

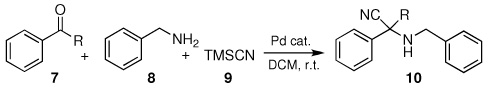
Abstract

A simple and efficient one-pot three-component method has been developed for the synthesis of α-aminonitriles. This Strecker reaction is applicable for aldehydes and ketones with aliphatic or aromatic amines and trimethyl siliyl cyanide in the presence of a palladium Lewis aid catalyst in dichloromethane solvent at room temperature.
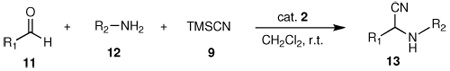
The Strecker reaction; which employs aldehydes or ketones, amines, and a cyanide source; is a well-established route for the preparation of α-aminonitriles, which are versatile intermediate compounds and are particularly useful in the preparation of α-amino acids and other biologically relevant molecules, such as nitrogen-containing heterocycles.1 Successful examples of this reaction have been demonstrated using titanium,2 iron,3 and zirconium4 catalysts, Schiff bases,5 Lewis bases,6 gallium triflate,7 ionic liquids,8 β-cyclodextrin,9 and other non-metal catalysts.10 However, most one-pot multi-component variations of the Strecker reaction involve aldehydes, and the Strecker synthesis applied to ketones and aliphatic amines remains a more difficult reaction. Often with these substrates, the reaction is carried out stepwise using premade imines or under high pressure conditions.11 Although recently one-pot procedures have been developed for the synthesis of α-aminonitriles using a variety of Lewis acids; such as lithium perchlorate,12 scandium triflate,13 vanadyl triflate,14 zinc halides,15 ytterbium triflate,16 and montmorillonite;17 most of these methods involve the use of strong acidic conditions, expensive reagents, extended reaction times, harsh conditions, fast hydrolysis, and tedious workup leading to the generation of a large amount of waste. Therefore, more general and milder reaction conditions for one-pot multi-component Strecker reactions, particularly those involving ketones, would be advantageous.
Recently, we found that N-heterocyclic carbene (NHC)-amidate palladium (II) complex 1a acts as an effective catalyst for asymmetric boron-Heck type carbon-carbon bond forming reactions under mild conditions.18 In addition, this palladium (II) complex 1a was converted to the palladium complex 1b by treating with aqueous AgBF4, and it was found that the subsequent monomer/dimer equilibrium process (1b↔1c) readily occurred in the aqueous solution (Scheme 1). Consequently, the catalytic reaction was not inhibited by coordination of water to palladium metal since the presence of strongly electron donating groups such as the NHC, amidate N, and O would increase the electron density of palladium and allow for a weak interaction between electrophilic Pd and water. Therefore, due to the stability towards aqueous condition and easy formation of a palladium open site, we prepared new NHC-amidate palladium (II) analogue 2 having an ester moiety as a portable chelating group. We herein report the results of its application in the synthesis of α-aminonitriles from the corresponding aldehydes or ketones and amines with trimethylsilyl cyanide (TMSCN) in dichloromethane solvent. These reactions required no further purification in most cases. The preparation of ligand precursor 6 was carried out as illustrated in Scheme 2. Treatment of 4, derived from the amidation of valine methyl ester and bromoacetyl bromide, with benzimidazole 3 in the presence of KOH in DMF provided compound 5 efficiently. The amido ester-substituted benzimidazolium iodine salt 6 was then obtained by allowing 5 to react with CH3I in refluxing THF. Formation of 6 was confirmed (1H NMR spectroscopy in CDCl3) by observation of the new peak assigned to the N-CH3 at 4.18 ppm, as well as the expected chemical shift change for the imidazole-H (7.95 to 10.16 ppm) as an iodine salt.
SCHEME 1.

SCHEME 2.

Preparation of the palladium complex 2
For coordination of 6 as an NHC to palladium, compound 6 was reacted with Ag2O in dichloromethane at room temperature for 3 h, and then the solvent was filtered under reduced pressure to give the silver NHC complex as a light gray color solid. This reaction could be carried out without any purification of the intermediate. Subsequent treatment of the silver compound with PdCl2(CH3CN)2 in CH3CN at room temperature for 3 h afforded palladium complex 2 in 83% yield. The structure was confirmed by 1H-NMR spectroscopic analysis and HRMS data (molecular peak at m/z 445.0371 [M+H]) As shown in Table 1, an initial screening of palladium catalysts was conducted. Using TMSCN as a cyanide source and sodium sulfate as a dessicant, reactions were conducted in dichloromethane solvent at room temperature. In the absence of a palladium catalyst, reactivity was poor and provided low conversion for the reaction between acetophenone and benzylamine at room temperature (entry 1). While the use of PdCl2 led to low conversion for reactions involving ketone and aldehyde (entries 2 and 5), optimal conversion (> 95%) was obtained using a 3 mol% loading of NHC-palladium complex 2 in reactions employing both carbonyl sources (entries 4 and 6).
TABLE 1.
Screening of palladium source and catalyst loading.a
 | ||||
|---|---|---|---|---|
| Entry | R | Catalyst (mol %) | Time (h) | Yield (%)b |
| 1 | Me | - | 72 | 15 |
| 2 | Me | PdCl2 (5 mol%) | 24 | 11 |
| 3 | Me | 2 (5 mol%) | 24 | 99 |
| 4 | Me | 2 (3 mol%) | 24 | 99 |
| 5 | H | PdCl2 (5 mol%) | 24 | 68 |
| 6 | H | 2 (3 mol%) | 24 | 95 |
To a mixture of palladium catalyst, sodium sulfate (100 mg, 0.7 mmol), 7 (0.2 mmol), and 8 (0.2 mmol) in 1 mL CH2Cl2 in Schlenk tube was added dropwise 9 (0.4 mmol). The mixture was stirred for 24 hrs at room temperature.
Conversion yield.
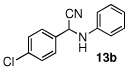
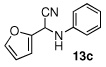
In the initial screening, both PdCl2 and 2 were demonstrated to be suitable catalysts for reactions involving aldehydes. However, due to its stability in water released during the course of the reaction, 2 appeared to promote better reactivity compared to PdCl2 for reactions employing ketones. Therefore, complex 2 was evaluated for potential application to a wider scope of substrates for the Strecker reaction, shown in Table 2. Reactions were carried out with aliphatic and aromatic aldehyde and amine substrates and in all cases, regardless of differences in electronic character of the aldehyde substrates, reactions were able to proceed with high yields. For example, an electron-withdrawing substituent (entries 2 and 9) were compatible to these conditions, as were heteroatom-containing aldehydes (entries 3–5 and 10–12) and aliphatic aldehydes (entries 7, 14, and 15). Similar to previous reports using other Lewis acid catalysts, aromatic (entries 1–7) and aliphatic (entries 8–15) amines were compatible with these reactions as well.7, 12–17 Without the use of a dessicating agent, the formation of the imine intermediate was hindered and low conversion to the α-aminonitrile produt was observed (not shown).
TABLE 2.
Strecker reactions of various aldehydes and amines in the presence of palladium complex 2.a
 | ||||
|---|---|---|---|---|
| Entry | Aldehyde | Amine | Product | Conv.(Yield) (%) |
| 1 |  |
 |
99 (79) | |
| 2 | 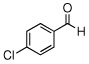 |
 |
99 (84) | |
| 3 | 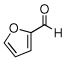 |
 |
99 (79) | |
| 4 | 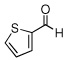 |
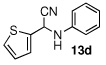 |
99 (91) | |
| 5 | 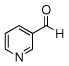 |
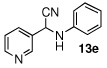 |
99 (95) | |
| 6 | 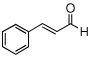 |
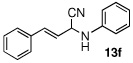 |
99 (83) | |
| 7 | 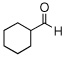 |
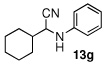 |
99 (88) | |
| 8 | 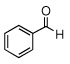 |
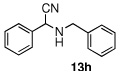 |
99 (95) | |
| 9 | 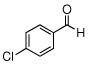 |
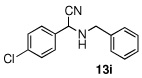 |
99 (90) | |
| 10 | 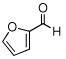 |
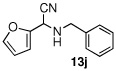 |
99 (86) | |
| 11 | 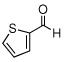 |
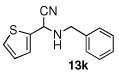 |
99 (91) | |
| 12 | 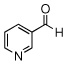 |
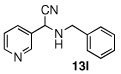 |
99 (87) | |
| 13 | 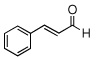 |
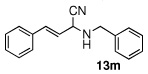 |
99 (98) | |
| 14 | 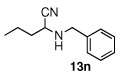 |
99 (70) | ||
| 15 | 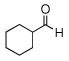 |
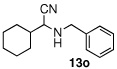 |
99 (62) | |
To a mixture of palladium catalyst, sodium sulfate (100 mg, 0.7 mmol), 7 (0.2 mmol), and 8 (0.2 mmol) in 1 mL CH2Cl2 in Schlenk tube was added dropwise 9 (0.4 mmol). The mixture was stirred for 24 hrs at room temperature.
Isolated yields.
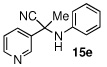
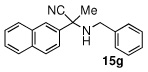
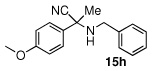
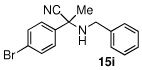
Due to the encouraging reactivity of complex 2 as a Lewis acid catalysts for the Strecker reaction employing aldehydes, its feasibility to promote reactions involving ketones was then evaluated. In the presence of 2, good reactivity was observed for reactions involving an aromatic amine and ketones with electron-donating (entry 2) substituents, as indicated in Table 3. A bromo-substituent was tolerated as well (entry 3), but the electron-withdrawing nitro group (entry 4) was not a feasible substrate. Lower yield with this substrate may be attributed to low conversion to the imine intermediate. In general, reactions employing aniline as the amine provided the α-aminonitrile products in better conversion than did those using benzylamine. Again, electron-withdrawing substituents on the ketone substrate were not well-tolerated, although electron-donating (entry 8), and heteroatom (entry 10) substituents provided modest reactivities.
TABLE 3.
Strecker reactions of various ketones and amines in the presence of palladium complex 2.a
 | ||||
|---|---|---|---|---|
| Entry | Aldehyde | Amine | Product | Conv.(Yield) (%) |
| 1 | 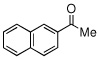 |
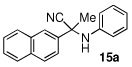 |
99 (92) | |
| 2 | 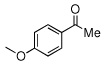 |
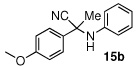 |
86 (74) | |
| 3 | 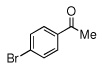 |
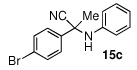 |
95 (88) | |
| 4 | 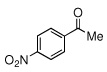 |
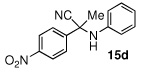 |
17 (15) | |
| 5 | 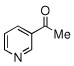 |
 |
44 (40) | |
| 6 | 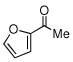 |
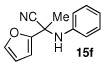 |
85 (83) | |
| 7 | 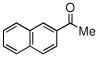 |
 |
49 (33) | |
| 8 | 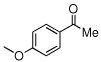 |
 |
58 (35) | |
| 9 | 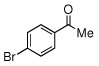 |
 |
62 (55) | |
| 10 | 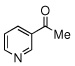 |
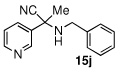 |
68 (60) | |
To a mixture of palladium catalyst, sodium sulfate (100mg, 0.7 mmol), 7 (0.2 mmol), and 8 (0.52 mmol) in 1 mL CH2Cl2 in Schlenk tube was added dropwise 9 (0.4 mmol). The mixture was stirred for 24 hrs at room temperature. If necessary, compounds were purified by column chromatography on silica gel with a gradient elution of hexanes/ethyl acetate.
Isolated yields.
In summary, NHC-amidate ester palladium (II) complex 2 has been demonstrated to be an useful Lewis acid catalyst to promote one-pot multicomponent Strecker reactions for the synthesis of α-aminonitriles. Additionally, the application of 2 in reactions involving ketone substrates allowed for the formation of α-aminonitrile products containing a quaternary carbon. The benefit of this methodology is the simplicity of the procedure involved, which often avoided the use of tedious chromatographic purification of products. Future studies involving optically active forms of 2 may allow for the formation of α-aminonitriles in an enantioselective manner.
Experimental Section
Palladium(II) complex (2)
The suspension of benzimidazolium iodine salt 6 (500 mg, 1.16 mmol) and silver(I) oxide (185 mg, 0.8 mmol) in CH2Cl2 (20 mL) was stirred for 3 hours in the dark at room temperature. The reaction mixture was concentrated under reduced pressure to give a gray solid. To a suspension of the silver complex in CH3CN (20 mL), was added PdCl2(CH3CN) (301 mg, 1.16 mmol) in the dark at room temperature. Then, the resulting suspension was stirred for 2 hours and filtered through a plug of glass fiber filter paper. The filtrate was evaporated to dryness in vacuo to afford product 2 (418 mg, 81% yield) as an orange solid. 1H NMR (CD3OD, 250 MHz) δ 7.63-7.59 (m, 1H), 7.51-7.48 (m, 1H), 7.38-7.33 (m, 2H), 5.82 (d, J = 16.5 Hz, 1H), 5.63 (d, J = 16.5 Hz, 1H), 4.41 (brs, 1H), 4.35 (s, 3H), 3.66 (s, 3H), 2.23-2.09 (m, 1H), 0.95 (d, J = 2.5 Hz, 3H), 0.92 (d, J = 2.5 Hz, 3H); 13C NMR (CD3OD, 63 MHz) δ 18.6, 19.4, 31.7, 35.3, 52.0, 52.4, 59.6; Elemental Anal. calcd. for C16H21ClN3O3Pd: C 43.16, H 4.75, N 9.44, Cl 7.96, found: C 42.98, H 4.81, N 7.59, Cl 7.38; HRMS-ESI (m/z) [M+H+] calcd. for C16H22ClN3O3Pd: 445.0385, found: 445.0371.
Representative Procedure for the Synthesis of 13a
To a mixture of 2 (3 mol %), sodium sulfate (100 mg, 0.7 mmol), benzaldehyde (0.020 mL, 0.2 mmol), and aniline (0.018 mL, 0.2 mmol) in 1 mL CH2Cl2 in a pressure tube was added dropwise TMSCN (0.053 mL, 0.4 mmol). The pressure tube was closed and stirred for 24 h at 23 °C. The mixture was then filtered and the residue was washed with CH2Cl2 (10 mL). The filtrate was collected and the solvent was removed under reduced pressure to obtain 2-phenyl-2-(phenylamino) acetonitrile 13a (33 mg, 79% yield) as a light yellow solid. m.p. 76~79 °C. 1H NMR (250 MHz) δ 4.67 (br s, 1H), 5.36 (s, 1H), 6.72 (d, J = 7.8Hz, 2H), 6.84 (t, J = 7.4 Hz, 1H), 7.22 (t, J = 8.0 Hz, 2 H), 7.37–7.44 (m, 3H), 7.52–7.55 (m, 2H); 13C NMR (63 MHz) δ 50.1, 114.1, 118.1, 120.2, 127.2, 128.4, 129.2, 129.4, 134.0, 144.7.
Supplementary Material
Experimental procedures, compound characterization data and copies of 1H NMR and 13C NMR spectra. This material free of charge via the Internet at http://pubs.acs.org
Acknowledgement
We acknowledge generous financial support from the National Institute of General Medical Sciences of the National Institutes of Health (RO1 GM71496).
References
- 1.(a) Yet L. Angew. Chem., Int. Ed. 2001;40:875–877. [PubMed] [Google Scholar]; (b) Gröger H. Chem. Rev. 2003;103:2795–2827. doi: 10.1021/cr020038p. [DOI] [PubMed] [Google Scholar]; (c) Spino C. Angew. Chem., Int. Ed. 2004;43:1764–1766. doi: 10.1002/anie.200301686. [DOI] [PubMed] [Google Scholar]; (d) Vilaivan T, Bhanthumnavin W, Sritana-Anant Y. Curr. Org. Chem. 2005;9:1315. [Google Scholar]; (e) Ohfune Y, Shinada T. Eur. J. Org. Chem. 2005;24:5127–5143. [Google Scholar]; (f) Friestad GK, Mathies AK. Tetrahedron. 2007;63:2541–2569. doi: 10.1016/j.tet.2007.06.117. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Connon SJ. Angew. Chem., Int. Ed. 2008;47:1176–1178. doi: 10.1002/anie.200703879. [DOI] [PubMed] [Google Scholar]
- 2.(a) Corey EJ, Grogan M. Org. Lett. 1999;1:157–160. doi: 10.1021/ol990623l. [DOI] [PubMed] [Google Scholar]; (b) Josephsohn NS, Kuntz KW, Snapper ML, Hoveyda AH. J. Am. Chem. Soc. 2001;123:11594–11599. doi: 10.1021/ja011875w. [DOI] [PubMed] [Google Scholar]; (c) Banphavichit V, Mansawat W, Bhanthumnavin W, Vilaivan T. Tetrahedron. 2004;60:10559–10568. [Google Scholar]; (d) Blacker J, Clutterbuck LA, Crampton MR, Grosjean C, North M. Tetrahedron: Asymmetry. 2006;17:1449–1456. [Google Scholar]
- 3.Khan NH, Agrawal S, Kureshy RI, Abdi SHR, Singh S, Suresh E, Jasra RV. Tetrahedron Lett. 2008;49:640–644. [Google Scholar]
- 4.Ishitani H, Komiyama S, Hasegawa Y, Kobayashi S. J. Am. Chem. Soc. 2000;122:762–766. [Google Scholar]
- 5.(a) Sigman MS, Jacobsen EN. J. Am. Chem. Soc. 1998;120:4901–4902. [Google Scholar]; (b) Vachal P, Jacobsen EN. J. Am. Chem. Soc. 2002;124:10012–10014. doi: 10.1021/ja027246j. [DOI] [PubMed] [Google Scholar]
- 6.Takahashi E, Fujisawa H, Yanai T, Mukaiyama T. Chem. Lett. 2005;34:604–605. [Google Scholar]
- 7.Prakash GKS, Mathew T, Panja C, Alconcel S, Vaghoo H, Do C, Olah GA. Proc. Nat. Acad. Sci. 2007;104:3703–3706. doi: 10.1073/pnas.0611316104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yadav JS, Reddy BVS, Eshwaraiah B, Srinivas M, Vishnumurthy P. New J. Chem. 2003;27:462–465. [Google Scholar]
- 9.Surendra K, Krishnaveni NS, Mahesh A, Rao KR. J. Org. Chem. 2006;71:2532–2534. doi: 10.1021/jo052510n. [DOI] [PubMed] [Google Scholar]
- 10.(a) Iyer MS, Gigstad KM, Namdev ND, Lipton M. J. Am. Chem. Soc. 1996;118:4910–4911. [Google Scholar]; (b) Sigman M, Jacobsen EN. J. Am. Chem. Soc. 1998;120:5315–5316. [Google Scholar]; (c) Sigman MS, Vachal P, Jacobsen EN. Angew. Chem., Int. Ed. 2000;39:1279–1281. doi: 10.1002/(sici)1521-3773(20000403)39:7<1279::aid-anie1279>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]; (d) Liu B, Feng X, Chen F, Zhang G, Cui X, Jiang Y. Synlett. 2001;10:1551–1554. [Google Scholar]
- 11.(a) Warmuth R, Munsch TE, Stalker RA, Li B, Beattey A. Tetrahedron. 2001;57:6383–6397. [Google Scholar]; (b) Matsumoto K, Kim JC, Iida H, Hamana H, Kumamoto K, Kotsuki H, Jenner G. Helv. Chim. Acta. 2005;88:1734–1754. [Google Scholar]; (c) Kumamoto K, Iida H, Hamana H, Kotsuki H, Matsumoto K. Heterocycles. 2005;66:675–681. [Google Scholar]
- 12.Prasad BA, Bhanu, Bisai A, Singh VK. Tetrahdron Lett. 2004;45:9565–9567. [Google Scholar]
- 13.Kobayashi S, Busujima T, Nagayama S. Chem. Commun. 1998;9:981–982. [Google Scholar]
- 14.De SK. Synth. Comm. 2005;35:1577–1582. [Google Scholar]
- 15.Kazemeini A, Azizi N, Saidi MR. Russ. J. Org. Chem. 2006;42:48–51. [Google Scholar]
- 16.Huguenot F, Brigaud T. J. Org. Chem. 2006;71:7075–7078. doi: 10.1021/jo0607717. [DOI] [PubMed] [Google Scholar]
- 17.Yadav JS, Reddy BVS, Eeshwaraian B, Srinivas M. Tetrahedron. 2004;60:1767–1771. [Google Scholar]
- 18.Sakaguchi S, Yoo KS, O’Neill J, Lee JH, Stewart T, Jung KW. Angew. Chem., Int. Ed. 2008;47:9326–9329. doi: 10.1002/anie.200803793. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental procedures, compound characterization data and copies of 1H NMR and 13C NMR spectra. This material free of charge via the Internet at http://pubs.acs.org