

Abstract
The pyruvate dehydrogenase multienzyme complex (PDC) is a key regulatory point in cellular metabolism linking glycolysis to the citric acid cycle and lipogenesis. Reversible phosphorylation of the pyruvate dehydrogenase enzyme is a critical regulatory mechanism and an important point for monitoring metabolic activity. To directly determine the regulation of the PDC by phosphorylation, we developed a complete set of phospho-antibodies against the three known phosphorylation sites on the E1 alpha subunit of pyruvate dehydrogenase (PDHE1α). We demonstrate phospho-site specificity of each antibody in a variety of cultured cells and tissue extracts. In addition, we show sensitivity of these antibodies to PDH activity using the pyruvate dehydrogenase kinase specific inhibitor dichloroacetate. We go on to utilize these antibodies to assess PDH phosphorylation in a patient suffering from Leigh's Syndrome. Finally, we observed changes in individual phosphorylation states following a small molecule screen, demonstrating that these reagents should be useful for monitoring phosphorylation of PDHE1α and, therefore, overall metabolism in both the disease state as well as in response to a myriad of physiological and pharmacological stimuli.
Keywords: Pyruvate dehydrogenase, Mitochondria, Dichloroacetate, Dichloroacetic acid, Leigh's Syndrome, Reversible phosphorylation, Phospho-peptide, Phospho-antibody
Introduction
The mammalian pyruvate dehydrogenase complex (PDC) links glycolysis to the tricarboxylic acid cycle by catalyzing the irreversible oxidative decarboxylation of pyruvate leading to the generation of CO2, NADH, and acetyl-CoA. Located within the matrix compartment of mitochondria, the PDC is one of the largest multienzyme complexes in eukaryotic cells and consists of multiple copies of three catalytic components [1]. The dihyrolipoamide acetyl-transferase (E2) makes up the core of the enzyme and contains binding domains for the α2β2 heterotetramer pyruvate dehdyrogenase (PDHE1), and the E2/E3 binding protein which links the dihyrolipoamide dehydrogenase (E3) to E2 [2]. The PDC is a major switch regulating glucose and fatty acid oxidation.
PDH regulation via phosphorylation is currently implicated in the altered patterns of metabolism in cancer, obesity and insulin resistance [3; 4]. The enzymatic activity of the complex is regulated allosterically, as well as being tightly regulated by reversible phosphorylation in response to the availability of glucose. The seminal work of Reed and coworkers [5] provided the first evidence that mitochondrial function could be regulated by phosphorylation. PDH activity is inhibited in response to site-specific phosphorylation at three sites on PDHE1α (Ser232, Ser293, or Ser300). Phosphorylation is catalyzed by one of four pyruvate dehydrogenase kinases (PDK) [6]. In opposition to the four PDKs, there are two isoforms of pyruvate dehydrogenase phosphatases (PDP1 and PDP2) present in mammalian cells [7; 8]. The acute control of PDH is mediated by end product (acetyl-CoA, NADH, ATP) activation of kinase activity, leading to the inhibition of PDH activity. In contrast, substrate (pyruvate, ADP) availability leads to kinase inhibition and reactivation of the complex by the PDPs [9; 10; 11]. Interestingly, phosphorylation at any one site leads to inhibition of the complex in vitro [12]. Additional site-specific regulation may occur as PDK1 is the only isoform reported to phosphorylate all three sites, while PDK2, PDK3, and PDK4 are reported to phosphorylate Ser293 and Ser300 in vitro [6; 13]. Furthermore, in studies done on PDH isolated from mammalian tissues, Ser293 has been shown to be phosphorylated at a faster rate than Ser300 and Ser232 [14]. PDP1 and PDP2 can dephosphorylate all three sites with similar preferences (Ser300> Ser232 > Ser293) yet show slight disparities in their specific activity for each of the three sites [15]. Variation in site preference and kinetic activity of each PDK and PDP isoform for each of the three sites infers yet another level of PDC regulation [6; 13].
In addition to having differences in site specificity, the PDKs and PDPs are differentially expressed in tissues [16]. Whilst PDK2 is widely expressed in tissues, PDK1 is highly expressed in heart, but only moderately expressed in skeletal muscle, pancreas, and liver [16]. PDK3 is found highly expressed in testis with lower levels of expression in lung, kidney, spleen, heart, and brain [16]. In contrast, PDK4 is highly expressed in skeletal muscle and heart, and to a lesser extent in kidney, liver and lung [16]. The PDPs are widely expressed in tissues with a notable absence of PDP2 in testis and skeletal muscle, but high levels in heart, liver and kidney [17]. PDP1 is widely expressed in tissues with high levels in brain, heart and testis [17]. Interestingly, PDK2 and PDK4 are upregulated at the transcriptional level, while PDP1 and PDP2 are downregulated in response to starvation and diabetes in a tissue-specific manner [18; 19; 20].
PDH regulation via phosphorylation is currently implicated in the altered patterns of metabolism in cancer, obesity and insulin resistance [3; 4]. The purpose of this study was to develop tools for studying the regulation of the PDH by reversible phosphorylation. We have developed the first complete set of phospho-specific antibodies against the known phosphorylation sites on PDHE1α (Ser232, Ser293, and Ser300). Moreover, we demonstrate that these antibodies are not only phospho-site specific, but sensitive to changes in PDH activity when the PDKs are inhibited. We also report, for the first time, the distribution of site-specific phosphorylation of the PDH across multiple tissues. Here we describe the development and validation of phospho-antibodies that will allow for assessment of changes in regulation of PDH by phosphorylation. Furthermore, these antibodies should provide an invaluable tool for monitoring changes in PDH regulation in response to changes in metabolism as well as in disease states such as diabetes and cancer.
Materials and methods
Cell culture, Immunocytochemistry (ICC) and Materials
COS-7 and HEK293A cells were maintained at 37°C at 5% CO2 in Dulbecco's Modified Eagle Medium (Invitrogen) containing 10% fetal bovine serum (FBS) and 50 U/ml each of penicillin and streptomycin (P/S). Primary human fibroblasts were isolated from foreskin biopsies and cultured in α-Modified Eagle Medium (Invitrogen) containing 10% FBS and P/S. For ICC, cells were incubated with 100 nM MitoTracker Red (Invitrogen) for 20 minutes before fixation with 3.7% formaldehyde and were then permeabilized in PBS containing 0.1% Tween 20, 0.3% Triton X-100, and 6% BSA. Cells were incubated with affinity purified anti-pSer293, anti-pSer300, or anti-pSer232 at 500 ng/mL or anti-PDHE1A (MitoSciences #A-2132) at 1:100 in PBS containing 0.1% Tween 20 and 6% BSA for one hour at room temperature, followed by Alexa Fluor 488 goat anti-rabbit conjugated secondary antibody (Invitrogen) at 1:500. Nuclei were stained with 300 nM DAPI (Molecular Probes) for 1 min before viewing. Fluorescent images were taken using a light microscope (DMR; Leica) with a PL APO 63× 1.32 NA oil objective (Leica) at room temperature, and images were captured with a CCD camera (C4742-95; Hamamatsu) using OpenLab 4.0.1 software (Improvision). Antibodies against total PDHE1α (EMD Biosciences, MitoSciences), E2/E3 binding protein (MitoSciences), α-tubulin (Sigma), NDUFB6 (MitoSciences), and GAPDH (Ambion) were used at concentrations as recommended by manufacturers protocol.
Phosphatase Treatment
100 μg of crude (see below) rat kidney mitochondria were treated with or without 20 U of calf intestinal alkaline phosphatase (Roche) for 30 min at 37°C in MSHE buffer (210 mM mannitol, 70 mM sucrose, 5 mM HEPES pH 7.4 with KOH, 2 mM EGTA) containing 1% Triton X-100, 2 mM MgCl2, and 2 mM CaCl2.
Generation of Polyclonal Phospho-Antibodies
The following synthetic phosphopeptides from PDHE1α were conjugated to KLH and injected into rabbits: Ser293 RYHGHpSMSDPG, Ser300 SDPGVpSYRTREC, Ser232 RYGMGTpSVEAAC. Antibodies were affinity purified over the appropriate phospho-peptide column, and then eluted. They were then passed over an unphosphorylated peptide column and the flow through was collected. Antibodies were used at a working concentration of 200 ng/mL for western blot and 500 ng/mL for immunofluorescence. Antibodies were produced by and will be distributed by EMD Biosciences.
Animals and Tissue Distribution
12-week-old male mice (C57 BL/6J) were purchased from the Jackson Laboratories and starved for 4 hours prior to tissue harvest (Bar Harbor, ME). Tissues were harvested from a male mouse, flash frozen in liquid nitrogen and homogenized using a T10 Basic S1 Disperser 115V, 50/60Hz (IKA Works, Inc) in PLC buffer (50 mM HEPES [pH 7.5]; 150 mM NaCl; 10% glycerol; 1% Triton-X100; 1.5 mM MgCl2; 1mM EGTA; 10 mM NaP2O7; 100 mM NaF; 500 μM Na3VO4). Protein levels were roughly normalized to total levels of PDHE1α by Western blot and compared to levels of phosphorylation at each of the three sites by densitometry. Although no comparisons can be made between antibodies because of the variability in antibody affinity, we can make semi-quantitative comparisons between tissues from antibodies against individual phosphorylation sites.
Purification of Crude Mitochondria
Mitochondria were purified as described [21; 22; 23; 24] with minor modifications. Fresh tissue was harvested and placed in ice-cold MSHE+BSA buffer (210 mM mannitol, 70 mM sucrose, 5 mM HEPES pH 7.4 with KOH, 2 mM EGTA, 0.5% fatty acid free BSA, and EDTA-free Complete protease inhibitor cocktail [Roche]). Tissues were minced, and then washed three times in MSHE+BSA buffer. Tissue was disrupted with 10-15 strokes using a tight fitting Potter-Elvehjem tissue homogenizer. Homogenate was centrifuged for 10 min at 600 × g to remove unbroken cells and nuclei. The pellet was re-homogenized and spun at 600 × g. Supernatants were combined and spun at 15,000 × g for 10 min to pellet mitochondria. Pellets were washed two times in MSHE+BSA, followed by one wash in BSA-free MSHE buffer.
Inhibition of PDKs by DCA in tissue extracts
Dichloroacetate was purchased from Sigma-Aldrich (634522). 0.5 M stocks were generated in 10 mM Hepes (pH 7.2). Crude mitochondria were isolated as indicated above. 200 μg of mitochondrial protein was incubated with 5 mM DCA for 2 hours at room temperature in MSHE buffer plus EDTA-free Complete protease inhibitor cocktail (Roche).
Leigh's Syndrome patient history
The patient was born after a full term pregnancy without complications and developed normally until 1 year of age, when he developed acute weakness and hypotonia. He became ataxic and developed carbohydrate sensitive lactic acidemia, lower extremity spastic paraparesis, dysarthria, and ophthalmoplegia. Brain MRI revealed symmetric T2 lesions in the basal ganglia. The patient received the diagnosis of LS of unknown cause at age 8, and an enzymatic diagnosis of PDH deficiency at 11 years of age. He died at the age of 20 years.
Results
Conservation of phosphorylation sites across species
The PDC is tightly regulated by reversible phosphorylation at three known phosphorylation sites on PDHE1α: Ser293, Ser300, and Ser232 (numbering relative to start methionine). To obtain a complete set of tools for studying reversible phosphorylation of PDHE1α and, by extension, the regulation of the PDH in intact cells, we generated antibodies against phospho-peptides corresponding to the three phosphorylation sites found on PDHE1α (Fig. 1A). We performed PSI-BLAST database searches using the human form of PDHE1α (Accession # NP_000275) and found that it is highly homologous across species. Moreover, the sites of phosphorylation are invariant in most vertebrates suggesting that these antibodies will have widespread utility across species (Fig. 1B).
Fig. 1.

Schematic of phospho-peptides used to generate antisera. (A) Phospho-peptides corresponding to each of these sites were synthesized and used to immunize rabbits for the production of phospho-specific antibodies (numbers correspond to start methionine). (B) Amino acid comparison of PDHE1α orthologs across several species, with identical amino acids highlighted in gray. Colored bars and residues correspond to phospho-sites and peptides utilized to generate anti-sera (site 1-pSer293 in yellow, site 2-pSer300 in orange, and site 3-pSer232 in green). Accession numbers follow the common name of each species.
PDHE1α phospho-antibodies are phospho-site specific
To confirm that our antibodies were phospho-specific, we isolated crude mitochondria from rat kidney tissue. Mitochondrial extracts were incubated in the presence or absence of alkaline phosphatase. All three phosphorylation sites showed a dramatic loss of signal following treatment with the phosphatase when compared to controls (Fig. 2A). In contrast, there was no change in the total amount of PDHE1α protein. Similar results were seen in the tissue culture cell line HEK293A as well as mouse liver tissue (data not shown). These data demonstrate that our antibodies are phospho-specific. In addition, these antibodies are able to detect the phosphorylation states in tissue extracts from mice and rats, as well as cell lines derived human tissues, demonstrating their utility in monitoring phosphorylation states in a number of species.
Fig. 2.
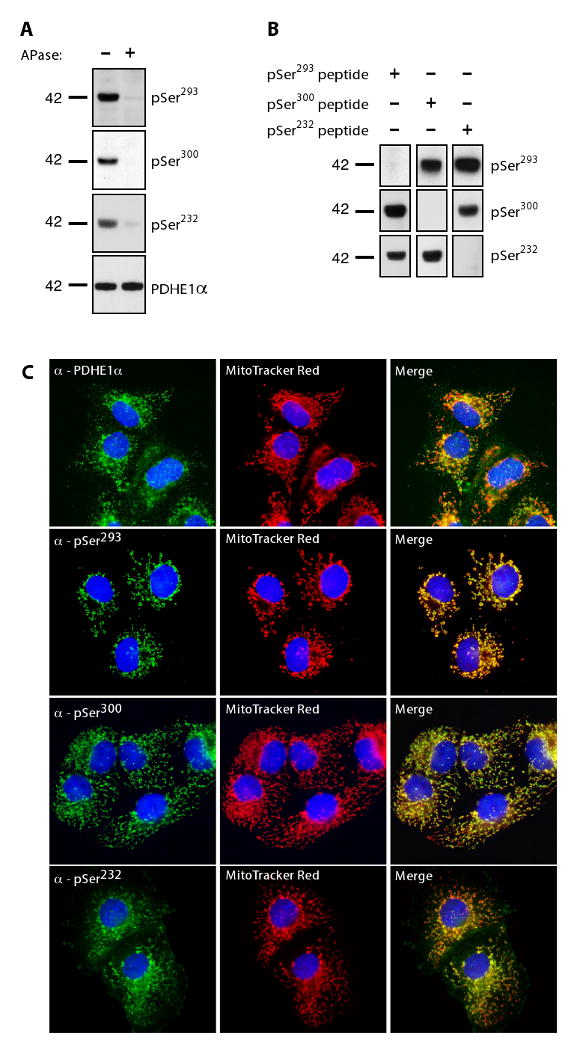
Phospho-PDHE1α antibody specificity. (A) 100 μg of crude rat kidney mitochondria were incubated with or without calf intestinal alkaline phosphatase (APase) for 30 min at 37°C. 5 μg of protein was separated by SDS-PAGE, and immunoblotted with the indicated antibodies. (B) Phospho-antibodies were incubated with a 20-fold excess of each of the phosphopeptides used for immunization. Crude mouse heart mitochondria were isolated; 5 μg of protein was separated by SDS-PAGE and immunoblotted with indicated phospho-antibodies (blots shown were exposed equally). (C) Immunofluorescence analysis of phosphorylation of endogenous PDHE1α in COS7 cells (green). In addition, cells were stained with the mitochondrial marker, MitoTracker Red, and the nuclear marker, DAPI (blue). Co-localization is represented in the merged images in yellow.
After determining that our antibodies were phospho-specific, we wanted to establish their sequence specificity. Therefore, we preincubated our phospho-antibodies with each of the three phospho-peptides used to generate antisera. Western blot analysis of mitochondria from mouse heart tissue using the anti-pSer293 antibody blocked with its corresponding phospho-peptide showed a complete loss of signal. In contrast, this antibody was not blocked with phospho-peptides against Ser300 and Ser232 of PDHE1α (Fig. 2B). Similar results were seen using the phospho-antibodies against pSer300 and pSer232 (Fig. 2B).
We also examined the use of our phospho-antibodies in assessing the phosphorylation levels of PDHE1α in fixed tissue. We first analyzed the monkey kidney fibroblast cell line COS7 using immunofluorescence analysis. As expected, PDHE1α immunofluorescence colocalized with MitoTracker Red staining (Fig. 2C). Similar results were observed with each phospho-antibody directed against each site, demonstrating that all three sites of phosphorylation can be detected in fixed cells. Surprisingly, pSer232 also showed a strong signal in a cell type (kidney) where PDK1, the only kinase known to phosphorylate Ser232, has not been reported to be expressed [25]. Similar results were seen in other cultured cell lines (e.g. C2C12, HeLa; data not shown). Taken together, these data demonstrate that we have developed phospho-site specific antibodies against each of the three known regulatory sites of PDHE1α and that phosphorylation levels can be detected using a variety of applications in a wide variety of cell types and tissues.
Monitoring PDH activity through pharmacological inhibition of PDKs
Classically, the PDK specific inhibitor dichloroacetic acid (DCA) is utilized to modulate PDH activity. Inhibition of the PDKs leads to the dephosphorylation of PDHE1α by the PDPs, thereby activating the complex [26]. DCA is a pyruvate analog currently under investigation for use in the treatment of genetic mitochondrial diseases, as well as cancer, for its ability to reverse the Warburg effect by shifting metabolism from glycolysis back to glucose oxidation [27; 28]. Structurally, DCA was shown to cause a conformational change in both the nucleotide and lipoyl binding pockets [29]. To confirm that our phospho-antibodies accurately reflected the phosphorylation state, and thus the activity, of the PDH, we treated mitochondrial extracts from mouse liver tissue with DCA. Samples treated with DCA showed a dramatic decrease in signal for each of the three phosphorylation sites (Fig. 3A). However, no change was observed in levels of total PDHE1α. Similar results were seen using rat kidney tissues extracts (data not shown). HEK293A cells treated with DCA showed a decrease in phosphorylation after 10 minutes (Fig. 3B). In immunocytochemical analysis, COS-7 cells treated with DCA and probed with antibodies against pSer293, pSer300, and pSer232 had a dramatic loss of phospho-signal at all three sites, with no change in total PDHE1α (Fig. 3C). These data demonstrate that the phospho-specific antibodies against PDHE1α can identify changes in phosphorylation following treatment with the PDK specific inhibitor DCA and are, therefore, sensitive enough to detect alteration in the phosphorylation status of PDH.
Fig. 3.
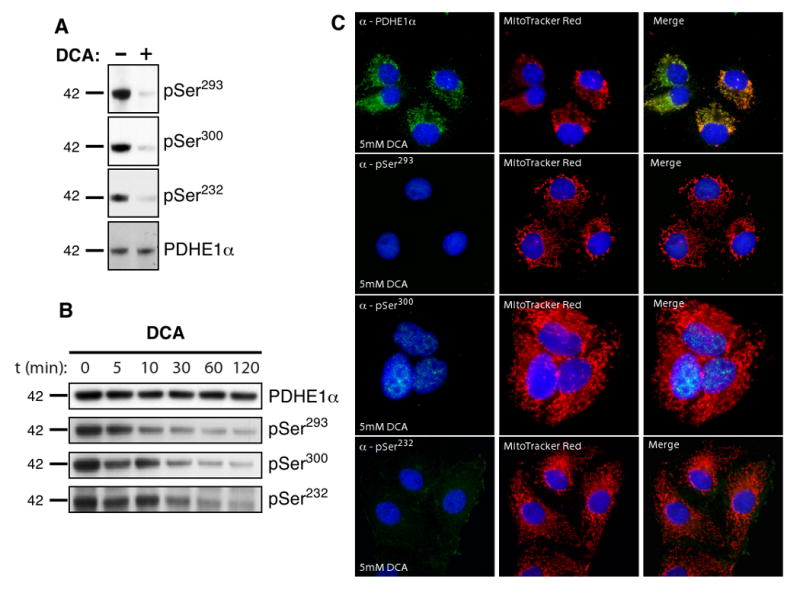
Detecting changes in PDC activity. (A) Mouse liver extracts were treated with 5mM DCA for 4 hours and then analyzed by Western blot with the indicated phospho-antibodies and compared to total PDHE1α. (B) Phosphorylation status of PDHE1α in HEK293A cells over time treated with 5mM DCA. (C) Analysis of PDH phosphorylation by immunofluorescence in COS-7 cells treated with 5 mM DCA for 2 hours. Images were taken following equal exposure to those taken in Fig. 2C.
Differential distribution of phosphorylation across tissues
The development of phospho-site specific antibodies against PDHE1α gave us the unique opportunity to explore the distribution of phosphorylation across different tissues and, therefore, assess the regulation of PDH in those tissues. Protein levels were normalized using PDHE1α as an internal control and compared to levels of phosphorylation at each of the three sites in mouse tissues (Table I). There was a wide distribution of phosphorylation not only between tissues but also between the different phospho-sites. Phosphorylation at Ser293 was detected in all tissues examined with the highest degree of phosphorylation seen in white adipose tissue (fat). Although no comparisons can be made between antibodies because of the variability in antibody affinity, we can draw semi-quantitative data between tissues from antibodies against individual phosphorylation sites. There was widespread distribution of pSer300 across all tissues except testis. The ratio of pSer300 to total PDHE1α shows the highest amount of phosphorylation in lung, minimal amounts in brain and skeletal muscle, and moderate levels in the rest of the tissues except testis. The distribution of pSer232 was also widespread across tissues with no detectable level in testis. Collectively, these data demonstrate that phosphorylation at site Ser232 is more widespread then has been previously reported [25], and our antibodies can be used to monitor individual phosphorylation sites on PDHE1α in a wide variety of tissues.
Assessing mitochondrial dysfunction in a patient suffering from Leigh's Syndrome
To demonstrate that our phospho-antibodies could be used as a diagnostic tool to assess the degree of PDH activity in patients with metabolic diseases, we obtained human primary fibroblasts from a previously uncharacterized patient (P1) suffering from Leigh's syndrome (LS). LS is a progressive neurological disorder with incidence of 1:77,000 births caused by mutations found in subunits of the PDC as well as the electron transport chain [30; 31]. We isolated cDNA from cultured human primary foreskin fibroblasts (HPFF) from the LS patient as well as from control cells. Analysis of P1's PDHE1α sequence revealed a missense mutation in nucleotide 412 (C → T) that results in an amino acid change from leucine to phenylalanine (L138F) (Fig. 4A). in vitro PDH activity assays of a patient with the same mutation showed a severe reduction in activity [32]. To examine the activity of the PDH utilizing our phospho-antibodies, we screened HPFF from P1 by immunofluorescence analysis. The P1 HPFF showed no staining of pSer293 as compared to wildtype cells (Fig. 4B), nor could we detect levels of pSer300 or pSer232 (see Supplementary Fig. 1 online). Similar results were seen with total PDHE1α, suggesting total levels of the protein were down in P1 (Fig. 4C). In contrast, levels of the PDC E2/E3 binding protein appeared normal under immunofluorescence (Fig. 4D). To confirm these results, we analyzed protein levels by Western blot and found that total levels of PDHE1α were dramatically reduced, while levels of pSer293 were barely detectable even during long exposures (Fig. 4E). When assessed by Western blot, levels of the E2/E3 binding protein were similar to the control (Fig. 4E). This suggests that total protein levels of PDHE1α in P1 were decreased overall and that the L138F mutation most likely causes instability in the protein leading to its degradation. We determined that the total amount of PDHE1α protein observed in the Leigh's patient was decreased, and as expected low levels of phosphorylation were also observed. We believe that our results suggest that these antibodies will be useful in the rapid assessment of metabolic disorders involving PDC.
Fig. 4.
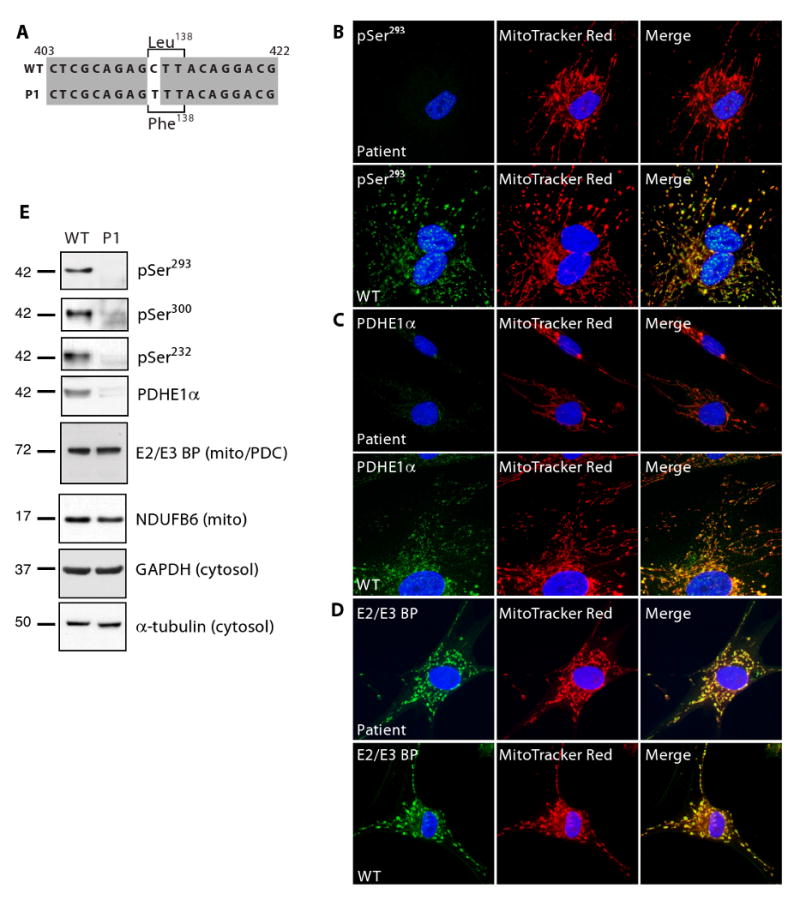
Use of phospho-antibodies to detect changes in phosphorylation in a patient diagnosed with Leigh's Syndrome. (A) Sequence analysis of PDHE1α cDNA isolated from cultured HPFF from the LS patient and a control revealed a missense mutation (C412T) resulting in an amino acid substitution of L138F. Fluorescent images of cultured human primary fibroblasts from the LS patient versus the control (WT) probed with antibodies against pSer293 (B), total PDHE1α (C), and the PDC E2/E3 binding protein (D); equal exposure between samples. (E) 20 μg of protein was isolated from cultured fibroblasts, separated by SDS-PAGE and analyzed by Western blot for levels of pSer293, pSer300, pSer232, total PDHE1α, and PDC E2/E3 binding protein. α-Tubulin and GAPDH were used as cytosolic loading controls, while NDUFB6 was used as an indicator of mitochondrial content.
Pharmacological modulation of PDHE1α phosphorylation
The central role of PDC in regulating glucose and fat metabolism makes it an attractive target for the development of specific small molecules able to modulate PDH activity. As a proof-of-principle, we treated human primary foreskin fibroblasts with a variety of compounds known to modulate mitochondrial metabolism and signaling pathways including oligomycin (an inhibitor of ATP synthase), rotenone (Complex I inhibitor), carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP; membrane uncoupler), doxorubicin (cancer chemotherapeutic that inhibits topoisomerase II), Bay 11-7082 (inhibitor of IκBα phosphorylation), UV light (activator of stress pathways), and observed changes in PDH phosphorylation status (Fig. 5). The molecular basis for the apparent increase in molecular weight of PDHE1α detected by α-pSer300 after treatment with BAY-11-7082 and UV light is currently unknown. These data demonstrate both increases and decreases in phosphorylation at different sites in response to treatment, establishing these antibodies as metabolic reporters of altered PDH phosphorylation status. Our antibodies should be useful for monitoring PDH activity in screens for small molecules which may modulate its phosphorylation status.
Fig. 5.
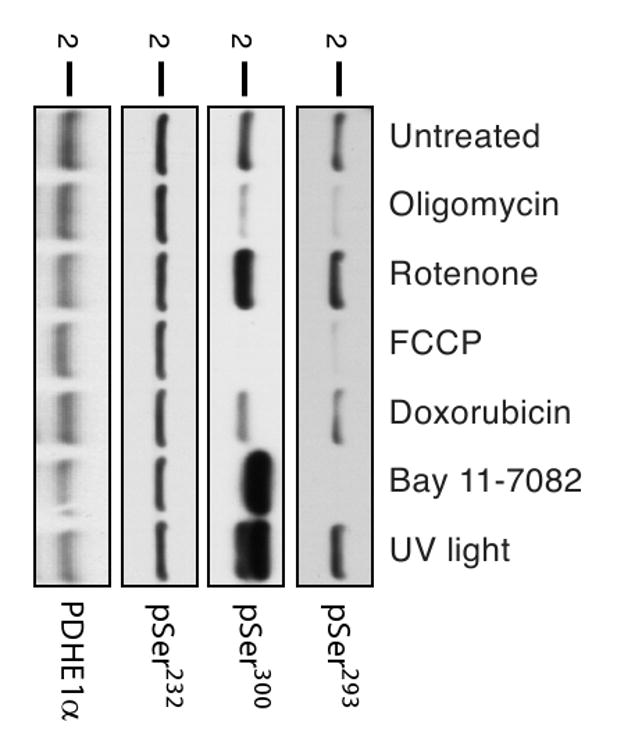
Pharmacological manipulation of the phospho-status of PDHE1α. Human primary fibroblasts were either left untreated or treated for 1 hour with various known inhibitors of oxidative phosphorylation or signaling pathways: oligomycin (2 μg/ml), rotenone (10 μM), FCCP (1 μM), doxorubicin (8 μM), BAY 11-7082 (20 μM), and UV light (4,000 μjoules). Whole cell lysates were prepared and 20 μg of protein were separated by SDS-PAGE. Western blot analysis was performed with indicated antibodies.
Discussion
Regulation of mitochondrial function by reversible phosphorylation is a rapidly emerging theme in cellular signaling. In the present study, we have developed the first complete set of phospho-specific antibodies against the PDH as a method for monitoring its regulation by phosphorylation. Previously, the phosphorylation state could be assessed through metabolic labeling, which was then correlated with cumbersome PDH assays involving either the capture of radiolabeled CO2 or prior purification of the complex. We demonstrate that these antibodies are phospho-site specific for the phosphorylation sites found on PDHE1α from a number of different organisms (human, mouse, monkey, rat). In addition, these antibodies are sensitive to changes in PDC activity as seen following treatment of cells and tissue extracts with DCA. Using these tools, we were able to assess the phospho-regulation of PDH in multiple mouse tissues and demonstrate that these sites appear to be differentially regulated in the extent of phosphorylation across tissues. Lastly, we show that these antibodies can be used to assess PDH activity in diseased patients.
The central role of PDC in regulating glucose and fat metabolism makes it an attractive target for the development of specific small molecules which could modulate PDH activity in diseases such as diabetes or cancer. We screened a variety of small molecules that are known affectors of mitochondrial function and signal transduction pathways. Although we do not currently understand the modulation of the different phosphorylation sites in response to these treatments, we believe this is a proof-of-principal for the utility of these antibodies in small molecule screens where monitoring PDH regulation is desired. In addition, these antibodies should prove useful in assessing cellular metabolism under different physiological conditions such as cell division and differentiation.
Regulation of mammalian PDC by reversible phosphorylation has been shown to occur at three sites on the PDHE1α subunit. Mutational analysis has shown that in vitro all four PDK isoforms have differential activity against Ser293 and Ser300; however, Ser232 phosphorylation is PDK1 dependent [6; 13; 33]. Interestingly, PDK1 is reported to be highly expressed in heart tissue, with moderate levels in skeletal muscle, pancreas, and liver [25]. Yet, our results suggest that phosphorylation of Ser232 is more widespread across tissues than previously thought with the highest amount of phosphorylation actually occurring in adipose tissue. Moreover, the phosphorylation of Ser232 is unexpectedly low in liver and pancreas where PDK1 is expressed at moderate levels. The production of phospho-specific antibodies has allowed, for the first time, examination of in vivo levels of relative phosphorylation at each of the three sites. Furthermore, tissue distribution of the relative levels of the phosphorylation of Ser232 did not directly correlate with high levels of phosphorylation at Ser293 or Ser300 (Table 1). Taken together, these data suggest that Ser232 phosphorylation may not be strictly PDK1 dependent. It is possible that PDK1 expression may be more widespread than previously thought. Alternatively, in vitro phosphorylation assays may have lacked an unknown component necessary for phosphorylation by other PDK isoforms, or perhaps there is another, as yet unidentified, kinase that phosphorylates Ser232. In addition, the levels of phosphorylation at Ser293 and Ser300 did not correlate across tissues, demonstrating that in vitro kinase activity against PDHE1α may not directly compare with in vivo PDC phosphorylation [6; 13; 16]. Therefore, the development of these tools to study reversible phosphorylation allows us to suggest that in vivo regulation of PDHE1α phosphorylation in different tissues may be more complex than in vitro kinase activity assays were able to demonstrate.
Table 1.
Distribution of PDHE1α phosphorylation levels across tissues.
| Tissue | Site 1 pSer293 |
Site 2 pSer300 |
Site 3 pSer232 |
|---|---|---|---|
| Brain | +a | + | + |
| Heart | ++ | +++ | +++ |
| Sk. Muscle | ++ | + | + |
| Pancreas | +++ | ++ | ++ |
| Liver | + | ++ | ++ |
| Kidney | + | +++ | + |
| Testis | ++ | NDb | ND |
| Spleen | + | ++ | ++ |
| Lung | + | ++++ | +++ |
| Fat (white adipose) | ++++ | +++ | ++++ |
Scale indicates levels of phosphorylation lowest (+) to highest (++++).
Not Detected
Levels of phosphorylation were calculated as a ratio of phospho-PDHE1α:PDHE1α by densitometry.
The development of phospho-specific antibodies are an invaluable tool for studying signal transduction pathways in response to extra-cellular stimuli such as insulin, or in a disease state as found in several forms of cancer [34; 35; 36]. Although regulation of the mammalian PDH by reversible phosphorylation has been studied since the late 1960's, the development of phospho-specific antibodies to PDHE1α has raised intriguing questions concerning the regulation of PDH in vivo [37; 38]. In addition, these tools should prove invaluable for future studies of PDH activity given the technically difficult activity assays currently performed using [1-C14]pyruvate which require significant amounts of tissue. The specificity and versatility of these antibodies will allow for monitoring PDH activity by immunohistochemistry (allowing for the assay of biopsy samples), as well as Western blot analysis permitting detection from small amounts of tissue. The central role of PDC in energy production makes it an attractive target for assessing cellular metabolism and these antibodies should allow for easy monitoring of PDH regulation by phosphorylation.
Supplementary Material
Use of phospho-antibodies to detect changes in phosphorylation in a patient diagnosed with Leigh's Syndrome. Fluorescent images of cultured human primary fibroblasts from the LS patient versus the control (WT) probed with antibodies against (A) pSer300 and (B) pSer232 (equal exposure between samples).
Acknowledgments
The authors thank Dr. Kevin Harvey at EMD Biosciences for his help in antibody production and purification. This work was supported by National Institutes of Health (NIH) Grant 18849 (to J.E.D.) and DK054441 (to A.N.M.), and by the NIH Pharmacology Training Grant NIH 2 T32 GM07752-25 (M.J.R.). R.K.N. was supported by the UCSD Foundation Chrisitini Fund, the Lennox Foundation, Hailey's Wish Foundation, and the Wright Family Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Zhou ZH, McCarthy DB, O'Connor CM, Reed LJ, Stoops JK. The remarkable structural and functional organization of the eukaryotic pyruvate dehydrogenase complexes. Proc Natl Acad Sci U S A. 2001;98:14802–7. doi: 10.1073/pnas.011597698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Harris RA, Bowker-Kinley MM, Huang B, Wu P. Regulation of the activity of the pyruvate dehydrogenase complex. Adv Enzyme Regul. 2002;42:249–59. doi: 10.1016/s0065-2571(01)00061-9. [DOI] [PubMed] [Google Scholar]
- 3.McFate T, Mohyeldin A, Lu H, Thakar J, Henriques J, Halim ND, Wu H, Schell MJ, Tsang TM, Teahan O, Zhou S, Califano JA, Jeoung NH, Harris RA, Verma A. Pyruvate dehydrogenase complex activity controls metabolic and malignant phenotype in cancer cells. J Biol Chem. 2008;283:22700–8. doi: 10.1074/jbc.M801765200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jeoung NH, Harris RA. Pyruvate dehydrogenase kinase-4 deficiency lowers blood glucose and improves glucose tolerance in diet-induced obese mice. Am J Physiol Endocrinol Metab. 2008;295:E46–54. doi: 10.1152/ajpendo.00536.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Linn TC, Pettit FH, Reed LJ. Alpha-keto acid dehydrogenase complexes. X. Regulation of the activity of the pyruvate dehydrogenase complex from beef kidney mitochondria by phosphorylation and dephosphorylation. Proc Natl Acad Sci U S A. 1969;62:234–41. doi: 10.1073/pnas.62.1.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Korotchkina LG, Patel MS. Site specificity of four pyruvate dehydrogenase kinase isoenzymes toward the three phosphorylation sites of human pyruvate dehydrogenase. J Biol Chem. 2001;276:37223–9. doi: 10.1074/jbc.M103069200. [DOI] [PubMed] [Google Scholar]
- 7.Teague WM, Pettit FH, Wu TL, Silberman SR, Reed LJ. Purification and properties of pyruvate dehydrogenase phosphatase from bovine heart and kidney. Biochemistry. 1982;21:5585–92. doi: 10.1021/bi00265a031. [DOI] [PubMed] [Google Scholar]
- 8.Huang B, Gudi R, Wu P, Harris RA, Hamilton J, Popov KM. Isoenzymes of pyruvate dehydrogenase phosphatase. DNA-derived amino acid sequences, expression, and regulation. J Biol Chem. 1998;273:17680–8. doi: 10.1074/jbc.273.28.17680. [DOI] [PubMed] [Google Scholar]
- 9.Pratt ML, Roche TE. Mechanism of pyruvate inhibition of kidney pyruvate dehydrogenasea kinase and synergistic inhibition by pyruvate and ADP. J Biol Chem. 1979;254:7191–6. [PubMed] [Google Scholar]
- 10.Hansford RG. Studies on the effects of coenzyme A-SH: acetyl coenzyme A, nicotinamide adenine dinucleotide: reduced nicotinamide adenine dinucleotide, and adenosine diphosphate: adenosine triphosphate ratios on the interconversion of active and inactive pyruvate dehydrogenase in isolated rat heart mitochondria. J Biol Chem. 1976;251:5483–9. [PubMed] [Google Scholar]
- 11.Cate RL, Roche TE. A unifying mechanism for stimulation of mammalian pyruvate dehydrogenase(a) kinase by reduced nicotinamide adenine dinucleotide, dihydrolipoamide, acetyl coenzyme A, or pyruvate. J Biol Chem. 1978;253:496–503. [PubMed] [Google Scholar]
- 12.Korotchkina LG, Patel MS. Mutagenesis studies of the phosphorylation sites of recombinant human pyruvate dehydrogenase. Site-specific regulation. J Biol Chem. 1995;270:14297–304. doi: 10.1074/jbc.270.24.14297. [DOI] [PubMed] [Google Scholar]
- 13.Kolobova E, Tuganova A, Boulatnikov I, Popov KM. Regulation of pyruvate dehydrogenase activity through phosphorylation at multiple sites. Biochem J. 2001;358:69–77. doi: 10.1042/0264-6021:3580069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yeaman SJ, Hutcheson ET, Roche TE, Pettit FH, Brown JR, Reed LJ, Watson DC, Dixon GH. Sites of phosphorylation on pyruvate dehydrogenase from bovine kidney and heart. Biochemistry. 1978;17:2364–70. doi: 10.1021/bi00605a017. [DOI] [PubMed] [Google Scholar]
- 15.Karpova T, Danchuk S, Kolobova E, Popov KM. Characterization of the isozymes of pyruvate dehydrogenase phosphatase: implications for the regulation of pyruvate dehydrogenase activity. Biochim Biophys Acta. 2003;1652:126–35. doi: 10.1016/j.bbapap.2003.08.010. [DOI] [PubMed] [Google Scholar]
- 16.Bowker-Kinley MM, Davis WI, Wu P, Harris RA, Popov KM. Evidence for existence of tissue-specific regulation of the mammalian pyruvate dehydrogenase complex. Biochem J. 1998;329(Pt 1):191–6. doi: 10.1042/bj3290191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang B, Wu P, Popov KM, Harris RA. Starvation and diabetes reduce the amount of pyruvate dehydrogenase phosphatase in rat heart and kidney. Diabetes. 2003;52:1371–6. doi: 10.2337/diabetes.52.6.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu P, Blair PV, Sato J, Jaskiewicz J, Popov KM, Harris RA. Starvation increases the amount of pyruvate dehydrogenase kinase in several mammalian tissues. Arch Biochem Biophys. 2000;381:1–7. doi: 10.1006/abbi.2000.1946. [DOI] [PubMed] [Google Scholar]
- 19.Wu P, Inskeep K, Bowker-Kinley MM, Popov KM, Harris RA. Mechanism responsible for inactivation of skeletal muscle pyruvate dehydrogenase complex in starvation and diabetes. Diabetes. 1999;48:1593–9. doi: 10.2337/diabetes.48.8.1593. [DOI] [PubMed] [Google Scholar]
- 20.Jeoung NH, Wu P, Joshi MA, Jaskiewicz J, Bock CB, Depaoli-Roach AA, Harris RA. Role of pyruvate dehydrogenase kinase isoenzyme 4 (PDHK4) in glucose homoeostasis during starvation. Biochem J. 2006;397:417–25. doi: 10.1042/BJ20060125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lapidus RG, Sokolove PM. Spermine inhibition of the permeability transition of isolated rat liver mitochondria: an investigation of mechanism. Arch Biochem Biophys. 1993;306:246–53. doi: 10.1006/abbi.1993.1507. [DOI] [PubMed] [Google Scholar]
- 22.Piergiacomi VA, Palacios A, Catala A. Vitamin A inhibits lipoperoxidation ascorbate-Fe++ dependent of rat kidney microsomes and mitochondria. Mol Cell Biochem. 1996;165:121–5. doi: 10.1007/BF00229473. [DOI] [PubMed] [Google Scholar]
- 23.Johnson D, Lardy H. Isolation of Liver or Kidney Mitochondria. Methods Enzymol. 1967;10:94–96. [Google Scholar]
- 24.Rardin MJ, Wiley SE, Murphy AN, Pagliarini DJ, Dixon JE. Dual specificity phosphatases 18 and 21 target to opposing sides of the mitochondrial inner membrane. J Biol Chem. 2008;283:15440–50. doi: 10.1074/jbc.M709547200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Patel MS, Korotchkina LG. Regulation of the pyruvate dehydrogenase complex. Biochem Soc Trans. 2006;34:217–22. doi: 10.1042/BST20060217. [DOI] [PubMed] [Google Scholar]
- 26.Whitehouse S, Cooper RH, Randle PJ. Mechanism of activation of pyruvate dehydrogenase by dichloroacetate and other halogenated carboxylic acids. Biochem J. 1974;141:761–74. doi: 10.1042/bj1410761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Michelakis ED, Webster L, Mackey JR. Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer. Br J Cancer. 2008;99:989–94. doi: 10.1038/sj.bjc.6604554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stacpoole PW, Kurtz TL, Han Z, Langaee T. Role of dichloroacetate in the treatment of genetic mitochondrial diseases. Adv Drug Deliv Rev. 2008;60:1478–1487. doi: 10.1016/j.addr.2008.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kato M, Li J, Chuang JL, Chuang DT. Distinct structural mechanisms for inhibition of pyruvate dehydrogenase kinase isoforms by AZD7545, dichloroacetate, and radicicol. Structure. 2007;15:992–1004. doi: 10.1016/j.str.2007.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maj MC, Cameron JM, Robinson BH. Pyruvate dehydrogenase phosphatase deficiency: orphan disease or an under-diagnosed condition? Mol Cell Endocrinol. 2006;249:1–9. doi: 10.1016/j.mce.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 31.Rahman S, Blok RB, Dahl HH, Danks DM, Kirby DM, Chow CW, Christodoulou J, Thorburn DR. Leigh syndrome: clinical features and biochemical and DNA abnormalities. Ann Neurol. 1996;39:343–51. doi: 10.1002/ana.410390311. [DOI] [PubMed] [Google Scholar]
- 32.Cameron JM, Levandovskiy V, Mackay N, Tein I, Robinson BH. Deficiency of pyruvate dehydrogenase caused by novel and known mutations in the E1alpha subunit. Am J Med Genet A. 2004;131:59–66. doi: 10.1002/ajmg.a.30287. [DOI] [PubMed] [Google Scholar]
- 33.Sale GJ, Randle PJ. Occupancy of phosphorylation sites in pyruvate dehydrogenase phosphate complex in rat heart in vivo. Relation to proportion of inactive complex and rate of re-activation by phosphatase. Biochem J. 1982;206:221–9. doi: 10.1042/bj2060221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chu EC, Tarnawski AS. PTEN regulatory functions in tumor suppression and cell biology. Med Sci Monit. 2004;10:RA235–41. [PubMed] [Google Scholar]
- 35.Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, Puc J, Miliaresis C, Rodgers L, McCombie R, Bigner SH, Giovanella BC, Ittmann M, Tycko B, Hibshoosh H, Wigler MH, Parsons R. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275:1943–7. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- 36.Kurose K, Zhou XP, Araki T, Cannistra SA, Maher ER, Eng C. Frequent loss of PTEN expression is linked to elevated phosphorylated Akt levels, but not associated with p27 and cyclin D1 expression, in primary epithelial ovarian carcinomas. Am J Pathol. 2001;158:2097–106. doi: 10.1016/S0002-9440(10)64681-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pilegaard H, Birk JB, Sacchetti M, Mourtzakis M, Hardie DG, Stewart G, Neufer PD, Saltin B, van Hall G, Wojtaszewski JF. PDH-E1alpha dephosphorylation and activation in human skeletal muscle during exercise: effect of intralipid infusion. Diabetes. 2006;55:3020–7. doi: 10.2337/db06-0152. [DOI] [PubMed] [Google Scholar]
- 38.Samavati L, Lee I, Mathes I, Lottspeich F, Huttemann M. Tumor necrosis factor alpha inhibits oxidative phosphorylation through tyrosine phosphorylation at subunit I of cytochrome c oxidase. J Biol Chem. 2008;283:21134–44. doi: 10.1074/jbc.M801954200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Use of phospho-antibodies to detect changes in phosphorylation in a patient diagnosed with Leigh's Syndrome. Fluorescent images of cultured human primary fibroblasts from the LS patient versus the control (WT) probed with antibodies against (A) pSer300 and (B) pSer232 (equal exposure between samples).