

Summary
The trace amine-associated receptor 1 (TAAR1) is a biogenic amine G-protein coupled receptor (GPCR) that is potently activated by 3-iodothyronamine (1, T1AM) in vitro. Compound 1 is an endogenous derivative of the thyroid hormone thyroxine that rapidly induces hypothermia, anergia, and bradycardia when administered to mice. To explore the role of TAAR1 in mediating the effects of 1, we rationally designed and synthesized rat TAAR1 superagonists and lead antagonists using the rotamer toggle switch model of aminergic GPCR activation. The functional activity of a ligand was found to be correlated to the nature of its interactions with the rotamer switch residues. Allowing the rotamer switch residues to toggle to their active conformation was associated with agonism while interfering with this conformational transition resulted in antagonism. These agonist and antagonist design principles provide a conceptual model for understanding the relationship between the molecular structure of a drug and its pharmacological properties.
INTRODUCTION
3-Iodothyronamine (1, T1AM; Fig. 1a) is an endogenous, decarboxylated, and deiodinated metabolite of the thyroid hormone thyroxine (T4; Fig. 1a) that is found in the brain, heart, liver and blood (Scanlan et al., 2004). When administered to mice intraperitoneally, 1 rapidly induces hypothermia, anergia, and bradycardia; effects of which are opposite those observed with hyperthyroidism. In vitro, 1 induces the production of cAMP (adenosine 3′,5′-cyclic monophosphate) in HEK293 (human embryonic kidney 293) cells stably transfected with the GPCR known as TAAR1 (Hart et al., 2006; Scanlan et al., 2004; Wainscott et al., 2007; Zucchi et al., 2006). Additionally, 1 has been found to inhibit neurotransmitter reuptake by the dopamine (DAT) and norepinephrine transporter (NET), and inhibits vesicular packaging by the vesicular monoamine transporter 2 (VMAT2) (Snead et al., 2007). To understand the role of TAAR1 in mediating the effects of 1, we sought to develop small molecules that regulate the activity of TAAR1.
Figure 1.
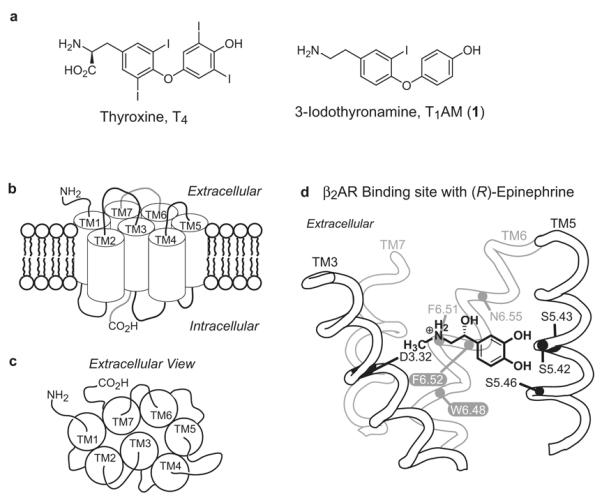
Hormones, metabolites, and biogenic amine GPCR. (a) Structures of thyroxine (T4) and 3-iodothyronamine (1, T1AM). (b) Schematic representations of the helical arrangement of GPCRs viewed from the cell membrane and (c) extracellular surface. (d) Binding orientation of (R)-epinephrine in the binding site of the β2AR viewed from the perspective of TM4. The location of the rotamer switch residues (white letters) (see Fig. 2) and residues known to interact with (R)-epinephrine are labeled. The residue indexing system is described in the Experimental Procedures section.
Rat TAAR1 (rTAAR1) is homologous to the β2 adrenergic (β2AR), dopamine, and serotonin receptors and belongs to the biogenic amine subfamily of class A rhodopsin like GPCRs (Borowsky et al., 2001; Bunzow et al., 2001; Lindemann et al., 2005). GPCRs are seven transmembrane (TM) proteins with an extracellular amino terminus and an intracellular carboxy terminus (Fig. 1b-c) (Gether, 2000; Wess, 1998). The binding site of aminergic GPCRs is located within the TM region and is primarily composed of the extracellular half of transmembranes 3, 5, 6, and 7 (Cherezov et al., 2007; Rasmussen et al., 2007; Rosenbaum et al., 2007; Tota et al., 1991). Elegant pharmacological and mutagenesis studies on β2AR suggest that epinephrine binds to β2AR with aspartic acid 3.32 (D3.32) acting as the counterion for the charged amine, serine residues 5.42, 5.43, and 5.46 (S5.42, S5.43, and S5.46, respectively) interacting with the catechol hydroxyls, phenylalanines 6.51 and 6.52 (F6.51 and F6.52) interacting with the catechol ring, and asparagine 6.55 (N6.55) as the partner for the β-hydroxy group (Fig. 1d) (see the Experimental Procedures section for a description of the residue indexing system) (Liapakis et al., 2000; Shi et al., 2002; Strader et al., 1989; Strader et al., 1994; Strader et al., 1988; Strader et al., 1989; Wieland et al., 1996; Zuurmond et al., 1999).
Previous work with the β2AR suggests that agonist binding toggles a rotamer switch to its active configuration and induces a conformational change in TM6 (Fig. 2) (Shi et al., 2002). The movement of the cytoplasmic end of TM6 away from TM3 is thought to break an ionic lock interaction that is present in the inactive state of the receptor (Fig. 2a). This exposes G-protein recognition sites in the intracellular surface of the receptor that activate G-proteins and initiate the signaling cascade (Ballesteros et al., 2001; Yao et al., 2006). The rotamer switch is partly composed of a tryptophan (W6.48) and phenylalanine (F6.52) residues in TM6 that toggle concertedly between their inactive (Fig. 2a) and active (Fig. 2b) rotamer configurations to modulate the bend angle of the kink in TM6 formed by proline 6.50 (P6.50). The ionic lock involves highly conserved aspartic acid (D3.49) and arginine (R3.50) residues in TM3 and a glutamic acid (E6.30) residue in TM6. The absolute conservation of the rotamer switch and ionic lock residues in rTAAR1 suggests a mechanism of activation for rTAAR1 similar to β2AR.
Figure 2.

Rotamer toggle switch model of aminergic GPCR activation. (a) Inactive state of the receptor with an antagonist sterically occluding the rotamer switch residues (W6.48 and F6.52) from assuming their active conformation. (b) Agonist binding toggles the rotamer switch to its active conformation and induces a conformational change in TM6 that breaks the ionic lock interaction (D3.49, R3.50, & E6.30) present in the inactive state of the receptor. (a) and (b) are viewed from the perspective of TM7, see Fig. 1b-c.
Studies probing the mechanism of agonist induced conformational changes in the β2AR have found that agonist binding occurs in a sequential process involving a series of conformational intermediates that have increasing numbers of interactions with the agonist as the receptor moves toward the fully active state (Kobilka et al., 2007). The binding site of β2AR is not prearranged to simultaneously interact with all of the functional groups of a given agonist like epinephrine (Fig. 1d). Upon binding, only a few structural elements of epinephrine (i.e. the amine and catechol moiety) are proposed to be engaged with the β2AR. These initial interactions induce a conformation transition to an intermediate that reveals additional contact points that interact with the β-hydroxyl and/or N-methyl groups. The functional groups of epinephrine have a synergistic effect on binding affinity and receptor activation and collectively influence the overall conformation of the active receptor (Liapakis et al., 2004). The ensemble of active receptor states induced by different agonists may have disparate functional properties and have different capacity to activate downstream effector molecules such as Gs protein, GPCR receptor kinase, and/or arrestin (Swaminath et al., 2004).
Despite being a major drug target and having insights into the molecular mechanism of GPCR activation and agonist induced conformational changes, the nature of the ligand-receptor interaction is not fully understood. Although there have been many successful campaigns into GPCR drug design, it is surprising to find that there are no general postulates that can serve as guiding principles in the process of agonist and/or antagonist development without requiring extensive structure activity relationship (SAR) data to develop a pharmacophore for the receptor of interest. Even with pharmacophore models in hand, the code to aminergic GPCR drug design is still unknown. Presently, it is unclear what inherent structural features of a ligand are responsible for endowing agonistic or antagonistic properties or how and why those structural elements lead to receptor activation or inhibition.
Based on the rotamer toggle switch model, we hypothesized that the functional properties of a compound are determined by the nature of its interaction with the rotamer switch residues. If a compound allows the rotamer switch to toggle and/or has more favorable interactions with the active state of the receptor, it will act as an agonist (Fig. 2b). In contrast, a compound will behave as an antagonist if it can sterically occlude the rotamer switch and/or has more favorable interactions with the inactive state of the receptor (Fig. 2a). Herein we describe the rational design and synthesis of rTAAR1 superagonists (agonists that are more potent and/or more efficacious than 1) and lead antagonists guided by the rotamer toggle switch model of aminergic GPCR activation.
RESULTS
Development of rTAAR1 superagonists
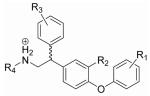
The ligand binding site of rTAAR1 differs from that of the β2AR in that two hydrophobic residues, alanine (A5.42) and phenylalanine (F5.43) (Fig. 3b), replace the serine residues S5.42 and S5.43 in TM5 (Fig. 1d). By analogy to the catecholamines (epinephrine, norepinephrine, and dopamine), we speculate that 2 (Fig. 3a) (Tan et al., 2007), a potent rTAAR1 agonist, is anchored into the binding site by the salt bridge interaction between the charged amine and D3.32, and the hydrogen bond interaction between the biaryl ether oxygen and S5.46 (Fig. 3b). To experimentally test this hypothesis, a series of derivatives of 2 containing functional groups at the β-phenyl ring (ring C in Fig. 3a) were synthesized. We specifically incorporated polar functional groups (3-7) (Table 1) capable of forming hydrogen bond interactions because our homology model of rTAAR1 (see Experimental Procedures section for a description of how the model was generated and how we analyzed the ligand-rTAAR1 interaction), which was based on the crystal structure of bovine rhodopsin, showed that the surrounding residues around the β-phenyl ring are asparagines (N7.35 & N7.39), a methionine (M6.55) and a cysteine (C6.54) (Fig. 3b). Therefore, if 2 binds in this orientation, having functional groups that can interact with these residues should theoretically enhance binding affinity and thus increase potency. Additionally, fluorine substituted analogs of 2 (8, 9) (Table 1) were also synthesized to determine the effects of decreasing the electron density of the β-phenyl ring on rTAAR1 activation. Compounds 3-9 were synthesized from 4-bromodiphenylether and a mono-substituted benzaldehyde in 4 to 7 steps (Supplementary Schemes 1 and 2). Detailed synthetic procedures for compounds 2-56 are described in the Supplementary Data.
Figure 3.

SAR of rTAAR1 ligands and their proposed binding mode in rTAAR1. (a) Structure of 2. The A, B, and C rings correspond to its outer, inner, and β-phenyl rings, respectively. (b) Proposed binding orientation of 2 in the binding site of rTAAR1, viewed from the perspective of TM4. The rotamer switch residues (white letters), proposed binding and specificity determinant residues are labeled. (c) Agonist dose response curves of 2 (○), 27 (□), 29 (△), 34 (■), 50 (●), and 51 (▲). (d) Proposed binding orientation of 34 in the binding site of rTAAR1, viewed from the perspective of TM4.
Table 1.
Agonist activity of compounds 1-13 on rTAAR1.
 | |||||||
|---|---|---|---|---|---|---|---|
| Compd | R1 | R2 | R3 | R4 | EC50a ± SEM (nM) |
Emaxb ± SEM (%) |
Nc |
| 1 (T1AM) | See Fig. 1 | 33 ± 3 | 100 ± 0 | 5 | |||
| 2 (ET-13) | H | H | H | H | 28 ± 2 | 103 ± 4 | 3 |
| 3 (ET-34) | H | H | p-OMe | H | 142 ± 40 | 68 ± 8 | 3 |
| 4 (ET-35) | H | H | o-OMe | H | 163 ± 18 | 84 ± 2 | 3 |
| 5 (ET-36) | H | H | p-OH | H | 6 ± 1 | 114 ± 9 | 4 |
| 6 (ET-37) | H | H | o-OH | H | 467 ± 107 | 70 ± 6 | 3 |
| 7 (ET-65) | H | H | m-OH | H | 212 ± 39 | 106 ± 7 | 3 |
| 8 (ET-66) | H | H | p-F | H | 28 ± 6 | 99 ± 9 | 3 |
| 9 (ET-67) | H | H | m-F | H | 57 ± 6 | 110 ± 7 | 3 |
| 10 (ET-64) | H | H | p-OH | Me | 5 ± 1 | 127 ± 2 | 4 |
| 11 (ET-68) | H | I | p-OH | H | 17 ± 2 | 107 ± 8 | 4 |
| 12 (ET-69) | p-OH | I | p-OH | H | 4 ± 1 | 115 ± 2 | 6 |
| 13 (ET-70) | m-OH | I | p-OH | H | 22 ± 2 | 111 ± 9 | 4 |
EC50 is the half-maximal effective concentration of a compound.
Emax is the maximum stimulation achieved at a concentration of 10 μM and was calculated by use of Prism software. EC50 and Emax values represent the average of N independent experiments in triplicate and were calculated by use of Prism software as described in the Experimental Procedures section. Emax = 100 % is defined as the activity of 1 at 10 μM.
N is the number of independent experiments in triplicate that were performed and used to calculate the EC50 and Emax values.
In HEK293 stable cell lines, 2 (EC50 = 28 ± 2 nM, Emax = 103 ± 4 %) activates the stimulatory G protein coupled rTAAR1 at the same level as 1 (EC50 = 33 ± 3 nM, Emax = 100 ± 0 %) (Table 1) (Tan et al., 2007). Representative dose-response curves of agonists for rTAAR1 are shown in Supplementary Figure 1a. Appending a methoxy group at the para (3) or ortho (4) position of the β-phenyl ring in 2 was detrimental, decreasing the potency ∼5-6-fold and the efficacy 19-35 % (3, EC50 = 142 ± 40 nM, Emax = 68 ± 8 %, and 4, EC50 = 163 ± 18 nM, Emax = 84 ± 2 %) (Table 1). A hydroxyl group at the β-phenyl ring was well tolerated by rTAAR1 but only at the para position. The potency of the para hydroxy derivative 5 increased ∼4.5-fold (EC50 = 6 ± 1 nM) and its efficacy was slightly enhanced (Emax = 114 ± 9 %). When the hydroxyl substituent was located at the ortho (6) or meta (7) position, the potency decreased ∼7.5-16.5-fold (EC50 = 467 ± 107 nM and 212 ± 39 nM, respectively) while the efficacy either decreased or was unaffected (Emax = 70 ± 6 % and 106 ± 7 %, respectively). Similarly, rTAAR1 somewhat prefers a fluorine group at the para over the meta position as the potency was the same for 8 (EC50 = 28 ± 6 nM) but decreased 2-fold for 9 (EC50 = 57 ± 6 nM). The efficacy of 8 and 9 (Emax = 99 ± 9 % and 110 ± 2 %, respectively) were unaffected by fluorination and were similar to that of 2. All compounds with stereogenic centers (2-50 and 52-56) were evaluated as racemic mixtures. The observed activities of all compounds tested (1-56) were found to be rTAAR1-dependent, as all compounds showed no cAMP accumulation when exposed to an empty vector control cell line (data not shown).
In an effort to improve the potency of 5, we explored its tolerance for methylation at the amine, iodination of the inner ring, and hydroxylation of the outer ring. These modifications, individually or in combination, have previously been found to be beneficial for rTAAR1 activation (Hart et al., 2006). Mono-methylation of the amine in 5 provided 10 while mono-iodination of the inner ring yielded 11 (Supplementary Schemes 3 and 4). Adding a hydroxyl group to the para or meta position of the outer ring in 11 gave 12 and 13, respectively (Supplementary Scheme 4).
When screened for agonist activity, some of the 5 derivatives were more efficacious but none were more potent. N-Methylation of 5 (10) was beneficial increasing the efficacy 13 % (Emax = 127 ± 2 %) but it did not improve potency (EC50 = 5 ± 1 nM) (Table 1). Mono-iodination of the inner ring (11) was unfavorable decreasing potency ∼3-fold (EC50 = 17 ± 2 nM) without significantly affecting efficacy (Emax = 107 ± 8 %). In the presence of an outer ring para hydroxyl group (12), the rTAAR1 activity improved back to the level of 5 (EC50 = 4 ± 1 nM, Emax = 115 ± 2 %). In contrast, a meta hydroxyl group on the outer ring of 11 (13) had no effect on potency and efficacy (EC50 = 22 ± 2 nM and Emax = 111 ± 9 %).
Development of rTAAR1 lead antagonist
According to our proposed binding orientation of 2 in rTAAR1 (Fig. 3b), the rotamer switch residues are located in the vicinity of position 2 of the inner ring (ring B in Fig. 3a). Using the toggle switch model of aminergic GPCR activation as a guideline (Fig. 2), we attempted to convert 2 into an antagonist by appending functional groups at the 2 position to theoretically interfere with the rotamer switch residues. An alcohol group was installed into the 2 position (R5, Table 2) of 2 (14) to serve as a handle for synthesizing a panel of ethers (15-24) and esters (25 and 26) varying in steric bulk, rigidity, topology and polarity (Table 2, Supplementary Schemes 5 and 6).
Table 2.
Agonist activity of compounds 14-26 on rTAAR1.
| Compd | R5 | EC50a-c ± SEM (nM) | Emaxa-c ± SEM (%) | Na-c |
|---|---|---|---|---|
| 14 (ET-51) | OH | 96 ± 10 | 108 ± 1 | 3 |
| 15 (ET-52) | OMe | 35 ± 4 | 82 ± 8 | 3 |
| 16 (ET-53) | OEt | 144 ± 31 | 95 ± 5 | 3 |
| 17 (ET-54) | OPr | >1000 | 69 ± 5 | 2 |
| 18 (ET-55) | OBu | >1000 | 31 ± 1 | 2 |
| 19 (ET-56) | OBn | >1000 | 58 ± 2 | 2 |
| 20 (ET-57) | O-iPr | >1000 | 62 ± 2 | 2 |
| 21 (ET-58) | O-iBu | >1000 | 15 ± 4 | 2 |
| 22 (ET-59) | OCH2CHCH2 | 169 ± 6 | 71 ± 4 | 2 |
| 23 (ET-60) | OCH2CCH | 138 ± 37 | 78 ± 1 | 2 |
| 24 (ET-61) | OCH2CO2CH3 | >1000 | 56 ± 0 | 2 |
| 25 (ET-62) | O2CCH2CH2Cl | 143 ± 4 | 57 ± 5 | 2 |
| 26 (ET-63) | O2CCH3 | 234 ± 43 | 74 ± 3 | 2 |
See footnotes for Table 1.
The effects of the ether and ester substituents on receptor agonist activity were variable. The core scaffold 14 and ethyl ether 16 were decent agonists activating to the same efficacy level as 2 (Emax = 108 ± 1 % and 95 ± 5 %, respectively) but at ∼3-5-fold lower potency (EC50 = 96 ± 10 nM and 144 ± 31 nM, respectively) (Table 2). On the other hand, the methyl ether 15 showed the opposite trend being equipotent to 2 (EC50 = 35 ± 4 nM) but less efficacious (Emax = 82 ± 8 %). The unsaturated alkene and alkyne counterparts of the propyl ether 17 appear to be well tolerated by rTAAR1 as 22 (EC50 169 ± 6 nM) and 23 (EC50 = 138 ± 37 nM) were at least 6-fold more potent than 17 (EC50 >1 μM). The efficacies of 17, 22, and 23 were comparable to each other (Emax = 69 ± 5 %, 71 ± 4 %, and 78 ± 1 %, respectively). Further increasing the size of the ether substituents (18-21 and 24) desirably decreased potency (EC50 > 1 μM) but it did not completely abolish the agonist activity (Emax ≤ 10 %) of the compounds. These compounds activated rTAAR1 between 15 % and 62 % efficacy. Similarly the ester substituents (25 and 26) decreased the potency of 2 (EC50 = 143 ± 4 nM and 234 ± 43 nM, respectively) but did not reduce its efficacy below 10 % (Emax = 57 ± 5 % and 74 ± 3 %, respectively) (Table 2).
The observed agonist activities of 14-26 were consistent with the idea that the inner ring functional groups of these compounds were not properly interfering with the rotamer switch residues. In compound 14, rotation of the inner ring about the β carbon and the biaryl ether oxygen axis renders position 2 and 6 indistinguishable (Table 2). Within the binding site, it’s possible that the inner rings of 15-26 have rotated 180° and are actually orienting the position 2 functional group towards the extracellular surface of rTAAR1 around methionine 6.55 (M6.55) instead of the intracellular region near the rotamer switch residues. In this alternate binding orientation, these compounds would be predicted to have some agonist activity as the ether or ester appendage would not be able to interfere with the rotamer switch residues.
To test this hypothesis, the core scaffold of 14 was modified to have the phenoxy group moved one carbon over to the meta position with respect to the ethylamine chain (28) (Table 3, Supplementary Scheme 7). In this orientation, the 2 and 6 positions of the inner ring are now structurally distinct. Having a meta phenoxy group should not be detrimental to binding affinity because the isomer of 2 with the phenoxy group at the meta position (27) was found to be a slightly better agonist than 2 for rTAAR1 (EC50 = 19 ± 2 nM, Emax = 131 ± 7 %) (Fig. 3c) (Tan et al., 2007). With this modification we synthesized 21 compounds (29-49) with an ether or ester appendage at the 2 position that again varied in steric bulk, rigidity, topology, and polarity (Table 3, Supplementary Schemes 7-9).
Table 3.
Agonist and antagonist activity of compounds 27-49 on rTAAR1.
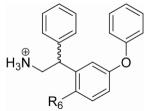 | |||||||
|---|---|---|---|---|---|---|---|
| Agonist Activity | Antagonist Activity | ||||||
| Compd | R6 | EC50a-c ± SEM (nM) |
Emaxa-c ± SEM (%) |
Na-c | IC50d ± SEM (μM) |
Imaxe ± SEM (%) |
Na-c |
| 27 (ET-14) | H | 19 ± 2 | 131 ± 7 | 3 | - | - | - |
| 28 (ET-72) | OH | 232 ± 8 | 88 ± 9 | 2 | - | - | - |
| 29 (ET-73) | OMe | 102 ± 26 | 88 ± 1 | 3 | - | - | - |
| 30 (ET-74) | OEt | >1000 | 66 ± 3 | 2 | - | - | - |
| 31 (ET-75) | OPr | >1000 | 41 ± 0 | 2 | - | - | - |
| 32 (ET-76) | OBu | >1000 | 3 ± 0 | 2 | 8 ± 2 | 12 ± 3 | 2 |
| 33 (ET-77) | OPent | >1000 | 0 ± 3 | 2 | 5 ± 0 | 6 ± 1 | 2 |
| 34 (ET-78) | OHex | >1000 | 0 ± 1 | 2 | 4 ± 0 | 3 ± 1 | 4 |
| 35 (ET-79) | OBn | >1000 | 9 ± 1 | 2 | 5 ± 1 | 6 ± 3 | 2 |
| 36 (ET-80) | O-iPr | >1000 | 40 ± 1 | 2 | - | - | - |
| 37 (ET-81) | O-iBu | >1000 | 6 ± 1 | 2 | >10 | 23 ± 6 | 2 |
| 38 (ET-82) | OCH2CHCH2 | 602 ± 10 | 79 ± 5 | 2 | - | - | - |
| 39 (ET-83) | OCH2CCH | 182 ± 46 | 103 ± 0 | 2 | - | - | - |
| 40 (ET-95) | OCH2-Cyclopropyl | >1000 | 26 ± 7 | 3 | - | - | - |
| 41 (ET-96) | OCH2-Cyclohexyl | >1000 | 0 ± 3 | 2 | 5 ± 1 | 9 ± 1 | - |
| 42 (ET-97) | OCH2CH2CH2CN | >1000 | 3 ± 4 | 2 | >10 | 30 ± 2 | 3 |
| 43 (ET-98) | OCH2CH2CH2CH2CN | >1000 | 2 ± 2 | 2 | >10 | 23 ± 4 | 3 |
| 44 (ET-99) | OCH2-(4-Pyridinyl) | >1000 | 4 ± 2 | 2 | 7 ± 1 | 15 ± 2 | 3 |
| 45 (ET-100) | OCH2-(3-Pyridinyl) | >1000 | 9 ± 0 | 2 | >10 | 33 ± 1 | 3 |
| 46 (ET-101) | OCH2-(2-Pyridinyl) | >1000 | 6 ± 1 | 2 | 7 ± 0 | 16 ± 2 | 3 |
| 47 (ET-84) | O2CCH2CH2Cl | >1000 | 50 ± 8 | 2 | - | - | - |
| 48 (ET-85) | O2CCH(CH3)2 | 599 ± 165 |
53 ± 6 | 2 | - | - | - |
| 49 (ET-86) | O2CCH2CH(CH3)2 | >1000 | 33 ± 7 | 2 | - | - | - |
See footnotes for Table 1.
IC50 is the half-maximal inhibitory concentration of a compound at inhibiting the signal of fixed concentration of 1 (33 nM) in a competition assay.
Imax is the maximum stimulation achieved by a fixed concentration of 1 (33 nM) when competed with a 10 μM dose of a compound. IC50 and Imax values represent the average of N independent experiments in triplicate and were calculated by use of Prism software as described in the Experimental Procedures section. Imax = 100 % is defined as the activity of 1 at 10 μM. Imax of T1AM at 33 nM was 45 ± 5 %. cN is the number of independent experiments in triplicate that were performed and used to calculate the EC50, IC50, Emax, and Imax values.
For the ether series (29-46), an interesting correlation was observed between the size of the position 2 substituent (R6, Table 3) and the agonist activity of the compound. The core scaffold 28 was ∼12-fold less potent (EC50 = 232 ± 8 nM) and 43 % less efficacious (Emax 88 ± 9 %) compared to 27 (Table 3). Methylating the phenol of 28 (29) increased the potency ∼2-fold (EC50 = 102 ± 26 nM) but had no effect on efficacy (Emax = 88 ± 1 %). When the ether group was an ethyl ether or larger (30-37), the potency of the compound was poor (>1 μM). The efficacy showed a different profile. When the ether group was less than 5 atom units long (30, 31, 36, and 40), the compound still had some degree of agonist activity (Emax = 26 % to 66 %). As the ether group increased in size equal to or greater than 5 atom units long (32-35 and 41-46), the compounds became non-agonists activating rTAAR1 at less than 10 % efficacy. An exception to this trend was 37. Although its isobutoxy group is only four atom units long, 37 activated below 10 % efficacy (Emax = 6 ± 1 %). Compared to 31 (EC50 = >1 μM, Emax = 41 ± 0 %), introducing an unsaturated alkene (38) or alkyne (39) into the position 2 group increased both potency (EC50 = 602 ± 10 nM and 182 ± 46 nM, respectively) and efficacy (Emax = 79 ± 5 % and 103 ± 0 %, respectively).
In the ester series (47-49), the potency of the compounds was greater than 1 μM when the position 2 functional group was 5 atom units long (47 and 49) but less than 1 μM when 4 atom units long (48, EC50 = 599 ± 165 nM) (Table 3). The efficacy of the 47-49 were between 33-53 %.
Since there are currently no binding assays available for rTAAR1, the antagonist activity of the 11 non-agonists (32-35 and 41-46) was determined by testing for the inhibition of cAMP production of rTAAR1 in stably transfected HEK293 cells treated with EC50 concentration (33 nM) of 1. Representative dose response curves of antagonists against rTAAR1 are shown in Supplementary Figure 1b. This competition assay was validated in the β2AR where the antagonist propranolol was able to inhibit the cAMP production induced by the agonist isoproterenol (data not shown).
The 11 non-agonists antagonized 1 induced rTAAR1 activation to varying degrees. The butyl ether 32 showed ca. 75 % antagonism with a half maximal inhibitory concentration (IC50) of 8 ± 2 μM (Table 3). Isobutyl ether 37 was also a weak antagonist showing 50 % inhibition and a potency of >10 μM. The longer pentyl and hexyl ethers (33 and 34, respectively) were better antagonists reducing the 1 signal to 3-6 % at a potency of ∼4-5 μM. The cyclohexylmethyl ether 41 was equally potent (IC50 = 5 ± 1 μM) but somewhat less inhibitory (Imax = 9 ± 1 %). Compared to the benzyl ether 35 (IC50 = 5 ± 1 μM, Imax = 6 ± 3 %), the heterocyclic pyridine methyl ethers (44-46) were less potent (IC50 ≥ 7 μM) and inhibitory (Imax ≥ 15 %). The cyanoalkyl ethers 42 and 43 were poor antagonists inhibiting the 1 signal no lower than 23 % with an IC50 value >10 μM. The inhibitory effects of these compounds were neither due to inhibition of adenyl cylcase nor cytotoxicty (data not shown); suggesting that these compounds are bona fide rTAAR1 antagonists.
Structure activity relationship of rTAAR1 lead antagonist
The agonist and antagonist properties of 27 and 34, respectively, suggested that the hexyloxy group is essential for antagonism. To determine if the outer ring (ring A in Fig. 3a) and β-phenyl ring are also necessary for antagonism, we synthesized analogs of 34 lacking the outer ring (50) or the β-phenyl ring (51) (Table 4, Supplementary Schemes 10 and 11). In an attempt to improve the potency of 34, we also explored the effects of N-methylation (52) and functionalization of the outer ring (53-56) (Table 4, Supplementary Schemes 8 and 10).
Table 4.
Agonist and antagonist activity of compounds 50-56 on rTAAR1.
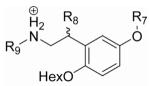 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Agonist Activity | Antagonist Activity | ||||||||
| Compd | R7 | R8 | R9 | EC50a-e ± SEM (nM) |
Emaxa-e ± SEM (%) |
Na-e | IC50a-e ± SEM (μM) |
Imaxa-e ± SEM (%) |
Na-e |
| 50 (ET-88) | H | Ph | H | >1000 | 37 ± 9 | 2 | - | - | - |
| 51 (ET-89) | Ph | H | H | 201 ± 23 | 59 ± 6 | 2 | - | - | - |
| 52 (ET-90) | Ph | Ph | CH3 | >1000 | 0 ± 3 | 2 | 5 ± 1 | 10 ± 4 | 3 |
| 53 (ET-91) | p-OH-Ph | Ph | H | >1000 | 16 ± 3 | 3 | - | - | - |
| 54 (ET-92) | p-F-Ph | Ph | H | >1000 | 0 ± 3 | 2 | 3 ± 0 | 0 ± 3 | 3 |
| 55 (ET-93) | m-F-Ph | Ph | H | >1000 | 0 ± 4 | 2 | 3 ± 1 | 0 ± 5 | 3 |
| 56 (ET-94) | m-CN-Ph | Ph | H | >1000 | 0 ± 4 | 2 | 3 ± 1 | 2 ± 4 | 3 |
See footnotes for Table 3.
Removing the outer ring or β-phenyl ring of 34 was detrimental to rTAAR1 antagonism. In the absence of the outer ring (50), 34 was converted into a weak agonist (Table 4, Fig 3c). Similarly, 34 became an agonist without the β-phenyl ring (51, EC50 = 201 ± 23 nM, EC50 = 59 ± 6 %) (Fig. 3c).
Mono-methylating the amine (52) or inserting electron withdrawing groups on the outer ring (54-56) preserved the antagonist activity of 34. When screened for agonist activity, these compounds did not activate rTAAR1 (Table 4). In the antagonist assay, the potency of 52 was unaffected (IC50 = 5 ± 1 μM) but the antagonist activity slightly decreased (IC50 = 10 ± 4 %). The potency and inhibitory capacity of 34 was also not significantly affected by introducing a para-fluoro, meta-fluoro, or meta-cyano group into the outer ring (54, 55, and 56 respectively). The IC50 and Imax values of these compounds were ∼3 μM and ≤ 2 %, respectively. Interestingly, inserting a para-hydroxy group into the outer ring (53) endowed some agonist activity to 34 activating rTAAR1 at >1 μM potency and 16 ± 3 % efficacy.
DISCUSSION
The rotamer toggle switch model of aminergic GPCR activation (Fig. 2) has proven to be a useful guideline in the design and synthesis of rTAAR1 agonists and antagonists. Previous SAR studies on the ethylamine portion of 1 for rTAAR1 provided 2 as a promising scaffold for developing rTAAR11 superagonists, which we define as compounds that are more potent and/or efficacious than 1 (Tan et al., 2007). In addition to being as potent and efficacious as 1, 2 provides the added benefit of having many potential sites for derivitization. By analogy to the assumed binding mode of epinephrine to β2AR (Fig. 1d), we deduced 2 to bind to rTAAR1 in a similar fashion with the charged amine forming a salt bridge interaction with D3.32 and the biaryl ether oxygen hydrogen bonding to S5.46 (Fig. 3b). The β-phenyl ring is proposed to occupy a pocket near the interface of TM6 and TM7.
In the context of the rotamer toggle switch model, our analysis of the ligand-receptor interaction of β2AR agonists showed that agonists generally lack functional groups in the region of the molecule that is predicted to be located in the vicinity of the rotamer switch residues. Structurally, most of these agonists appear to have functional groups that complement the physicochemical properties of the residues within the binding site. Following this lead, we attempted to improve the agonist properties of 2 by incorporating functional groups in the regions of the molecule (e.g. β-phenyl ring, charged amine, outer ring, and position 5 of the inner ring Fig. 3a,b)) away from rotamer switch residues. In the β-phenyl ring, SAR studies presented here showed a clear preference for a hydroxyl group at the para position. The para hydroxyl analog (5) was 24-78-fold and 8-46 % more potent and efficacious, respectively, compared to the ortho or meta hydroxyl analogs (6 and 7) and ortho or para methoxy (3 and 4) analogs. Additionally, the para hydroxyl improved the potency and efficacy of 2 ∼4.5-fold and 11 %, respectively. We believe that this enhancement in agonist activity is a reflection of an increase in the binding affinity of 2 for rTAAR1 due to hydrogen bond interactions of the para hydroxyl with N7.39 and/or N7.35 (Fig. 3b). Mutating residue 7.39 in the β2AR has previously been found to perturb the binding affinity of agonists and antagonists (Suryanarayana et al., 1991). In the recently determined crystal structure of the β2AR, N7.39 of β2AR was involved in hydrogen bond interactions with the β-carbon hydroxyl group of the partial inverse agonist carazolol (Cherezov et al., 2007; Rasmussen et al., 2007; Rosenbaum et al., 2007).
In the presence of the para hydroxyl, mono-methylating the charged amine (10) or incorporating a 1 moiety into the molecule (12) was tolerated but it had modest effects on agonist activity, if any at all. N-methyl 10 was equipotent to 5 but 13 % more efficacious. On the other hand, 12 essentially has the same potency and efficacy as 5. The comparable levels of agonist activity of 5, 10, and 12, suggest that these compounds have similar interactions with rTAAR1 and possibly elicit the same final active conformation of the receptor.
In contrast to the β2AR agonists, our analysis of the SAR and potential binding modes of antagonists for the dopamine 1-like and 2-like receptors revealed the presence of structural moieties within these compounds that could conceivably sterically occlude the rotamer toggle switch residues from assuming their active conformation. Applying this hypothesis to rTAAR1 we attempted to convert 2 into an antagonist by installing ethers and esters at the 2 position of the inner ring that varied in steric bulk, rigidity, topology, and polarity (15-26). Based on our proposed binding orientation of 2 (Fig. 3b), this position was identified to be the prime location for presenting groups that could interfere with the rotamer switch residues in rTAAR1. Unfortunately, none of these compounds turned out to be antagonists. Presumably 15-26 were still able to activate rTAAR1 between 15 % and 95 % efficacy because the variable position 2 groups (R5, Table 2) are positioned away from the rotamer switch residues within the binding site due to rotation of the inner ring about the β-carbon and biaryl ether oxygen axis.
To circumvent this problem, we modified the core scaffold by moving the phenoxy group from the para (14) to the meta (28) position (Tables 2 and 3). With this modification, the agonist activity of the compound decreased as the size of the ether substituent increased (Fig. 3c). When the ether group was ≥ 5 atom units long (32-35, and 41-46), the agonist activity of the compound was completely abolished (≤ 10 % efficacy). Compounds with substituents less than 5 atom units long (29-31) were weak agonists activating rTAAR1 between 41 to 88 % efficacy. The composition of the substituent appears to be important as an ester group that is 5 atom units long (47 and 49) was still an agonist (EC50 = 33 to 53 %). When the non-agonists (32-35 and 41-46) were screened for antagonist activity in a competition assay with 1 at its EC50 concentration (33 nM), all compounds were found to inhibit 1 induced cAMP production to varying degrees at 10 μM. Compound 34 was the best antagonist showing >90 % inhibition of rTAAR1 activation with an IC50 value of 4 μM. The antagonist activity of 32-35 and 41-46 are thought to arise from the ether substituents sterically occluding F6.52 and/or W6.48 of the rotamer switch residues from assuming their active conformation.
The hexyloxy group, outer ring, and β-phenyl ring of 34 are all necessary for antagonism. In the absence of any one of these groups, the resulting compounds lose their antagonist activity and become agonists. Since the transformation of 34 to 27 yielded the greatest increase in agonist potency and efficacy, the hexyloxy group is the most important of the three structural elements in terms of decreasing agonist activity and conferring antagonist properties to 34 (Fig. 3c). This is consistent with the notion that the outer ring and β-phenyl ring are essential scaffolding elements that assures 34 docks into the rTAAR1 binding site in the proper orientation to position the hexyloxy group, the molecular basis of antagonism, to interfere with the rotamer switch residues (Fig. 3d).
SIGNIFICANCE
The rotamer toggle switch model of aminergic GPCR activation is a useful model for understanding the molecular basis of rTAAR1 activation by 1 and related analogs. It has proven helpful in the development of rTAAR1 agonists and antagonists providing superagonists 5, 10, and 12 and lead antagonists 34, 54 and 55. This proof of concept study shows that agonist or antagonist properties of aminergic GPCR drugs arise from drug interactions with the rotamer switch residues. Agonists allow the rotamer switch to toggle and/or have more favorable interactions with the active state of the receptor while antagonists sterically occlude the rotamer switch and/or have more favorable interactions with the inactive state of the receptor.
These agonist and antagonist design principles have the potential to accelerate and increase the efficiency of the drug discovery and development process for GPCRs. Having insights into the critical ligand-receptor interactions important for receptor activation or inhibition facilitates the interpretation of SAR data and correlation of pharmacophore models with the molecular properties of the receptor binding site. This information then provides a map of the binding site landscape and presents a drug design blueprint for identifying promising scaffolds, recognizing compatible functional groups to incorporate, and evaluating the contribution of individual structural elements of a given compound towards its binding affinity, selectivity, and functional properties. We envision these principles to supplement all current GPCR drug design strategies (e.g. ligand-based drug design, focused library screening, virtual screening, structure based drug design, etc.) (Evers et al., 2005; Evers et al., 2005; Klabunde et al., 2005; Klabunde et al., 2002) and help generate predictive rules and guidelines that would prove to be a useful and general method for designing activators or inhibitors for biogenic amine GPCRs and possibly other rhodopsin like GPCRs.
EXPERIMENTAL PROCEDURES
Residue indexing system
Residues are labeled relative to the most conserved amino acid in the transmembrane segment in which it is located (Ballesteros et al., 1995). Tryptophan 6.48, for example, is located in transmembrane 6 and precedes the most conserved residue by 2 positions. Arginine 3.50 is the most conserve residue in TM3. This system simplifies the identification of corresponding residues in different GPCRs.
Homology model of rTAAR1
The sequence of rTAAR1 was aligned to 26 human biogenic amine GPCRs (i.e. dopamine, α-adrenergic, β-adrenergic, and serotonin receptors) and the sequence for bovine rhodopsin (Protein Data Bank accession code 1F88) (Palczewski et al., 2000) using the program MUSCLE (Edgar, 2004). We constructed our homology model of rTAAR1 based on the crystal structure of the inactive state bovine rhodopsin as a template and used our in house software PLOP (commercially available as Prime from Schrödinger Inc). The modeling program did not modify conserved residues, leaving each atom in these residues at their original PDB coordinates. Non-conserved side chains were built onto the structure using the backbone coordinates for bovine rhodopsin as a reference point. All chain breaks or gaps were closed using a previously published loop building and optimization algorithm (Jacobson et al., 2004). After building the complete model, side chain optimization, followed by backbone and side chain energy minimization, was performed on all non-conserved residues. The homology modeling program relies on the OPLS all atom force field (Jacobson et al., 2002; Jorgensen et al., 1996; Kaminski et al., 2001) and a Generalized Born solvent model (Gallicchio et al., 2002; Ghosh et al., 1998) to evaluate the energy of different conformations and select the lowest energy structure as the final model.
Ligand-rTAAR1 interaction analysis
Ligands were manually docked into the binding site of rTAAR1 with the following constraints: (1) D3.32 is the counterion for the charged amine, (2) S5.46 forms a hydrogen bond interaction with the biaryl ether oxygen, and (3) the β-phenyl ring is located in the vicinity of N7.39. The binding orientations of compounds were then manually adjusted to try to satisfy as many of the proposed binding and specificity determinant residues in the binding site as possible.
Synthesis
Detailed synthetic procedures and chemical compound information are described in the supplementary information.
In Vitro cAMP Assays-Agonist activity assay
After incubating in fresh medium for at least 2 h, HEK293 cells stably transfected with rTAAR1 were harvested in Krebs-Ringer-HEPES buffer (KRH) and preincubated with 200 μM 3-isobutyl-1-methylxanthine (IBMX) for 20-30 minutes. Cells were incubated in KRH with 133 μM IBMX and 3μL of the test compound, forskolin (10 μM), or vehicle (dimethyl sulfoxide, DMSO) for 1 h at 37 °C (300 μL total volume). The cells were boiled for 20 min after addition of 100 μL 0.5 mM sodium acetate buffer. The cell lysate was centrifuged to remove cellular debris, and an aliquot (30 μL) was transferred to an opaque, flat bottom 96-well plate (Corning #3917). The cAMP content of the aliquot was measured by use of the Hithunter™ cAMP XS kit (DiscoveRX, Fremont, CA). The plate was shaken on a titer plate shaker for 2 min after addition of 20 μL of cAMP XS antibody/lysis mix. After incubation in the dark for 1h, 20 μL of cAMP XS ED reagent was added and the plate was shaken for 2 min. After another hour of incubation in the dark, 40 μL of cAMP XS EA/CL substrate mix was added and the plate was shaken for 2 min. The plate was sealed with an acetate plate sealer (Thermo Scientific #3501) and allowed to incubate in the dark for 15-18 h before luminescence was measured (3 readings/well at 0.33 s/reading) on an Analyst™ AD Assay Detection System (LJL Biosystems) or a Packard Fusion Microplate Reader. Data were reported relative to 1 and expressed as %T1AM. The activity of 1 at 10μM was set as 100 %T1AM. Concentration-response 1 curves were plotted and EC50 values were calculated with Prism software (GraphPad, San Diego, CA). Standard error of the mean was calculated from the EC50 and Emax values of each independent triplicate experiment by use of Prism Software.
In Vitro cAMP Assays-Antagonist activity assay
Same as the agonist activity assay procedure described above with the following changes: cells that were harvested in KRH buffer and preincubated with IBMX for 20-30 minutes were incubated in KRH with 133 μM IBMX and 3μL of the putative antagonist, or vehicle (DMSO) for 30 min at 37 °C (300 μL total volume). 3μL of the competing agonist (T1AM, EC50 concentration (33 nM) as the final concentration), T1AM (10 μM), forskolin (10 μM) or vehicle (DMSO) was then added to the reactions before incubating for 1h at 37°C. The cells were then processed as described in the agonist activity assay. Concentration-response curves were plotted and IC50 values were calculated with Prism software (GraphPad, San Diego, CA).
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by grants from the National Institutes of Health (Grant DK52798 to T.S.S.) and Ikaria, Inc.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Ballesteros J, Jensen A, Liapakis G, Rasmussen S, Shi L, Gether U, Javitch J. Activation of the beta(2)-adrenergic receptor involves disruption of an ionic lock between the cytoplasmic ends of transmembrane segments 3 and 6. J. Biol. Chem. 2001;276:29171–29177. doi: 10.1074/jbc.M103747200. [DOI] [PubMed] [Google Scholar]
- Ballesteros J, Weinstein H. Integrated methods for the construction of three-dimensional models of structure-function relations in G protein-coupled receptors. Methods Neuroscience. 1995;25:366–428. [Google Scholar]
- Borowsky B, Adham N, Jones K, Raddatz R, Artymyshyn R, Ogozalek K, Durkin M, Lakhlani P, Bonini J, Pathirana S, et al. Trace amines: Identification of a family of mammalian G protein-coupled receptors. Proc. Natl. Acad. Sci. U. S. A. 2001;98:8966–8971. doi: 10.1073/pnas.151105198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunzow J, Sonders M, Arttamangkul S, Harrison L, Zhang G, Quigley D, Darland T, Suchland K, Pasumamula S, Kennedy J, et al. Amphetamine, 3,4-methylenedioxymethamphetamine, lysergic acid diethylamide, and metabolites of the catecholamine neurotransmitters are agonists of a rat trace amine receptor. Mol. Pharmacol. 2001;60:1181–1188. doi: 10.1124/mol.60.6.1181. [DOI] [PubMed] [Google Scholar]
- Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, et al. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evers A, Hessler G, Matter H, Klabunde T. Virtual screening of biogenic amine-binding G-protein coupled receptors: Comparative evaluation of protein- and ligand-based virtual screening protocols. J. Med. Chem. 2005;48:5448–5465. doi: 10.1021/jm050090o. [DOI] [PubMed] [Google Scholar]
- Evers A, Klabunde T. Structure-based drug discovery using GPCR homology modeling: Successful virtual screening for antagonists of the Alpha1A adrenergic receptor. J. Med. Chem. 2005;48:1088–1097. doi: 10.1021/jm0491804. [DOI] [PubMed] [Google Scholar]
- Gallicchio E, Zhang LY, Levy RM. The SGB/NP hydration free energy model based on the surface generalized born solvent reaction field and novel nonpolar hydration free energy estimators. J. Comput. Chem. 2002;23:517–529. doi: 10.1002/jcc.10045. [DOI] [PubMed] [Google Scholar]
- Gether U. Uncovering molecular mechanisms involved in activation of G protein-coupled receptors. Endocr. Rev. 2000;21:90–113. doi: 10.1210/edrv.21.1.0390. [DOI] [PubMed] [Google Scholar]
- Ghosh A, Rapp C, Friesner R. Generalized born model based on a surface integral formulation. J. Phys. Chem. B. 1998;112:10983–10990. [Google Scholar]
- Hart M, Suchland K, Miyakawa M, Bunzow J, Grandy D, Scanlan T. Trace amine-associated receptor agonists: Synthesis and evaluation of thyronamines and related analogues. J. Med. Chem. 2006;49:1101–1112. doi: 10.1021/jm0505718. [DOI] [PubMed] [Google Scholar]
- Jacobson M, Kaminski G, Friesner R, Rapp C. Force field validation using protein side chain prediction. J. Phys. Chem. B. 2002;106:11673–11680. [Google Scholar]
- Jacobson MP, Pincus DL, Rapp CS, Day TJ, Honig B, Shaw DE, Friesner RA. A hierarchical approach to all-atom protein loop prediction. Proteins: Struct., Funct., Bioinf. 2004;55:351–367. doi: 10.1002/prot.10613. [DOI] [PubMed] [Google Scholar]
- Jorgensen W, Maxwell D, Tirado-Rives J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996;118:11225–11236. [Google Scholar]
- Kaminski G, Friesner R, Tirado-Rives J, Jorgensen W. Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J. Phys. Chem. B. 2001;105:6474–6487. [Google Scholar]
- Klabunde T, Evers A. GPCR antitarget modeling: Pharmacophore models for biogenic amine binding GPCRs to avoid GPCR-mediated side effects. ChemBioChem. 2005;6:876–889. doi: 10.1002/cbic.200400369. [DOI] [PubMed] [Google Scholar]
- Klabunde T, Hessler G. Drug design strategies for targeting G-protein-coupled receptors. ChemBioChem. 2002;3:929–944. doi: 10.1002/1439-7633(20021004)3:10<928::AID-CBIC928>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Kobilka B, Deupi X. Conformational complexity of G-protein-coupled receptors. Trends Pharmacol. Sci. 2007;28:397–406. doi: 10.1016/j.tips.2007.06.003. [DOI] [PubMed] [Google Scholar]
- Liapakis G, Ballesteros J, Papachristou S, Chan W, Chen X, Javitch J. The forgotten serine - A critical role for Ser-203(5.42) in ligand binding to and activation of the beta(2)-adrenergic receptor. J. Biol. Chem. 2000;275:37779–37788. doi: 10.1074/jbc.M002092200. [DOI] [PubMed] [Google Scholar]
- Liapakis G, Chan W, Papadokostaki M, Javitch J. Synergistic contributions of the functional groups of epinephrine to its affinity and efficacy at the beta(2) adrenergic receptor. Mol. Pharmacol. 2004;65:1181–1190. doi: 10.1124/mol.65.5.1181. [DOI] [PubMed] [Google Scholar]
- Lindemann L, Ebeling M, Kratochwil N, Bunzow J, Grandy D, Hoener M. Trace amine-associated receptors form structurally and functionally distinct subfamilies of novel G protein-coupled receptors. Genomics. 2005;85:372–385. doi: 10.1016/j.ygeno.2004.11.010. [DOI] [PubMed] [Google Scholar]
- Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le Trong I, Teller DC, Okada T, Stenkamp RE, et al. Crystal structure of rhodopsin: A G protein-coupled receptor. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- Rasmussen SG, Choi HJ, Rosenbaum DM, Kobilka TS, Thian FS, Edwards PC, Burghammer M, Ratnala VR, Sanishvili R, Fischetti RF, et al. Crystal structure of the human beta2 adrenergic G-protein-coupled receptor. Nature. 2007;450:383–387. doi: 10.1038/nature06325. [DOI] [PubMed] [Google Scholar]
- Rosenbaum DM, Cherezov V, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Yao XJ, Weis WI, Stevens RC, et al. GPCR engineering yields high-resolution structural insights into beta2-adrenergic receptor function. Science. 2007;318:1266–1273. doi: 10.1126/science.1150609. [DOI] [PubMed] [Google Scholar]
- Scanlan T, Suchland K, Hart M, Chiellini G, Huang Y, Kruzich P, Frascarelli S, Crossley D, Bunzow J, Ronca-Testoni S, et al. 3-iodothyronamine is an endogenous and rapid-acting derivative of thyroid hormone. Nat. Med. 2004;10:638–642. doi: 10.1038/nm1051. [DOI] [PubMed] [Google Scholar]
- Shi L, Javitch J. The binding site of aminergic G protein-coupled receptors: The transmembrane segments and second extracellular loop. Annu. Rev. Pharmacol. Toxicol. 2002;42:437–467. doi: 10.1146/annurev.pharmtox.42.091101.144224. [DOI] [PubMed] [Google Scholar]
- Shi L, Liapakis G, Xu R, Guarnieri F, Ballesteros J, Javitch J. Beta(2) adrenergic receptor activation - Modulation of the proline kink in transmembrane 6 by a rotamer toggle switch. J. Biol. Chem. 2002;277:40989–40996. doi: 10.1074/jbc.M206801200. [DOI] [PubMed] [Google Scholar]
- Snead AN, Santos MS, Seal RP, Miyakawa M, Edwards RH, Scanlan TS. Thyronamines inhibit plasma membrane and vesicular monoamine transport. ACS Chem. Biol. 2007;2:390–398. doi: 10.1021/cb700057b. [DOI] [PubMed] [Google Scholar]
- Strader C, Candelore M, Hill W, Sigal I, Dixon R. Identification of 2 serine residues involved in agonist activation of the beta-adrenergic-receptor. J. Biol. Chem. 1989;264:13572–13578. [PubMed] [Google Scholar]
- Strader C, Fong T, Tota M, Underwood D, Dixon R. Structure and function of G-protein coupled receptors. Annu. Rev. Biochem. 1994;63:101–132. doi: 10.1146/annurev.bi.63.070194.000533. [DOI] [PubMed] [Google Scholar]
- Strader C, Sigal I, Candelore M, Rands E, Hill W, Dixon R. Conserved aspartic-acid residue-79 and residue-113 of the beta-adrenergic-receptor have different roles in receptor function. J. Biol. Chem. 1988;263:10267–10271. [PubMed] [Google Scholar]
- Strader C, Sigal I, Dixon R. Structural basis of beta-adrenergic-receptor function. FASEB J. 1989;3:1825–1832. doi: 10.1096/fasebj.3.7.2541037. [DOI] [PubMed] [Google Scholar]
- Suryanarayana S, Daunt D, von Zastrow M, Kobilka B. A point mutation in the 7th hydrophobic domain of the alpha-2-adrenergic-receptor increases its affinity for a family of beta-receptor-antagonists. J. Biol. Chem. 1991;266:15488–15492. [PubMed] [Google Scholar]
- Swaminath G, Xiang Y, Lee T, Steenhuis J, Parnot C, Kobilka B. Sequential binding of agonists to the beta(2) adrenoceptor - Kinetic evidence for intermediate conformational states. J. Biol. Chem. 2004;279:686–691. doi: 10.1074/jbc.M310888200. [DOI] [PubMed] [Google Scholar]
- Tan E, Miyakawa M, Bunzow J, Grandy D, Scanlan T. Exploring the structure-activity relationship of the ethylamine portion of 3-iodothyronamine for rat and mouse trace amine-associated receptor 1. J. Med. Chem. 2007;50:2787–2798. doi: 10.1021/jm0700417. [DOI] [PubMed] [Google Scholar]
- Tota MR, Candelore MR, Dixon RA, Strader CD. Biophysical and genetic analysis of the ligand-binding site of the beta-adrenoceptor. Trends Pharmacol. Sci. 1991;12:4–6. doi: 10.1016/0165-6147(91)90479-c. [DOI] [PubMed] [Google Scholar]
- Wainscott D, Little S, Yin T, Tu Y, Rocco V, He J, Nelson D. Pharmacologic characterization of the cloned human trace amine-associated receptor1 (TAAR1) and evidence for species differences with the rat TAAR1. J. Pharmacol. Exp. Ther. 2007;320:475–485. doi: 10.1124/jpet.106.112532. [DOI] [PubMed] [Google Scholar]
- Wess J. Molecular basis of receptor/G-protein-coupling selectivity. Pharmacol. Ther. 1998;80:231–264. doi: 10.1016/s0163-7258(98)00030-8. [DOI] [PubMed] [Google Scholar]
- Wieland K, Zuurmond H, Krasel C, Ijzerman A, Lohse M. Involvement of Asn-293 in stereospecific agonist recognition and in activation of the beta(2)-adrenergic receptor. Proc. Natl. Acad. Sci. U. S. A. 1996;93:9276–9281. doi: 10.1073/pnas.93.17.9276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao X, Parnot C, Deupi X, Ratnala V, Swaminath G, Farrens D, Kobilka B. Coupling ligand structure to specific conformational switches in the beta(2)-adrenoceptor. Nat. Chem. Bio. 2006;2:417–422. doi: 10.1038/nchembio801. [DOI] [PubMed] [Google Scholar]
- Zucchi R, Chiellini G, Scanlan T, Grandy D. Trace amine-associated receptors and their ligands. Br. J. Pharmacol. 2006;149:967–978. doi: 10.1038/sj.bjp.0706948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuurmond H, Hessling J, Bluml K, Lohse M, Ijzerman A. Study of interaction between agonists and Asn293 in helix VI of human beta(2)-adrenergic receptor. Mol. Pharmacol. 1999;56:909–916. doi: 10.1124/mol.56.5.909. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.