 113– 136 [DOI] [PubMed] [Google Scholar]
- 28.Scheres S. H., Gao H., Valle M., Herman G. T., Eggermont P. P., Frank J., Carazo J. M. ( 2007) Nature Methods 4, 27– 29 [DOI] [PubMed] [Google Scholar]
- 29.Scheres S. H., Valle M., Carazo J. M. ( 2005) Bioinformatics 21, Suppl. 2, 243– 244 [DOI] [PubMed] [Google Scholar]
- 30.Scheres S. H., Valle M., Nuñez R., Sorzano C. O., Marabini R., Herman G. T., Carazo J. M. ( 2005) J. Mol. Biol. 348, 139– 149 [DOI] [PubMed] [Google Scholar]
- 31.Kühlbrandt W. ( 1992) Q. Rev. Biophys. 25, 1– 49 [DOI] [PubMed] [Google Scholar]
- 32.Yeager M., Unger V. M., Mitra A. K. ( 1999) Methods Enzymol. 294, 135– 180 [DOI] [PubMed] [Google Scholar]
- 33.Mel S. F., Stroud R. M. ( 1993) Biochemistry 32, 2082– 2089 [DOI] [PubMed] [Google Scholar]
- 34.Kienker P. K., Jakes K. S., Blaustein R. O., Miller C., Finkelstein A. ( 2003) J. Gen. Physiol. 122, 161– 176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jarvis L. J., Louis C. F. ( 1995) Curr. Eye Res. 14, 799– 808 [DOI] [PubMed] [Google Scholar]
- 36.Vinothkumar K. R., Raunser S., Jung H., Kühlbrandt W. ( 2006) J. Biol. Chem. 281, 4795– 4801 [DOI] [PubMed] [Google Scholar]
- 37.Minn A. J., Vélez P., Schendel S. L., Liang H., Muchmore S. W., Fesik S. W., Fill M., Thompson C. B. ( 1997) Nature 385, 353– 357 [DOI] [PubMed] [Google Scholar]
- 38.Antonsson B., Montessuit S., Lauper S., Eskes R., Martinou J. C. ( 2000) Biochem. J. 345, 271– 278 [PMC free article] [PubMed] [Google Scholar]
- 39.Fivaz M., Abrami L., Tsitrin Y., van der Goot F. G. ( 2001) Curr. Top. Microbiol. Immunol. 257, 35– 52 [DOI] [PubMed] [Google Scholar]
- 40.Jakes K. S., Kienker P. K., Finkelstein A. ( 1999) Q. Rev. Biophys. 32, 189– 205 [DOI] [PubMed] [Google Scholar]
- 41.Levinthal F., Todd A. P., Hubbell W. L., Levinthal C. ( 1991) Proteins 11, 254– 262 [DOI] [PubMed] [Google Scholar]
- 42.Slatin S. L. ( 1988) Int. J. Biochem. 20, 737– 744 [DOI] [PubMed] [Google Scholar]
- 43.Stroud R. M., Reiling K., Wiener M., Freymann D. ( 1998) Curr. Opin. Struct. Biol. 8, 525– 533 [DOI] [PubMed] [Google Scholar]
- 44.Koebnik R., Locher K. P., Van Gelder P. ( 2000) Mol. Microbiol. 37, 239– 253 [DOI] [PubMed] [Google Scholar]
- 45.Verkman A. S., Mitra A. K. ( 2000) Am. J. Physiol. 278, F13– 28 [DOI] [PubMed] [Google Scholar]
- 46.Dutzler R. ( 2007) FEBS Lett. 581, 2839– 2844 [DOI] [PubMed] [Google Scholar]
- 47.Buchanan S. K., Lukacik P., Grizot S., Ghirlando R., Ali M. M., Barnard T. J., Jakes K. S., Kienker P. K., Esser L. ( 2007) EMBO J. 26, 2594– 2604 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.