

SUMMARY
Insulin and leptin play complementary roles in regulating the consumption, uptake, oxidation and storage of nutrients. Chronic consumption of diets that contain a high proportion of calories from saturated fat induces a progressive deterioration in function of both hormones. Certain rat lines and strains of mice are particularly sensitive to the obesogenic and diabetogenic effects of high fat diets, and have been used extensively to study the developmental progression of insulin and leptin resistance in relation to the increasing adiposity that is characteristic of their response to these diets. Some aspects of the diminished efficacy of each hormone are secondary to increased adiposity but a consensus is emerging to support the view that direct effects of dietary components or their metabolites, independent of the resulting obesity, play important roles in development of insulin and leptin resistance. In this minireview, we will examine the implications of crosstalk between leptin and insulin signaling during the development of diet-induced obesity, emphasizing potential interactions between pathways that occur among target sites, and exploring how these interactions may influence the progression of obesity and diabetes.
Introduction
After the original description of variable diabetogenic effects of high fat diets among mouse strains by Surwit et al. [1; 2], the fat-sensitive C57BL/6J mouse emerged as a key model to study the developmental pathology of the obese/diabetic syndrome that develops after chronic consumption of high fat (HF) diets. Various rat lines respond similarly, and studies contrasting rats or mice on LF versus HF diets have illustrated the global and tissue-specific changes that occur during the time frame when defects in leptin and insulin sensitivity first become apparent. However, the question of whether the initial stages of glucose intolerance are the product of a uniform progression of IR across tissues or the result of a sequential but punctuated progression of IR among tissues remains open. HF diets also increase circulating leptin levels and promote the development of leptin resistance. Potential mechanisms of diet-induced leptin resistance include decreased access of leptin to hypothalamic targets, diminished responsiveness of hypothalamic neurons to leptin, modified translation of leptin-dependent signals in downstream target tissues, or some combination thereof [3]. The complex progression of events involved in the development of insulin and leptin resistance represents a significant impediment to understanding the underlying cause and effect mechanisms within each pathway. Given the overlap within the respective signaling pathways of each hormone, crosstalk between the pathways is likely to become significant and contribute to the progression of diet-induced obesity and diabetes.
I. Developmental Progression of Insulin and Leptin Resistance During Diet-Induced Obesity
The strong relationship between obesity and diabetes is well documented but the underlying mechanisms which link development of obesity to initiation and progression of insulin and leptin resistance are less well understood. This is in part due to the chronic nature of both conditions and the associated progressive deterioration in function of multiple, highly integrated organ systems which function in concert to maintain glucose and energy homeostasis. Because of the temporal nature of the process, it has been difficult to identify the tissue sites and events which constitute the initial steps of IR and link deterioration in insulin sensitivity to progression of diet-induced obesity and diabetes. For example, without careful design, it is difficult to establish that observed changes in tissue responses and function are not secondary to the initial events which alter insulin sensitivity, and thus a consequence rather than an underlying cause of IR.
The original experiments of Surwit et al. [1; 2] involved weaning male C57BL/6J mice onto a diet high in saturated fat (58 kcal%) derived primarily from coconut oil, and the mice developed fasting hyperglycemia and hyperinsulinemia after 6 wks on the diet. Variations of this initial design have used different amounts (58 vs 45 kcal%) and sources of fat (lard vs coconut oil) in the diet, as well as age when the diet was first introduced. The modified designs have also been used with various rat lines, and, in general, the responses have been similar in the sense that the animals increased fat deposition and developed some degree of IR, depending on length of time on the diet. Relatively few of the many studies in rats and mice have used longitudinal approaches coupled with in vivo measurements (hyperinsulinemic-euglycemic clamps) to evaluate whole body insulin sensitivity and identify the tissue sites where IR first appears. However, the consensus emerging from limited studies with young animals is that the very high fat (55–60 kcal%) diets produce a rapid deterioration (3 wks) in whole body insulin sensitivity that coincides with a simultaneous reduction in insulin-dependent glucose uptake in peripheral tissues coupled with a failure of insulin to suppress hepatic glucose production [4–6]. The response pattern is common to both rats and mice on the very high fat diet formulation (55–60 kcal%) and supports the view that whole body IR is the product of a rapid, uniform progression of IR among peripheral tissues [4–7]. In contrast, a different outcome is obtained when rats or mice are given HF diets with a lower proportion of calories from fat (ie 45 kcal% versus 55–60 kcal%). Consuming the 45 kcal% HF diet for 1–5 wks resulted in loss of suppression of hepatic glucose production by insulin without any change in insulin-sensitive glucose uptake in peripheral tissue [8; 9]. These findings argue that loss of insulin sensitivity in liver is responsible for the initial deterioration in whole body insulin sensitivity. Mice on the 45 kcal% HF diet for longer periods (12–14 wks) develop IR in peripheral tissues [10], but this outcome may be a reflection of longer term consumption of the 45 kcal% HF diet and indicative of later stages in the progression. It seems likely that the reported differences among studies are due to a combination of differences in amount, source, and saturation of dietary fat, as well as length of exposure and age of animals when the test diets were first introduced.
Leptin is a key component of the neuroendocrine circuitry that regulates food intake and energy utilization, and its absence in ob/ob mice produces a complex metabolic syndrome characterized by hyperphagia and lowered rates of energy expenditure [11–13]. However, diet-induced obesity is not associated with an absence of leptin, as circulating leptin is actually increased in proportion to the increase in body adiposity. The failure of this hyperleptinemia to produce any compensatory decrease in food intake or body weight was initially viewed as evidence of leptin resistance. The existence of high fat diet-induced leptin resistance has since been directly demonstrated as a failure of exogenous leptin to suppress food intake or induce weight loss [14–16]. Chronic consumption of HF diets promotes the development of leptin resistance as defined by both criteria, but understanding the temporal sequence of events involved in the development of diet-induced leptin resistance is complicated by the anatomical and functional complexity of the leptin sensing, signaling, and effector systems. Leptin functions to maintain body weight homeostasis by modulating appetite control centers in the hypothalamus and energy utilization in part by increasing sympathetic outflow to peripheral tissues [17–20]. Sympathetic outflow to adipose tissue enhances fat oxidation by coordinated transcriptional activation of genes favoring substrate oxidation [21; 22]. Thus in the broadest sense, leptin acts to regulate adipose tissue reserves through a coordinated suppression of energy intake and stimulation of fat oxidation. The consensus is that leptin initiates these signals by crossing the blood brain barrier and binding to receptors on hypothalamic NPY/AGRP and POMC neurons, which activate STAT-3 through a Jak2-dependent pathway [23–26]. Leptin receptors are expressed on neurons located primarily but not exclusively within the arcuate nuclei, but emerging evidence suggests that proper integration of the complex behavioral and metabolic responses to leptin requires input from these additional populations of leptin responsive neurons [3; 27].
Studies examining leptin sensitivity have typically measured the ability of exogenous leptin, given either peripherally or centrally, to activate hypothalamic STAT-3, reduce food intake, reduce body weight, or increase sympathetic nervous system (SNS) activity. In C57BL/6J mice weaned onto a 45 kcal% HF diet, Van Heek et al. [28] reported that the anorectic response to leptin was lost after 16 d on the HF diet. Using essentially the same experimental design, El Haschimi et al. [29] found that the 45 kcal% HF diet did not alter the ability of peripherally injected leptin to activate hypothalamic STAT3. In contrast, C57BL/6J mice weaned onto a higher percentage of dietary fat (58 kcal% vs 45 kcal%) became fully resistant to both the anorectic and metabolic effects of peripherally injected leptin after 4 wks on the diet [30]. However, the responses to leptin were not compromised when it was administered centrally [30], suggesting compromised access to hypothalamic target neurons as a basis for resistance to peripherally injected leptin [31; 32]. Although working with a different strain of mice (AKR/J), Van Heek et al [28] proposed a similar mechanism to explain resistance to peripheral leptin after 8 wks on a HF diet. Returning to the C57BL/6J model, longer term consumption (15 wks) of the 45 kcal% HF diet fully compromised the ability of peripherally injected leptin to activate STAT3, but also reduced the capacity of hypothalamic neurons to respond to centrally injected leptin [29]. The diminished response to central leptin argues that the HF diet produced a second defect compromising cellular signaling at some point upstream of STAT3 [29].
An important component of leptin’s metabolic effects is mediated through SNS stimulation of fat oxidation in adipose tissue [21; 22]. Therefore, the observed decrease in β-adrenergic receptor function in adipose tissue after long term (16 wks) consumption of the 58 kcal% HF diet [33] could diminish efficacy of SNS input and limit translation of this component of leptin’s actions. However, in recent studies with C57BL/6J mice consuming the 58 kcal% HF diet for 8 wks [30], centrally administered leptin activated hypothalamic STAT3, reduced food consumption, and increased UCP1 mRNA in BAT. The latter response indicates that leptin signaling to brown adipose tissue was intact, but in the same mice leptin was unable to increase UCP1 expression in retroperitoneal WAT but was able to decrease leptin mRNA. Given that both WAT responses are mediated by SNS input [17; 34], the intactness of the latter response indicates that SNS-dependent β-adrenergic activation was not compromised in retroperitoneal WAT. Additional studies showed that the defect in UCP1 induction induced by the HF diet was downstream of β-adrenergic receptor activation in retroperitoneal WAT [30]. Together, these studies illustrate how HF diets can induce leptin resistance in part by compromising the ability of peripheral target tissues to translate input from central effector systems (SNS) that are regulated by hypothalamic leptin signaling.
Lastly, recent work has shown that the time course of leptin signaling after peripheral injection of leptin is not synchronized within all regions of hypothalamic neurons expressing leptin receptors, whereas centrally administered leptin produces simultaneous leptin-dependent signaling among these neuronal populations [27; 35; 36]. This finding argues that peripherally injected leptin does not reach all leptin responsive neurons at the same time, and it suggests that a population of ARC neurons expressing leptin receptors may extend processes across the BBB to directly contact the circulation [27]. These findings also raise the interesting possibility that this population of leptin responsive neurons may be more subject to desensitization by high leptin levels or other circulating factors associated with chronic consumption of HF diets [32].
II. Mechanisms of Diet-Induced Insulin Resistance
A growing consensus supports the view that accumulation of lipid in tissues not designed for storage is a key initial step in the progression towards insulin resistance (IR) [37; 38]. Lipodystrophy is characterized by defective storage of triglyceride in adipose tissue, and the resulting oversupply of lipids to metabolic organs produces ectopic fat accumulation and the development of IR [39]. Chronic consumption of HF diets results in a similar oversupply of lipids to metabolic organs that is temporally related to periods of rapid weight gain, and associated with decreased insulin sensitivity and impaired glucose homeostasis [38; 40]. Muoio et al [41; 42] have presented evidence that the oversupply of lipids to mitochondria produces a mismatch between TCA cycle flux and fat oxidation that results in depletion of organic acid intermediates, incomplete fatty acid oxidation, and accumulation of lipotoxic short chain fatty acylcarnitines. Although the underlying mechanisms remain unclear, the accumulation of these lipotoxic metabolites compromises insulin signaling and is associated with the onset of IR in skeletal muscle of HF fed Wistar rats. A reversal of the mismatched metabolic flux could be obtained by increasing mitochondrial oxidative capacity or limiting entry of fatty acids into mitochondria [41–43], both of which restored complete fatty acid oxidation and insulin sensivity. It remains to be seen whether a similar mechanism is involved in compromising insulin sensitivity in other peripheral tissues such as the liver, where IR first appears with the 45 kcal% HF diet.
III. Cellular Mechanisms of Diet-Induced Leptin Resistance
While the existence of leptin resistance is widely accepted, the physiological and cellular mechanisms that underlie this resistance are less clear. This is due in part to the complexity of the system itself, and the fact that leptin resistance could occur at multiple points within this multifaceted cascade. It has been argued that high fat diets lead to reduced leptin transport across the blood brain barrier (BBB) [31], consistent with work demonstrating reduced sensitivity to peripherally administered leptin prior to loss of central leptin sensitivity [28; 30]. Nevertheless, the cellular mechanisms of leptin transport and its disruption in obesity are not fully resolved [31; 32; 44; 45], and some neurons within the hypothalamic arcuate nucleus may extend projections beyond the BBB to directly sample hormones in circulation [27]. It has also been established that HF diets compromise leptin signaling within target neurons [29; 46–48]. This resistance is manifest as a loss of leptin-dependent suppression of food intake and impaired activation of specific signaling pathways, namely Stat3 and PI3K [3; 47]. Leptin resistance could also occur indirectly, with loss of sensitivity in various effector pathways mediating downstream effects of the leptin receptor. For instance, some have detected a HF-induced loss of sensitivity to melanocortins, although others fail to detect this resistance [49–52]. Lastly, there is evidence for selective leptin resistance based on physiological endpoint, a loss of leptin-dependent suppression of food intake despite a maintenance of leptin-dependent stimulation of sympathetic outflow to peripheral tissues [30; 53; 54]. In summary, these examples illustrate the challenges that are inherent to understanding the origins and mechanisms of leptin resistance.
In assessing potential causes of leptin resistance, perhaps the most progress has been made in defining molecular mediators of cellular leptin resistance. This work has identified two molecules which inhibit leptin receptor signaling; suppressor of cytokine signaling 3 (Socs3) and protein tyrosine phosphatase 1B (PTP1B). Socs3 is one of a family of proteins produced in response to cytokine signaling which functions as an intracellular negative feedback signal [55]. Socs3 expression is induced by Stat3 signaling and binds to the leptin receptor, blocking Stat3 activation [56; 57]. Mice bearing genetic modifications which delete Socs3 or inhibit its ability to bind the leptin receptor exhibit reduced food intake and body weight and are resistant to diet induced obesity [58–60].
Protein tyrosine phosphatase 1B (PTP1B) is similarly implicated in leptin resistance due to its ability to limit the magnitude and duration of leptin receptor signaling. PTP1B binds to and dephosphorylates Janus Kinase 2 (Jak2), the initial tyrosine kinase activated by the leptin receptor [61; 62]. Overexpression of PTP1B in vitro dampens signaling from the leptin receptor [63], and mice genetically deficient for PTP1B are lean and hypersensitive to leptin [61; 62]. Neuron-specific deletion of PTP1B recapitulates the leptin hypersensitivity and resistance to diet-induced obesity [64], and local inhibition of PTP1B via pharmacological or adenoviral approaches enhances the effects of central leptin injection [65; 66]. Thus these data indicate that PTP1B, like Socs3, tonically inhibits signaling from the leptin receptor.
Interestingly, Socs3 and PTP1B are also associated with regulation of insulin signaling. PTP1B limits insulin signaling by binding to and dephosphorylating the insulin receptor, and mice with global PTP1B deficiency are hypersensitive to insulin [67]. Similarly, Socs3 inhibits insulin receptor signaling in vitro and in vivo [68; 69]. Given that leptin and insulin regulate a common set of hypothalamic neurons, the overlapping function of these two inhibitors has the potential to produce significant crosstalk between the signaling pathways, particularly during diet induced obesity as hyperinsulinemia and hyperleptinemia develop [70]. Thus PTP1B and/or Socs3 could underlie the combined loss of sensitivity to leptin and insulin that are observed in obesity.
IV. Direct and Indirect Effects of High Fat Diets on Leptin Signaling
Available evidence suggests that high fat diets induce leptin resistance via both direct and indirect mechanisms. Direct mechanisms refer to the ability of high fat diets to induce leptin resistance in the absence of obesity, presumably via direct effects of dietary fat or its metabolites on leptin-sensitive neurons. In contrast, indirect mechanisms reflect an induction of leptin resistance that is secondary to obesity and the associated hyperleptinemia and are thus not dependent on the presence of excess dietary fat.
Several lines of evidence indicate that HF diets can induce hypothalamic leptin (and insulin) resistance in the absence of either obesity or elevated circulating leptin. A rapid diet-induced leptin and insulin resistance has been detected prior to any change in body adiposity [71], while others have provided evidence for central insulin resistance in HF-fed animals, even when obesity is prevented via caloric restriction [72]. Obesity prone rat lines exhibit reduced central leptin and insulin sensitivity prior to becoming obese on HF diets [73–75], suggesting that some degree of basal resistance contributes to their propensity to become obese. We recently examined leptin signaling in ob/ob mice to determine whether HF diets could modify leptin sensitivity in the absence of leptin. Notwithstanding the high initial sensitivity of ob/ob mice to exogenous leptin (see below), consumption of the HF diet for 14 days attenuated leptin’s anorectic effects, indicating that leptin resistance can be induced in the absence of leptin and hyperleptinemia [76]. Lastly, these findings are also consistent with evidence that hypothalamic neurons are sensitive to and respond directly to fatty acids and other lipid metabolites ([77; 78] and see section below).
In addition to direct effects of dietary fat on leptin signaling efficacy, the evidence also supports mechanisms that are indirect and secondary to the development of obesity and chronic hyperleptinemia. Nearly all settings of leptin resistance are associated with a chronic elevation of circulating leptin, and it appears that chronic hyperleptinemia inhibits leptin sensitivity via the down regulation of leptin receptors and the stimulation of negative feedback molecules such as Socs3 and PTP1B [56; 68; 79]. Additional evidence supporting a role for hyperleptinemia stems from genetic models. Despite their massive obesity, leptin-deficient ob/ob mice are highly sensitive to leptin [80]. Similarly, the leptin resistance of agouti viable yellow (Ay) mice was diminished when leptin was removed by crossing the Ay mutation onto the ob/ob background [81]. Together these observations suggest that a component of the leptin resistance of AY mice is induced by hyperleptinemia. Chronic exposure to high leptin induces a form of leptin-induced leptin resistance [48; 82], and animals chronically exposed to elevated leptin levels exhibit increased weight gain on a HF diet [83; 84]. Together, these findings argue that this leptin-induced leptin resistance alters the regulation of energy homeostasis and increases the risk for obesity.
Viewed collectively, these findings indicate that leptin resistance is both a cause and a consequence of obesity [85]. Loss of leptin signaling clearly predisposes to weight gain, while interventions which enhance leptin signaling protect against diet induced obesity. These observations, coupled with evidence for HF-induced, obesity independent reductions in leptin sensitivity, support a causative role for leptin resistance in the development of obesity. However, a consequence of the expanding adipose mass of obesity is a chronic increase in circulating leptin, which acts on the hypothalamus to further dampen leptin signaling.
V. Direct Effects of Fatty Acids on Hypothalamic Neurons
In addition to responding to circulating hormones such as leptin and insulin, hypothalamic neurons also possess a nutrient sensing system that responds to circulating lipids and provides regulatory inputs into systemic glucose homeostasis. Increasing hypothalamic long chain fatty acids (LCFA-CoA) via either direct ICV administration of oleic acid [86] or inhibition of hypothalamic carnitine palmitoyltransferase-1 (CPT-1) [87; 88] causes a marked decrease in hepatic glucose production via suppression of glycogenolysis. This hypothalamic lipid sensing mechanism appears to require the opening of hypothalamic KATP channels and an intact vagus nerve [77]. While LCFAs suppress hepatic glucose output via the hypothalamus, they increase hepatic glucose production through a direct effect on the liver [89]. Thus, this opposing effect may serve an autoregulatory function to balance the centrally mediated inhibitory effects of LCFA. In addition to LCFAs, there is also evidence that triglycerides may act in the brain to reduce leptin transport across the blood brain barrier [32]. Lastly, given the substantial overlap between leptin and insulin signaling and the emerging consensus that dysregulation of lipid metabolism is at the core of peripheral insulin resistance, it appears likely that similar mechanisms occur in the brain and could contribute to central leptin resistance. Taken together, these data indicate that fatty acids, triglycerides and lipid molecules act locally in the brain to influence leptin transport, food intake and peripheral glucose metabolism, and as such provide additional mechanisms through which the chronic consumption of a HF diet might be compromising body weight and glucose homeostasis.
VI. Implications of Crosstalk between Leptin and Insulin During Development of Diet-Induced Obesity
Postweaning consumption of diets rich in saturated fatty acids results in the progressive development of insulin resistance, leptin resistance, and obesity in C57BL/6J mice. Maintenance of glucose and energy homeostasis requires the proper integration of complex communication and effector systems, and the collective evidence shows that chronic consumption of HF diets progressively compromises the function of essential components and steps within these systems. The difficulty in understanding the underlying sequence of events in this overall process is underscored by the fact that the progression is also dependent on the source and amount of fat in the diet. Therefore, the following comments are an attempt to summarize the key early responses of C57BL/6J mice to HF diets, suggesting potential points of interaction between insulin and leptin signaling that may contribute to or accentuate the progressive dysregulation of glucose and energy homeostasis.
Consumption of the very high fat (58 kcal%) diet formulation for 2–3 wks after weaning produces a rapid, uniform increase in IR among peripheral tissues [4; 5]. The IR, detected by hyperinsulinemic-euglycemic clamps, is a reflection of both compromised insulin-dependent glucose uptake in peripheral tissues and compromised insulin-dependent suppression of hepatic glucose production [4]. After 2 wks on the 58 kcal% HF diet, C57BL/6J mice are significantly fatter than LF-fed mice [90] and by 4 wks are completely resistant to peripherally injected leptin [30; 35]. Mice on the 58 kcal% HF diet retain sensitivity to centrally administered leptin at this time point, increasing hypothalamic STAT3 phosphorylation, reducing food intake and increasing UCP1 expression in BAT after ICV injection of leptin [30]. Together these data indicate that the 58 kcal% HF formulation produces a rapid and almost simultaneous increase in adiposity and deterioration of signaling through insulin and leptin. Moreover, it appears that beyond the 4 wk time point, it becomes difficult to argue that observed changes are not secondary to signaling defects developed earlier in the progression.
The progression of both IR and leptin resistance is significantly slower with the 45 kcal% versus the 58 kcal% HF diet. After 4 wks on the 45 kcal% formulation, activation of hypothalamic STAT3 by peripheral leptin was not diminished in C57BL/6J mice [29], and it was only after 15 wks on the HF diet that resistance to leptin was observed. Using the 45 kcal% HF diet, recent studies have shown that ambient temperature and rearing conditions also affect the progression of diet-induced leptin resistance [91–93]. However, it should be noted that Munzberg et al. [35] have proposed that leptin resistance may develop in a region-specific manner within the hypothalamus of diet-induced obese mice, with resistance developing initially within the arcuate nuclei. If different hypothalamic regions regulate different components of the leptin response, it could explain how the development of selective leptin resistance among subgroups of responsive nuclei could eliminate certain components of leptin’s actions while others are preserved [53].
IR develops sooner (1–3 wks) than leptin resistance in both rats [8; 9] and C57BL/6J mice [94] consuming the 45 kcal% HF diet. Using hyperinsulinemic-euglycemic clamps to measure in vivo insulin sensitivity, a modest decrease in glucose utilization is detected in C57BL/6J mice after 3 wks on the HF diet. This decrease in insulin sensitivity can be fully accounted for by failure of insulin to suppress hepatic glucose production, with no decrease in sensitivity of peripheral tissues to insulin. Hepatic IR becomes somewhat more severe between 3 and 8 wks on the 45 kcal% HF diet, but peripheral tissues retain full responsiveness to insulin during this period [94]. It is noteworthy that the increase in hepatic IR between 3 and 8 wks is sufficient to increase pancreatic insulin release such that fasting insulin levels are ~2 fold higher than levels in control mice on a LF diet. The establishment of hyperinsulinemia during a period when peripheral tissues retain full insulin sensitivity has important physiological implications. In addition to promoting glucose uptake in peripheral tissues, insulin also activates phosphodiesterase 3B [95–97], resulting in the degradation of cyclic AMP and the antagonism of glucagon signaling in the liver. If the liver was responsive to insulin during this period, the increase in insulin would have the corrective effect of suppressing hepatic glucose production. The hyperinsulinemia also could have important effects in adipose tissue, where the lipogenic and antilipolytic effects of insulin create conditions favoring fat deposition. We have shown that the time frame (3 to 8 wks) when hyperinsulinemia develops coincides with a significant increase in the rate of fat deposition. This may be a mechanism to compensate for the oversupply of lipids produced by the 45 kcal% HF diet, and effectively slow the progression of IR to peripheral tissues by preventing or delaying ectopic fat accumulation. If correct, this suggests that there may be a threshold of dietary fat that determines whether the early stage of IR is limited to the liver or extends to other peripheral tissues.
The development of hyperinsulinemia between 3 to 8 wks may also have important effects on the endocrine function of adipose tissue function. For example, previous studies have shown that insulin increases the release of leptin from adipose tissue [98] while cyclic AMP inhibits both leptin expression and release [17; 98]. In addition to promoting triglyceride accumulation, the higher insulin levels would favor increased leptin expression and release from adipose tissue during this period. Although beyond the scope of this minireview, it is also likely that other components of the adipocyte endocrine repertoire that are regulated by insulin and cyclic AMP would be similarly affected.
Lastly, leptin regulates SNS outflow to adipose tissue, which acts through cyclic AMP to mobilize triglyceride and transcriptionally remodel adipose tissue. However, the increased serum insulin levels would antagonize cyclic AMP-dependent signaling, effectively limiting the efficacy of leptin-dependent SNS input. Viewed collectively, the promotion of hepatic IR of sufficient magnitude to increase circulating insulin, but not induce IR in peripheral tissues, may be a critical characteristic of the response to the 45 kcal% version of the HF diet which promotes rapid fat deposition and the subsequent development of leptin resistance. It seems clear that detailed longitudinal studies at much earlier time points will be required to resolve how and where the signaling defects develop, where they interact, and how they impact the expansion of adipose tissue mass.
VII. Conclusions
It is well accepted that postweaning consumption of diets high in saturated fat produces obesity and metabolic dysfunction. The severity and developmental progression of this syndrome is dependent upon the amount of dietary fat (45–60 kcal%), the strain of mouse being studied, and the age at which exposure to the diet is initiated. Our theoretical model (Figure 1) is based on postweaning consumption of a 45 kcal% high fat diet by C57BL/6J mice, and proposes that the development of insulin resistance is progressive and tissue specific, with high-fat diets leading to hepatic insulin resistance prior to detectable changes in insulin sensitivity in peripheral tissues. The resulting increase in circulating insulin, in conjunction with retention of peripheral insulin sensitivity, produces an expansion of adipose mass and a concomitant increase in circulating leptin. This increase in leptin, though perhaps initially acting to limit body adiposity, leads to hypothalamic leptin resistance and a further dysregulation of energy balance and glucose homeostasis. With continued exposure to a high fat diet, the liver becomes progressively less responsive to insulin, vagal inhibition of hepatic glucose production is diminished, and the resulting oversupply of lipids to mitochondria of peripheral tissues produces a mismatch between TCA cycle flux and fat oxidation (Figure 1, Panel C). The result is incomplete fatty acid oxidation, accumulation of lipotoxic short chain fatty acylcarnitines, and peripheral insulin resistance. Adipose tissue in particular becomes resistant to inhibition of leptin release by SNS input, resulting in chronic hyperleptinemia and leptin resistance. Lastly, we recognize the possibility that excess dietary fat may also more directly induce hypothalamic leptin (and insulin) resistance, with this effect serving to exacerbate the initial deterioration peripheral insulin sensitivity and whole body energy homeostasis.
Figure 1. Interactions between insulin and leptin signaling in mice reared on low fat diets (Panel A), mice reared for 4–8 wks after weaning on 45 kcal% high fat diet (Panel B), and mice after chronic consumption (4–5 mo after weaning) of high fat diet (Panel C).
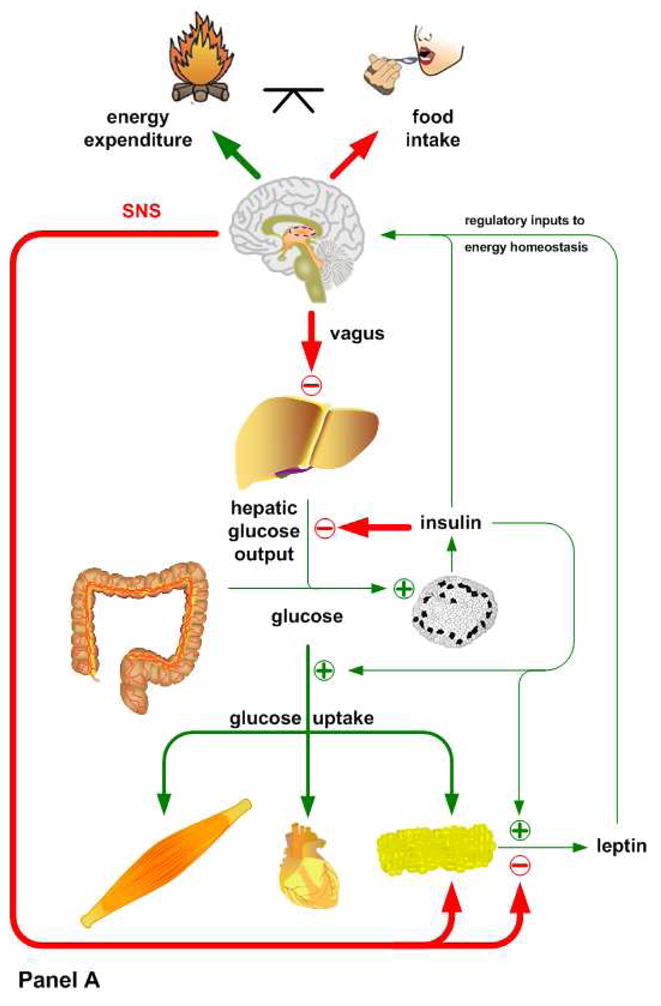
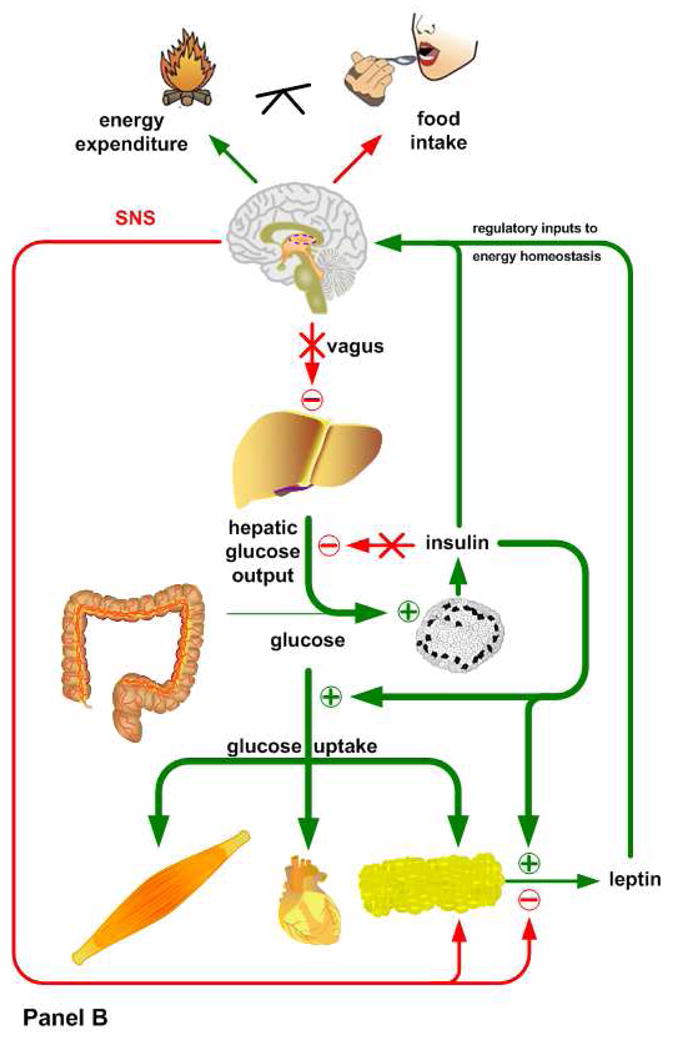
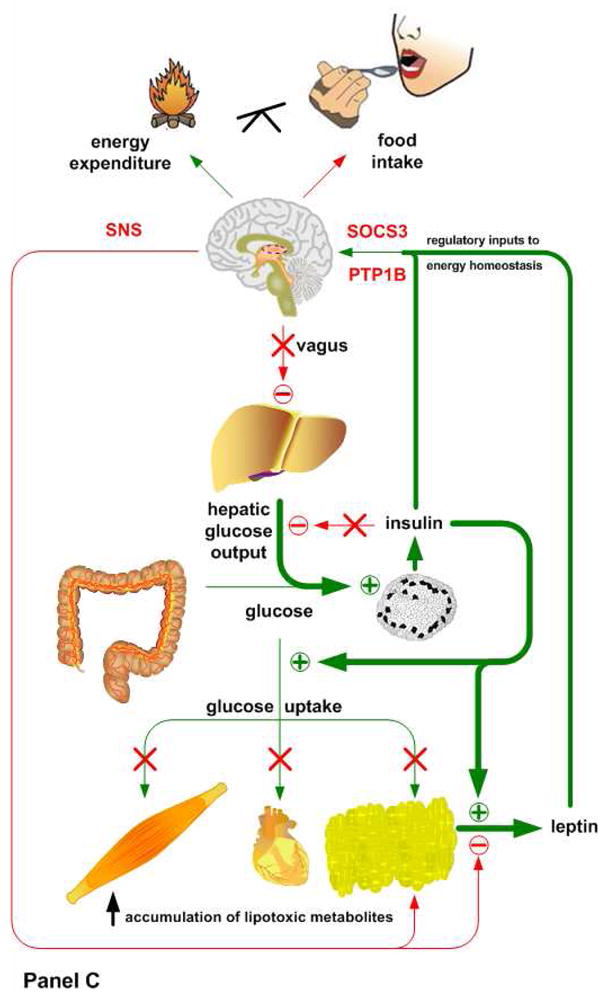
Animals placed on a low fat diet (Panel A) exhibit normal systemic insulin sensitivity. After a meal, insulin shuts down hepatic glucose production, via both direct and indirect pathways, and promotes glucose uptake in muscle and adipose tissue. The subsequent release of leptin from adipose tissue suppresses food intake and stimulates the sympathetic tonus, which ensures lipid mobilization and fatty acid oxidation during fasting. Mice fed a high fat diet for a short period of time (Panel B) display hepatic insulin resistance, despite preserved insulin sensitivity in muscle and adipose tissue. The loss of hepatic insulin sensitivity is most likely caused by the direct actions of high fat availability on gluconeogenesis or the hypothalamus, which would suppress hepatic glucose output under normal conditions via a vagal pathway. This period of selective hepatic insulin resistance is characterized by increased circulating glucose levels, hyperinsulinemia and a significant increase in fat deposition. When these mice are kept on a high fat diet for a longer period of time (Panel C), hepatic insulin resistance becomes more severe. Initially the peripheral tissues still maintain insulin sensitivity, but the exacerbated fat accumulation in adipose tissue and subsequent hyperleptinemia cause hypothalamic leptin resistance via the induction of SOCS3 and PTP1B. Basal hyperinsulinemia and chronic high fat availability have also been shown to promote a condition in which incomplete fatty acid oxidation and accumulation of lipotoxic short chain fatty acids occurs in peripheral tissues, ultimately leading to a complete loss of their insulin sensitivity.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Surwit RS, Seldin MF, Kuhn CM, Cochrane C, Feinglos MN. Control of expression of insulin resistance and hyperglycemia by different genetic factors in diabetic C57BL/6J mice. Diabetes. 1991;40:82–87. doi: 10.2337/diab.40.1.82. [DOI] [PubMed] [Google Scholar]
- 2.Surwit RS, Kuhn CM, Cochrane C, McCubbin JA, Feinglos MN. Diet-induced type II diabetes in C57BL/6J mice. Diabetes. 1988;37:1163–1167. doi: 10.2337/diab.37.9.1163. [DOI] [PubMed] [Google Scholar]
- 3.Myers MG, Cowley MA, Munzberg H. Mechanisms of leptin action and leptin resistance. Annu Rev Physiol. 2008;70:537–556. doi: 10.1146/annurev.physiol.70.113006.100707. [DOI] [PubMed] [Google Scholar]
- 4.Park SY, Cho YR, Kim HJ, Higashimori T, Danton C, Lee MK, Dey A, Rothermel B, Kim YB, Kalinowski A, Russell KS, Kim JK. Unraveling the temporal pattern of diet-induced insulin resistance in individual organs and cardiac dysfunction in C57BL/6 mice. Diabetes. 2005;54:3530–3540. doi: 10.2337/diabetes.54.12.3530. [DOI] [PubMed] [Google Scholar]
- 5.Storlien LH, James DE, Burleigh KM, Chisholm DJ, Kraegen EW. Fat feeding causes widespread in vivo insulin resistance, decreased energy expenditure, and obesity in rats. Am J Physiol. 1986;251:E576–E583. doi: 10.1152/ajpendo.1986.251.5.E576. [DOI] [PubMed] [Google Scholar]
- 6.Kraegen EW, James DE, Storlien LH, Burleigh KM, Chisholm DJ. In vivo insulin resistance in individual peripheral tissues of the high fat fed rat: assessment by euglycaemic clamp plus deoxyglucose administration. Diabetologia. 1986;29:192–198. doi: 10.1007/BF02427092. [DOI] [PubMed] [Google Scholar]
- 7.Oakes ND, Cooney GJ, Camilleri S, Chisholm DJ, Kraegen EW. Mechanisms of liver and muscle insulin resistance induced by chronic high-fat feeding. Diabetes. 1997;46:1768–1774. doi: 10.2337/diab.46.11.1768. [DOI] [PubMed] [Google Scholar]
- 8.Pagliassotti MJ, Horton TJ, Gayles EC, Koppenhafer TA, Rosenzweig TD, Hill JO. Reduced insulin suppression of glucose appearance is related to susceptibility to dietary obesity in rats. Am J Physiol Regul Integr Comp Physiol. 1997;272:R1264–R1270. doi: 10.1152/ajpregu.1997.272.4.R1264. [DOI] [PubMed] [Google Scholar]
- 9.Commerford SR, Bizeau ME, McRae H, Jampolis A, Thresher JS, Pagliassotti MJ. Hyperglycemia compensates for diet-induced insulin resistance in liver and skeletal muscle of rats. Am J Physiol Regul Integr Comp Physiol. 2001;281:R1380–R1389. doi: 10.1152/ajpregu.2001.281.5.R1380. [DOI] [PubMed] [Google Scholar]
- 10.Dhalla AK, Wong MY, Voshol PJ, Belardinelli L, Reaven GM. A1 adenosine receptor partial agonist lowers plasma FFA and improves insulin resistance induced by high-fat diet in rodents. Am J Physiol Endocrinol Metab. 2007;292:E1358–E1363. doi: 10.1152/ajpendo.00573.2006. [DOI] [PubMed] [Google Scholar]
- 11.Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- 12.Friedman JM. The function of leptin in nutrition, weight, and physiology. Nutr Rev. 2002;60:S1–14. doi: 10.1301/002966402320634878. [DOI] [PubMed] [Google Scholar]
- 13.Gettys TW. Leptin. In: William J, Lennarz MDL, editors. Encylopedia of Biological Chemistry. Elsevier; San Diego: 2004. pp. 541–545. [Google Scholar]
- 14.Maffei M, Halaas J, Ravussin E, Pratley RE, Lee GH, Zhang Y, Fei H, Kim S, Lallone R, Ranganathan S. Leptin levels in human and rodent: measurement of plasma leptin and ob RNA in obese and weight-reduced subjects. Nat Med. 1995;1:1155–1161. doi: 10.1038/nm1195-1155. [DOI] [PubMed] [Google Scholar]
- 15.Considine RV, Sinha MK, Heiman ML, Kriauciunas A, Stephens TW, Nyce MR, Ohannesian JP, Marco CC, McKee LJ, Bauer TL. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N Engl J Med. 1996;334:292–295. doi: 10.1056/NEJM199602013340503. [DOI] [PubMed] [Google Scholar]
- 16.Halaas JL, Boozer C, Blair-West J, Fidahusein N, Denton DA, Friedman JM. Physiological response to long-term peripheral and central leptin infusion in lean and obese mice. Proc Natl Acad Sci USA. 1997;94:8878–8883. doi: 10.1073/pnas.94.16.8878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Commins SP, Marsh DJ, Thomas SA, Watson PM, Padgett MA, Palmiter RD, Gettys TW. Norepinephrine is required for leptin effects on gene expression in brown and white adipose tissue. Endocrinology. 1999;140:4772–4776. doi: 10.1210/endo.140.10.7043. [DOI] [PubMed] [Google Scholar]
- 18.Collins S, Kuhn CM, Petro AE, Swick AG, Chrunyk BA, Surwit RS. Role of leptin in fat regulation. Nature. 1996;380:677. doi: 10.1038/380677a0. [DOI] [PubMed] [Google Scholar]
- 19.Haynes WG, Morgan DA, Walsh SA, Mark AL, Sivitz WI. Receptor-mediated regional sympathetic nerve activation by leptin. J Clin Invest. 1997;100:270–278. doi: 10.1172/JCI119532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scarpace PJ, Matheny M. Leptin induction of UCP1 gene expression is dependent on sympathetic innervation. Am J Physiol Endocrinol Metab. 1998;275:E259–E264. doi: 10.1152/ajpendo.1998.275.2.E259. [DOI] [PubMed] [Google Scholar]
- 21.Hwa JJ, Ghibaudi L, Compton D, Fawzi AB, Strader CD. Intracerebroventricular injection of leptin increases thermogenesis and mobilizes fat metabolism in ob/ob mice. Horm Metab Res. 1996;28:659–663. doi: 10.1055/s-2007-979873. [DOI] [PubMed] [Google Scholar]
- 22.Hwa JJ, Fawzi AB, Graziano MP, Ghibaudi L, Williams P, Van Heek M, Davis H, Rudinski M, Sybertz E, Strader CD. Leptin increases energy expenditure and selectively promotes fat metabolism in ob/ob mice. Am J Physiol Regul Integr Comp Physiol. 1997;272:R1204–R1209. doi: 10.1152/ajpregu.1997.272.4.R1204. [DOI] [PubMed] [Google Scholar]
- 23.Vaisse C, Halaas JL, Horvath CM, Darnell JE, Jr, Stoffel M, Friedman JM. Leptin activation of Stat3 in the hypothalamus of wildtype and ob/ob mice but not db/db mice. Nature Genet. 1996;14:95–97. doi: 10.1038/ng0996-95. [DOI] [PubMed] [Google Scholar]
- 24.Tartaglia LA, Dembski M, Weng X, Deng N, Culpepper J, Devos R, Richards GJ, Campfield LA, Clark FT, Deeds J, Muir C, Sanker S, Moriarty A, Moore KJ, Smutko JS, Mays GG, Woolf EA, Monroe CA, Tepper RI. Identification and expression cloning of a leptin receptor, OB-R. Cell. 1995;83:1263–1271. doi: 10.1016/0092-8674(95)90151-5. [DOI] [PubMed] [Google Scholar]
- 25.Cowley MA, Smart JL, Rubinstein M, Cerdan MG, Diano S, Horvath TL, Cone RD, Low MJ. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature. 2001;411:480–483. doi: 10.1038/35078085. [DOI] [PubMed] [Google Scholar]
- 26.Bjorbaek C, Uotani S, Da Silva B, Flier JS. Divergent signaling capacities of the long and short isoforms of the leptin receptor. J Biol Chem. 1997;272:32686–32695. doi: 10.1074/jbc.272.51.32686. [DOI] [PubMed] [Google Scholar]
- 27.Faouzi M, Leshan R, Bjornholm M, Hennessey T, Jones J, Munzberg H. Differential accessibility of circulating leptin to individual hypothalamic sites. Endocrinology. 2007;148:5414–5423. doi: 10.1210/en.2007-0655. [DOI] [PubMed] [Google Scholar]
- 28.Van Heek M, Compton DS, France CF, Tedesco RP, Fawzi AB, Graziano MP, Sybertz EJ, Strader CD, Davis HR., Jr Diet-induced obese mice develop peripheral, but not central, resistance to leptin. J Clin Invest. 1997;99:385–390. doi: 10.1172/JCI119171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.El-Haschimi K, Pierroz DD, Hileman S, Bjorbæk C, Flier JS. Two defects contribute to hypothalamic leptin resistance in mice with diet-induced obesity. J Clin Invest. 2000;105:1827–1832. doi: 10.1172/JCI9842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Prpic V, Watson PM, Frampton IC, Sabol MA, Jezek GE, Gettys TW. Differential mechanisms and development of leptin resistance in A/J vs C57BL/6J mice during diet-induced obesity. Endocrinology. 2003;144:1155–1163. doi: 10.1210/en.2002-220835. [DOI] [PubMed] [Google Scholar]
- 31.Banks WA, Farrell CL. Impaired transport of leptin across the blood-brain barrier in obesity is acquired and reversible. Am J Physiol Endocrinol Metab. 2003;285:E10–E15. doi: 10.1152/ajpendo.00468.2002. [DOI] [PubMed] [Google Scholar]
- 32.Banks WA, Coon AB, Robinson SM, Moinuddin A, Shultz JM, Nakaoke R, Morley JE. Triglycerides induce leptin resistance at the blood-brain barrier. Diabetes. 2004;53:1253–1260. doi: 10.2337/diabetes.53.5.1253. [DOI] [PubMed] [Google Scholar]
- 33.Collins S, Daniel KW, Petro AE, Surwit RS. Strain-specific response to β3-adrenergic receptor agonist treatment of diet-induced obesity in mice. Endocrinology. 1997;138:405–413. doi: 10.1210/endo.138.1.4829. [DOI] [PubMed] [Google Scholar]
- 34.Commins SP, Watson PM, Levin N, Beiler RJ, Gettys TW. Central leptin regulates the UCP1 and ob genes in brown and white adipose tissue via different β-adrenoceptor subtypes. J Biol Chem. 2000;275:33059–33067. doi: 10.1074/jbc.M006328200. [DOI] [PubMed] [Google Scholar]
- 35.Munzberg H, Flier JS, Bjorbaek C. Region-specific leptin resistance within the hypothalamus of diet-induced obese mice. Endocrinology. 2004;145:4880–4889. doi: 10.1210/en.2004-0726. [DOI] [PubMed] [Google Scholar]
- 36.Munzberg H. Differential leptin access into the brain - A hierarchical organization of hypothalamic leptin target sites? Physiol Behav. 2008;94:664–669. doi: 10.1016/j.physbeh.2008.04.020. [DOI] [PubMed] [Google Scholar]
- 37.Rasouli N, Molavi B, Elbein SC, Kern PA. Ectopic fat accumulation and metabolic syndrome. Diabetes Obes Metab. 2007;9:1–10. doi: 10.1111/j.1463-1326.2006.00590.x. [DOI] [PubMed] [Google Scholar]
- 38.Pagliassotti MJ, Prach PA, Koppenhafer TA, Pan DA. Changes in insulin action, triglycerides, and lipid composition during sucrose feeding in rats. Am J Physiol Regul Integr Comp Physiol. 1996;271:R1319–R1326. doi: 10.1152/ajpregu.1996.271.5.R1319. [DOI] [PubMed] [Google Scholar]
- 39.Kim JK, Gavrilova O, Chen Y, Reitman ML, Shulman GI. Mechanism of insulin resistance in A-ZIP/F-1 fatless mice. J Biol Chem. 2000;275:8456–8460. doi: 10.1074/jbc.275.12.8456. [DOI] [PubMed] [Google Scholar]
- 40.Yu C, Chen Y, Cline GW, Zhang D, Zong H, Wang Y, Bergeron R, Kim JK, Cushman SW, Cooney GJ, Atcheson B, White MF, Kraegen EW, Shulman GI. Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. J Biol Chem. 2002;277:50230–50236. doi: 10.1074/jbc.M200958200. [DOI] [PubMed] [Google Scholar]
- 41.An J, Muoio DM, Shiota M, Fujimoto Y, Cline GW, Shulman GI, Koves TR, Stevens R, Millington D, Newgard CB. Hepatic expression of malonyl-CoA decarboxylase reverses muscle, liver and whole-animal insulin resistance. Nat Med. 2004;10:268–274. doi: 10.1038/nm995. [DOI] [PubMed] [Google Scholar]
- 42.Koves TR, Ussher JR, Noland RC, Slentz D, Mosedale M, Ilkayeva O, Bain J, Stevens R, Dyck JR, Newgard CB, Lopaschuk GD, Muoio DM. Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab. 2008;7:45–56. doi: 10.1016/j.cmet.2007.10.013. [DOI] [PubMed] [Google Scholar]
- 43.Koves TR, Li P, An J, Akimoto T, Slentz D, Ilkayeva O, Dohm GL, Yan Z, Newgard CB, Muoio DM. PPARgamma coactivator-1alpha -mediated metabolic remodeling of skeletal myocytes mimics exercise training and reverses lipid-induced mitochondrial inefficiency. J Biol Chem. 2005;280:33588–33598. doi: 10.1074/jbc.M507621200. [DOI] [PubMed] [Google Scholar]
- 44.Kastin AJ, Pan W. Dynamic regulation of leptin entry into brain by the blood-brain barrier. Regul Pept. 2000;92:37–43. doi: 10.1016/s0167-0115(00)00147-6. [DOI] [PubMed] [Google Scholar]
- 45.Ayala JE, Bracy DP, McGuinness OP, Wasserman DH. Considerations in the design of hyperinsulinemic-euglycemic clamps in the conscious mouse. Diabetes. 2006;55:390–397. doi: 10.2337/diabetes.55.02.06.db05-0686. [DOI] [PubMed] [Google Scholar]
- 46.Enriori PJ, Evans AE, Sinnayah P, Jobst EE, Tonelli-Lemos L, Billes SK, Glavas MM, Grayson BE, Perello M, Nillni EA, Grove KL, Cowley MA. Diet-induced obesity causes severe but reversible leptin resistance in arcuate melanocortin neurons. Cell Metab. 2007;5:181–194. doi: 10.1016/j.cmet.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 47.Metlakunta AS, Sahu M, Sahu A. Hypothalamic phosphatidylinositol 3-kinase pathway of leptin signaling is impaired during the development of diet-induced obesity in FVB/N mice. Endocrinology. 2008;149:1121–1128. doi: 10.1210/en.2007-1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sahu A, Metlakunta AS. Hypothalamic phosphatidylinositol 3-kinase-phosphodiesterase 3B-cyclic AMP pathway of leptin signalling is impaired following chronic central leptin infusion. J Neuroendocrinol. 2005;17:720–726. doi: 10.1111/j.1365-2826.2005.01362.x. [DOI] [PubMed] [Google Scholar]
- 49.Chandler PC, Viana JB, Oswald KD, Wauford PK, Boggiano MM. Feeding response to melanocortin agonist predicts preference for and obesity from a high-fat diet. Physiol Behav. 2005;85:221–230. doi: 10.1016/j.physbeh.2005.04.011. [DOI] [PubMed] [Google Scholar]
- 50.Clegg DJ, Benoit SC, Air EL, Jackman A, Tso P, D’Alessio D, Woods SC, Seeley RJ. Increased dietary fat attenuates the anorexic effects of intracerebroventricular injections of MTII. Endocrinology. 2003;144:2941–2946. doi: 10.1210/en.2002-0218. [DOI] [PubMed] [Google Scholar]
- 51.Lu HQ, Buison A, Jen KLC, Dunbar JC. Leptin resistance in obesity is characterized by decreased sensitivity to proopiomelanocortin products. Peptides. 2000;21:1479–1485. doi: 10.1016/s0196-9781(00)00301-6. [DOI] [PubMed] [Google Scholar]
- 52.Pierroz DD, Ziotopoulou M, Ungsunan L, Moschos S, Flier JS, Mantzoros CS. Effects of acute and chronic administration of the melanocortin agonist MTII in mice with diet-induced obesity. Diabetes. 2002;51:1337–1345. doi: 10.2337/diabetes.51.5.1337. [DOI] [PubMed] [Google Scholar]
- 53.Carreia ML, Haynes WG, Rahmouni K, Morgan DA, Sivitz WI, Mark AL. The concept of selective leptin resistance: evidence from agouti yellow obese mice. Diabetes. 2002;51:439–442. doi: 10.2337/diabetes.51.2.439. [DOI] [PubMed] [Google Scholar]
- 54.Rahmouni K, Morgan DA, Morgan GM, Mark AL, Haynes WG. Role of selective leptin resistance in diet-induced obesity hypertension. Diabetes. 2005;54:2012–2018. doi: 10.2337/diabetes.54.7.2012. [DOI] [PubMed] [Google Scholar]
- 55.Howard JK, Flier JS. Attenuation of leptin and insulin signaling by SOCS proteins. Trends Endocrinol Metab. 2006;17:365–371. doi: 10.1016/j.tem.2006.09.007. [DOI] [PubMed] [Google Scholar]
- 56.Bjorbaek C, Elmquist JK, Frantz JD, Shoelson SE, Flier JS. Identification of SOCS-3 as a potential mediator of central leptin resistance. Mol Cell. 1999;1:819–825. doi: 10.1016/s1097-2765(00)80062-3. [DOI] [PubMed] [Google Scholar]
- 57.Bjorbaek C, El-Haschimi K, Frantz JD, Flier JS. The role of SOCS-3 in leptin signaling and leptin resistance. J Biol Chem. 1999;274:30059–30065. doi: 10.1074/jbc.274.42.30059. [DOI] [PubMed] [Google Scholar]
- 58.Bjornholm M, Munzberg H, Leshan RL, Villanueva EC, Bates SH, Louis GW, Jones JC. Ishida-Takahashi R., Bjorbaek C., Myers M.G., Jr., Mice lacking inhibitory leptin receptor signals are lean with normal endocrine function. J Clin Invest. 2007;117:1354–1360. doi: 10.1172/JCI30688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Howard JK, Cave BJ, Oksanen LJ, Tzameli I, Bjorbaek C, Flier JS. Enhanced leptin sensitivity and attenuation of diet-induced obesity in mice with haploinsufficiency of Socs3. Nat Med. 2004;10:734–738. doi: 10.1038/nm1072. [DOI] [PubMed] [Google Scholar]
- 60.Mori H, Hanada R, Hanada T, Aki D, Mashima R, Nishinakamura H, Torisu T, Chien KR, Yasukawa H, Yoshimura A. Socs3 deficiency in the brain elevates leptin sensitivity and confers resistance to diet-induced obesity. Nat Med. 2004;10:739–743. doi: 10.1038/nm1071. [DOI] [PubMed] [Google Scholar]
- 61.Zabolotny JM, Bence-Hanulec KK, Stricker-Krongrad A, Haj F, Wang Y, Minokoshi Y, Kim YB, Elmquist JK. Tartaglia L.A., Kahn B.B., Neel B.G., PTP1B regulates leptin signal transduction in vivo. Dev Cell. 2002;2:489–495. doi: 10.1016/s1534-5807(02)00148-x. [DOI] [PubMed] [Google Scholar]
- 62.Cheng A, Uetani N, Simoncic PD, Chaubey VP, Lee-Loy A, McGlade CJ, Kennedy BP, Tremblay ML. Attenuation of leptin action and regulation of obesity by protein tyrosine phosphatase 1B. Dev Cell. 2002;2:497–503. doi: 10.1016/s1534-5807(02)00149-1. [DOI] [PubMed] [Google Scholar]
- 63.Kaszubska W, Falls HD, Schaefer VG, Haasch D, Frost L, Hessler P, Kroeger PE, White DW, Jirousek MR, Trevillyan JM. Protein tyrosine phosphatase 1B negatively regulates leptin signaling in a hypothalamic cell line. Mol Cell Endocrinol. 2002;195:109–118. doi: 10.1016/s0303-7207(02)00178-8. [DOI] [PubMed] [Google Scholar]
- 64.Bence KK, Delibegovic M, Xue B, Gorgun CZ, Hotamisligil GS, Neel BG, Kahn BB. Neuronal PTP1B regulates body weight, adiposity and leptin action. Nat Med. 2006;12:917–924. doi: 10.1038/nm1435. [DOI] [PubMed] [Google Scholar]
- 65.Morrison CD, White CL, Wang Z, Lee SY, Lawrence DS, Cefalu WT, Zhang ZY, Gettys TW. Increased hypothalamic protein tyrosine phosphatase 1B contributes to leptin resistance with age. Endocrinology. 2007;148:433–440. doi: 10.1210/en.2006-0672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Picardi PK, Calegari VC, Prada PO, Moraes JC, Araujo E, Marcondes MC, Ueno M, Carvalheira JB, Velloso LA, Saad MJ. Reduction of hypothalamic protein tyrosine phosphatase improves insulin and leptin resistance in diet-induced obese rats. Endocrinology. 2008;149:3870–3880. doi: 10.1210/en.2007-1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Elchebly M, Payette P, Michaliszyn E, Cromlish W, Collins S, Loy AL, Normandin D, Cheng A, Himms-Hagen J, Chan CC, Ramachandran C, Gresser MJ, Tremblay ML, Kennedy BP. Increased insulin sensitivity and obesity resistance in mice lacking the protein tyrosine phosphatase-1B gene. Science. 1999;283:1544–1548. doi: 10.1126/science.283.5407.1544. [DOI] [PubMed] [Google Scholar]
- 68.Emanuelli B, Peraldi P, Filloux C, Sawka-Verhelhe D, Hilton DJ, Van Obberghen E. SOCS-3 is an insulin-induced negative regulator of insulin signaling. J Biol Chem. 2000;275:15985–15991. doi: 10.1074/jbc.275.21.15985. [DOI] [PubMed] [Google Scholar]
- 69.Shi H, Tzameli I, Bjorbaek C, Flier JS. Suppressor of cytokine signaling 3 is a physiological regulator of adipocyte insulin signaling. J Biol Chem. 2004;279:34733–34740. doi: 10.1074/jbc.M403886200. [DOI] [PubMed] [Google Scholar]
- 70.Niswender KD, Schwartz MW. Insulin and leptin revisited: adiposity signals with overlapping physiological and intracellular signaling capabilities. Front Neuroendocrinol. 2003;24:1–10. doi: 10.1016/s0091-3022(02)00105-x. [DOI] [PubMed] [Google Scholar]
- 71.Wang J, Obici S, Morgan K, Barzilai N, Feng Z, Rossetti L. Overfeeding rapidly induces leptin and insulin resistance. Diabetes. 2001;50:2786–2791. doi: 10.2337/diabetes.50.12.2786. [DOI] [PubMed] [Google Scholar]
- 72.Woods SC, D’Alessio DA, Tso P, Rushing PA, Clegg DJ, Benoit SC, Gotoh K, Liu M, Seeley RJ. Consumption of a high-fat diet alters the homeostatic regulation of energy balance. Physiol Behav. 2004;83:573–578. doi: 10.1016/j.physbeh.2004.07.026. [DOI] [PubMed] [Google Scholar]
- 73.Clegg DJ, Benoit SC, Reed JA, Woods SC, Dunn-Meynell A, Levin BE. Reduced anorexic effects of insulin in obesity-prone rats fed a moderate-fat diet. Am J Physiol Regul Integr Comp Physiol. 2005;288:R981–R986. doi: 10.1152/ajpregu.00675.2004. [DOI] [PubMed] [Google Scholar]
- 74.Levin BE, Dunn-Meynell AA, Banks WA. Obesity-prone rats have normal blood-brain barrier transport but defective central leptin signaling before obesity onset. AmJ Physiol Regul Integr Comp Physiol. 2004;286:R143–R150. doi: 10.1152/ajpregu.00393.2003. [DOI] [PubMed] [Google Scholar]
- 75.Levin BE, Magnan C, Migrenne S, Chua SC, Jr, Dunn-Meynell AA. F-DIO obesity-prone rat is insulin resistant before obesity onset. Am J Physiol Regul Integr Comp Physiol. 2005;289:R704–R711. doi: 10.1152/ajpregu.00216.2005. [DOI] [PubMed] [Google Scholar]
- 76.White CL, Whittington A, Barnes MJ, Wang Z, Bray GA, Morrison CD. Increased hypothalamic PTP1B in response to diet-induced obesity and chronic hyperleptinemia. Am J Physiol Endocrinol Metab. 2008 doi: 10.1152/ajpendo.90513.2008. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lam TK, Pocai A, Gutierrez-Juarez R, Obici S, Bryan J, guilar-Bryan L, Schwartz GJ, Rossetti L. Hypothalamic sensing of circulating fatty acids is required for glucose homeostasis. Nat Med. 2005;11:320–327. doi: 10.1038/nm1201. [DOI] [PubMed] [Google Scholar]
- 78.Morgan K, Obici S, Rossetti L. Hypothalamic responses to long-chain fatty acids are nutritionally regulated. J Biol Chem. 2004;279:31139–31148. doi: 10.1074/jbc.M400458200. [DOI] [PubMed] [Google Scholar]
- 79.Maness LM, Banks WA, Kastin AJ. Persistence of blood-to-brain transport of leptin in obese leptin-deficient and leptin receptor-deficient mice. Brain Res. 2000;873:165–167. doi: 10.1016/s0006-8993(00)02520-8. [DOI] [PubMed] [Google Scholar]
- 80.Harris RBS, Zhou J, Redmann SM, Jr, Smagin GN, Smith SR, Rodgers E, Zachwieja JJ. A leptin dose-response study in obese (ob/ob) and lean (+/?) mice. Endocrinology. 1998;139:8–19. doi: 10.1210/endo.139.1.5675. [DOI] [PubMed] [Google Scholar]
- 81.Boston BA, Blaydon KM, Varnerin J, Cone RD. Independent and additive effects of central POMC and leptin pathways on murine obesity. Science. 1997;278:1641–1644. doi: 10.1126/science.278.5343.1641. [DOI] [PubMed] [Google Scholar]
- 82.Benomar Y, Wetzler S, Larue-Achagiotis C, Djiane J, Tome D, Taouis M. In vivo leptin infusion impairs insulin and leptin signalling in liver and hypothalamus. Mol Cell Endocrinol. 2005;242:59–66. doi: 10.1016/j.mce.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 83.Shapiro A, Matheny M, Zhang Y, Tumer N, Cheng KY, Rogrigues E, Zolotukhin S, Scarpace PJ. Synergy between leptin therapy and a seemingly negligible amount of voluntary wheel running prevents progression of dietary obesity in leptin-resistant rats. Diabetes. 2008;57:614–622. doi: 10.2337/db07-0863. [DOI] [PubMed] [Google Scholar]
- 84.Scarpace PJ, Matheny M, Tumer N, Cheng KY, Zhang Y. Leptin resistance exacerbates diet-induced obesity and is associated with diminished maximal leptin signalling capacity in rats. Diabetologia. 2005;48:1075–1083. doi: 10.1007/s00125-005-1763-x. [DOI] [PubMed] [Google Scholar]
- 85.Scarpace PJ, Zhang Y. Elevated leptin: consequence or cause of obesity? Front Biosci. 2007;12:3531–3544. doi: 10.2741/2332. [DOI] [PubMed] [Google Scholar]
- 86.Obici S, Feng Z, Morgan K, Stein D, Karkanias G, Rossetti L. Central administration of oleic acid inhibits glucose production and food intake. Diabetes. 2002;51:271–275. doi: 10.2337/diabetes.51.2.271. [DOI] [PubMed] [Google Scholar]
- 87.Obici S, Feng Z, Arduini A, Conti R, Rossetti L. Inhibition of hypothalamic carnitine palmitoyltransferase-1 decreases food intake and glucose production. Nat Med. 2003;9:756–761. doi: 10.1038/nm873. [DOI] [PubMed] [Google Scholar]
- 88.Pocai A, Obici S, Schwartz GJ, Rossetti L. A brain-liver circuit regulates glucose homeostasis. Cell Metab. 2005;1:53–61. doi: 10.1016/j.cmet.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 89.Lam TK, van de WG, Giacca A. Free fatty acids increase basal hepatic glucose production and induce hepatic insulin resistance at different sites. Am J Physiol Endocrinol Metab. 2003;284:E281–E290. doi: 10.1152/ajpendo.00332.2002. [DOI] [PubMed] [Google Scholar]
- 90.Watson PM, Commins SP, Beiler RJ, Hatcher HC, Gettys TW. Differential regulation of leptin release and function in A/J versus C57BL/6J mice during diet-induced obesity. Am J Physiol. 2000;279:E356–E365. doi: 10.1152/ajpendo.2000.279.2.E356. [DOI] [PubMed] [Google Scholar]
- 91.Harris RBS, Mitchell TD, Hebert S. Leptin-induced changes in body composition in high fat-fed mice. Exp Biol Med. 2003;228:24–32. doi: 10.1177/153537020322800103. [DOI] [PubMed] [Google Scholar]
- 92.Bowen H, Mitchell TD, Harris RBS. Method of leptin dosing, strain, and group housing influence leptin sensitivity in high-fat-fed weanling mice. Am J Physiol Regul Integr Comp Physiol. 2003;284:R87–R100. doi: 10.1152/ajpregu.00431.2002. [DOI] [PubMed] [Google Scholar]
- 93.Haltiner AL, Mitchell TD, Harris RB. Leptin action is modified by an interaction between dietary fat content and ambient temperature. Am J Physiol Regul Integr Comp Physiol. 2004;287:R1250–R1255. doi: 10.1152/ajpregu.00313.2004. [DOI] [PubMed] [Google Scholar]
- 94.Stewart LK, Wang Z, Ribnicky D, Soileau JL, Cefalu WT, Gettys TW. Failure of dietary quercetin to alter the temporal progression of insulin resistance among tissues of C57BL/6J mice during the development of diet-induced obesity. Anonymous. 2008 doi: 10.1007/s00125-008-1252-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Beebe SJ, Redmon JB, Blackmore PF, Corbin JD. Discriminative insulin antagonism of stimulatory effects of various cAMP analogs on adipocyte lipolysis and heptocyte glycogenolysis. J Biol Chem. 1985;260:15781–15788. [PubMed] [Google Scholar]
- 96.Corbin JD, Beebe SJ, Blackmore PF, Redmon JB, Sheorain VS, Gettys TW. Discriminative insulin antagonism of effects of different cAMP analogs in intact mammalian cells. In: Belfrage P, Donner J, Stralfors P, editors. Mechanisms of Insulin Action. Elsevier Science Publishers; Amsterdam: 1986. pp. 167–174. [Google Scholar]
- 97.Loten EG, Assimacopoulos-Jeannet FD, Exton JH, Park CR. Stimulation of a low Km phosphodiesterase from liver by insulin and glucagon. J Biol Chem. 1978;253:746–757. [PubMed] [Google Scholar]
- 98.Gettys TW, Harkness PJ, Watson PM. The β3-adrenergic receptor inhibits insulin-stimulated leptin secretion from isolated rat adipocytes. Endocrinology. 1996;137:4054–4057. doi: 10.1210/endo.137.9.8756584. [DOI] [PubMed] [Google Scholar]