

Abstract
The replacement of a t-butyl group with a trifluoromethyl group has profound effects on the biological profile of 1α,25-dihydroxyvitamin D3 sulfone analogs. Investigation of whether the improved biological activities are due to steric and electronic factors of the trifluoromethyl group led to the design, synthesis and biological evaluation of two analogous alkyl sulfone molecules, methyl sulfone (AU-16-ene-25-SO2-CH3) and isopropyl sulfone (AU-16-ene-25-SO2-i-Pr). These alkyl sulfones are sterically comparable to, but electronically very different from a trifluoromethyl group. The syntheses, antiproliferative activities and calcemic activities of these new alkyl sulfones are presented herein. In comparing the in vitro antiproliferative profiles of the new alkyl sulfone 1α,25-dihydroxyvitamin D3 analogs with the trifluoromethylsulfone and an analogous t-butyl sulfone, the activities increase in the following order: CH3<t-Bu≅i-Pr<CF3. In contrast to the calcemic t-butyl sulfone, the novel alkyl sulfones and trifluoromethyl sulfone display desirable low calcemic levels.
A. Introduction
The biological importance of the hormone 1α,25-dihydroxyvitamin D3 is evident in the use of the secosteroid in the treatment of diseases such as osteoporosis, psoriasis, and various types of cancers.1 Although 1α,25-dihydroxyvitamin D3 is a well-known therapeutic, the favorable biological activity is coupled with undesirable hypercalcemia at high doses of the molecule.2-4 In attempts to create an optimal balance between favorable biological activities and low calcemic levels, thousands of analogs of the active metabolite, 1α,25-dihydroxyvitamin D3 otherwise called calcitriol (1) (Figure 1), have been synthesized.5, 6 Some of the most biologically successful 1α,25-dihydroxyvitamin D3 analogs include alterations of the side chain such as unsaturation, methylation, homologation, epimerization, and fluorination.5-8
Figure 1.

Incorporation of a fluorine atom is one of the most effective alterations leading to improved biological activity within a 1α,25-dihydroxyvitamin D3 molecule. Several years ago the Posner lab designed and prepared 1α,25-dihydroxyvitamin D3 compounds replacing t-butyl groups of antiproliferative sulfone analogs with trifluoromethyl groups.8 (Figure 1) We were not surprised that this alteration proved valuable in biological profile of 1α,25-dihydroxyvitamin D3 analogs8 seeing that the incorporation of fluorine into a molecule is oftentimes beneficial in medicinal chemistry.9-11 In attempts to explain the favorable increase in antiproliferative activity of the trifluoromethyl 1α,25-dihydroxyvitamin D3 analogs, we considered both the electronic and steric factors behind 1α,25-dihydroxyvitamin D3 drug design as well as the electronic and steric characteristics of the fluorine atom.
Electronic factors could play a significant role in the improved antiproliferative activity of the trifluoromethyl 1α,25-dihydroxyvitamin D3 analogs. The presence of fluorine, a strongly electronegative atom and a hydrogen bond acceptor,9-12 might increase the necessary vitamin D receptor interactions13 with the given analog. Fluorine's high electronegativity may also hinder metabolic degradation. A carbon-fluorine bond is the strongest covalent bond with a binding energy of 115.7 kcal/mol.9, 12 When comparing this binding energy to that of a carbon-hydrogen bond at 98.0 kcal/mol, or a carbon-carbon bond at 83 kcal/mol, it is clear that the carbon fluorine bond is much more difficult to cleave,9, 12 and therefore, less susceptible to p450 catabolic degradation.5,14 Carbon-carbon bonds with fluorines bound to one carbon is also known to be stronger than those with only hydrogens.9, 12 All of these stability enhancing characteristics would make for a more metabolically stable and perhaps more biologically active compound.
In addition to the favorable electronic differences that fluorine incorporation displays, there are also desirable steric factors. A trifluoromethyl group is most comparable in size to an isopropyl group,12 slightly smaller than a t-butyl group and slightly larger than a methyl group. The smaller size in comparison with t-butyl may allow for a better fit into the vitamin D receptor.13 Additionally, the fluorine van der waals radius is 1.47Å compared to the hydrogen van der waals radius of 1.20Å.9, 11, 12 Due to the similarity in size, fluorine is an excellent candidate to replace hydrogen without disruption of molecular geometry.
In order to discern whether the improved antiproliferative activities of the trifluoromethyl sulfone analogs are due to steric or electronic factors, we developed two new 1α,25-dihydroxyvitamin D3 analogs, methyl sulfone 3 and isopropyl sulfone 4. Both of these analogs are identical to the highly antiproliferative trifluoromethyl sulfone (2) except for the C,D-ring side chain terminus. These alkyl sulfones are similar in size to that of the trifluoromethyl group, but have significantly different electronic character. (Figure 1) If sterics is the major factor leading to the favorable antiproliferative activity of trifluoromethyl sulfone 2, the newly designed alkyl sulfones should both demonstrate high antiproliferative activity and should provide two new biologically valuable 1α,25-dihydroxyvitamin D3 analogs.
B. Chemistry
The isopropyl and methyl sulfones were prepared by synthetic methods illustrated in scheme 1. Iodide (+)-5 is prepared as previously reported by Posner.15 Displacement of iodide (+)-5 with the lithium anion of the given methyl sulfone installs the desired sidechain giving (+)-6. Subsequent deprotection of the C-8 alcohol, followed by oxidation yields ketone (+)-7. The ketone is then coupled in a Wittig-Horner reaction with enantiomerically pure A-ring (-)-8 which was prepared as reported by Posner.16 Following the Wittig-Horner coupling, hydroxyl groups 1 and 3 are deprotected to give the desired isopropyl and methyl sulfone analogs (+)-3 and (+)-4, respectively. It is important to point out that the desilylations of silyl ether (+)-6 and the silyl ether versions of (+)-3 and (+)-4 with the acid HF all proceed without compromising the olefinic bonds in the molecules.
Scheme 1.

The low yield of the Wittig-Horner coupling of A-ring (-)-8 with methyl sulfone ketone (+)-7a is most likely due to the instability of the ketone. Decomposition could occur upon standing at room temperature while the ketone and A-ring are drying for several days prior to the coupling. The decomposition byproducts could interfere with the reaction and the quantity of starting material will be less than calculated. The sterically open terminus of the methyl sulfone may also make it more prone to degradation in the presence of excess base.
C. Results and Discussion
Dose responses for the antiproliferative activities of trifluoromethyl sulfone 2, alkyl sulfones 3 and 4, and calcitriol (1), determined using our standard murine keratinocyte cell culture assay,17 are displayed in figure 2. Methyl sulfone 3 is less potent than the natural hormone calcitriol. In contrast, isopropyl sulfone 4 exhibits similar antiproliferative activity to calcitriol. Trifluoromethyl sulfone 2 is the most antiproliferative with activity twice that of the natural product at a concentration as low as 10 nM. The order of antiproliferative potency of the sulfone analogs increases from CH3<i-Pr<CF3.
Figure 2.
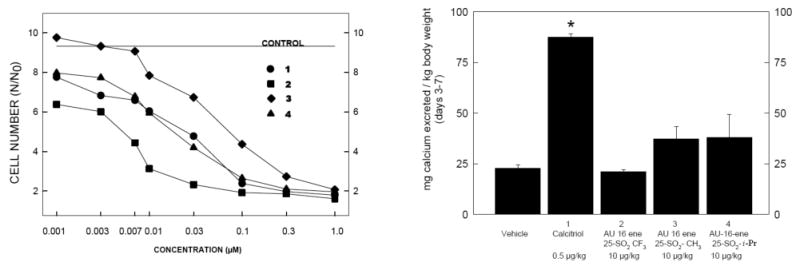
The isopropyl and trifluoromethyl groups are most similar in biological activity as well as size suggesting that sterics may be a reason for improved biological activity of the trifluoromethyl sulfone analogs. However, when including the antiproliferative activity of the analogous t-butyl sulfone15, which is the largest of the series, we find that this sterically cumbersome molecule exhibits activity comparable to isopropyl sulfone 4. This comparison leads to the following trend in antiproliferative activities: CH3<t-Bu≅i-Pr<CF3. The antiproliferative activity of the series does not directly correlate to size. Here it becomes obvious that sterics may not be the primary contributing factor in the improved antiproliferative potency of the trifluoromethyl sulfones. In attempts to better explain the trends in antiproliferative activites of the alkyl and trifluoromethyl sulfone analogs, we turn to electronic factors.
A large part of the activity differences could be attributed to stability and electronic factors. The strength of the carbon-fluorine bonds9, 11, 12 of the trifluoromethyl group compared to the carbon-hydrogen bonds of the methyl may extend the lifetime of the trifluoromethyl compound and make for a more efficacious molecule. Stability may actually be the reason why the methyl sulfone is inferior to all of the sulfone analogs. Methyl sulfone 3, containing three terminal α -hydrogens for abstraction, is most susceptible to catabolic p450 oxidation and subsequent degradation.5, 14 This analysis illustrates that stability due to electronic factors plays an important role in the observed antiproliferative trend of the alkyl and trifluoromethyl sulfones.
In addition to antiproliferative activity, we must also consider the calcemic effects of these biologically active molecules. Using our previously reported protocol15 in which rats are treated orally with 1α,25-dihydroxyvitamin D3 (1) or with new analogs (2-4) daily for one week, the trifluoromethyl (2), methyl (3), and isopropyl (4) sulfones all show calciuria levels far below that of calcitriol (1), even at twenty times the dosage of the natural hormone. The calcemic levels of animals treated with all three analogs (2-4) were comparable to that of the vehicle. Interestingly, treatment with these three novel 1α,25-dihydroxyvitamin D3 analogs resulted in lower calcemic levels than that of the analogous t-butyl sulfone which exhibited lower calciuric levels than calcitriol but significantly higher levels than the vehicle.
D. Conclusion
The design, synthesis, and biological evaluation of new alkyl sulfone analogs, based upon the trifluoromethyl sulfone 2, led to the discovery of the antiproliferative and low calcemic 1α,25-dihydroxyvitamin D3 analog, AU-16-ene-25-SO2-i-Pr (4). The novel study of alkyl and trifluoromethylsulfone analogs reveals that although steric factors affect the biological activity of the molecule, electronic factors and catabolic stability of an analog are essential when designing new 1α,25-dihydroxyvitamin D3 analogs. The extremely stable trifluoromethyl analog (2) and the novel isopropyl sulfone analog (4) are successful antiproliferative agents and low calciuric compounds. The newfound success and the future pursuit of trifluoromethyl and alkyl sulfone analogs may lead to highly antiproliferative and low calcemic 1α,25-dihydroxyvitamin D3 analogs creating optimal therapeutics for diseases such as psoriasis, osteoporosis, and cancer.
E. Experimental
The following experimental applies to all compounds in this research report: All air and moisture sensitive reactions were carried out in oven dried (at 120 °C) glassware under an inert atmosphere of argon. All solvents and reagents were used as received unless otherwise stated. 1H spectra were obtained on a Varian XL 400 and/or Bruker 300 MHz spectrometer at 400 MHz and/or 300 MHz respectively. 13C spectra were obtained on a Varian XL 400 at 100 MHz. Chemical shifts (δ) are reported in parts per million (ppm). Multiplicities of signals in the 1H spectra are reported as follows, s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), dd (doublet of doublets), etc. Infrared spectra were obtained on a Perkin Elmer 1600 FR-IR spectrometer as liquid films or as a thin layer with NaCl cells. Intensities were reported as s (strong 67-100%), m (medium 34-66%), and w (weak 0-33%) with the following notations, br (broadened), sh (shoulder), etc. UV-Vis absorbance measurements were made on a Hewlett-Packard 8453 diode array spectrophotometer. Optical rotations were recorded on JASCO, P-1100 model polarimeter (Japan Spectroscopic Co., Ltd.) with sodium D line at the temperatures as indicated in the experimental for the specific compounds. HRMS were obtained on a Micromass QTOF Electrospray mass spectroscopy with electronic or chemical ionization (EI or CI) at Ohio State University or FAB mass spectra were obtained using a VG70S double focusing magnetic sector mass spectrometer (VG Analytical, Manchester, UK, now Micromass/Waters) equipped with a Xe gas FAB gun (8kV @ 1.2mA), an off-axis electron multiplier and an MSS data system (MasCom, Bremen, Germany) at Johns Hopkins University.
AU-16-ene-25-SO2-Me (+)-3
16-ene-25-SO2-Me C-8 TBS-protected Alcohol (+)-6a
THF (1.5 ml) was added to methylsulfone (0.029g, 0.31mmol) in an oven-dried 10 mL pear-shaped bottom flask. To this solution, nBuLi (1.6M, 0.19mL, 0.31mmol) was added dropwise. The solution stirred at room temperature for one hour at which time 23-iodide (+)-5 (0.029g, 0.063mmol) was added via cannula in THF (1.00mL). The reaction stirred 16 hrs at which time TLC showed almost complete consumption of starting material. The reaction was quenched with H2O (2 mL) dropwise, then rinsed into a separatory funnel with ethyl acetate. The mixture was extracted with ethyl acetate (3×20 mL). The combined extracts were washed with ice water (1×20 mL) and dried over magnesium sulfate. The filtrate was concentrated in vacuo to give the crude product that was purified by column chromatography (70% hexanes/30% ethyl acetate), affording 0.013g of 25-methylsulfone (+)-6a as an oil in 50% yield.
Data for (+)-6a
[α]D21= +6.23 (c=0.65, CHCl3) 1H NMR (CDCl3, 400 MHz): δ 5.28 (m, 1H), 4.12 (m, 1H), 2.99 (t, 2H, J=7.8), 2.89 (s, 3H), 2.31-2.18(m, 1H), 2.09-2.05 (m, 1H), 1.91-1.76 (m, 4H), 1.71-1.63 (m, 3H), 1.51-1.17 (m, 5H), 1.02 -0.99 (m, 9H), 0.96-0.94 (m, 6H), 0.61-0.53 (m, 6H). 13C NMR (CDCl3 400MHz): δ 159.29, 120.36, 75.03, 68.90, 55.09, 46.69, 40.40, 35.77, 35.15, 34.89, 31.45, 30.75, 22.24, 20.80, 18.91, 18.05, 6.96, 4.92. IR: 2948(s), 2929(s), 2882(m), 1451(m), 1413(w), 1375(w), 1318(s), 1281(m), 1251(m), 1224(m), 1186(w), 1129(s), 1072(m), 968(m), 930(m), 921(m), 845(m), 797(w), 769(m), 741(m), 712(m).
16-ene 25-SO2-Me C-8 Ketone (+)-7a
C-8 TES protected alcohol (+)-6a (0.013g, 0.031 mmol) was dissolved in acetonitrile (1.00 mL) in an argon-purged 10 ml oven-dried round bottom flask equipped with a magnetic stir bar. After stirring for five minutes, HF (0.062 g, 3.10 mmol, 49% aqueous solution) was added at rt via syringe. The reaction mixture was stirred in the dark at rt for 1 hour. TLC showed consumption of starting material. The reaction was diluted with ethyl acetate (1.50 mL), and saturated NaHCO3 was added until CO2 liberation stopped. The reaction mixture was then rinsed into a separatory funnel and extracted with ethyl acetate (3×10 mL). The combined extracts were washed with H2O (1×10 mL), brine (1×10mL), and dried over magnesium sulfate. The filtrate was concentrated in vacuo to give the crude product which was purified by column chromatography (50% hexanes/50% ethyl acetate) affording 0.009g of deprotected product as an oil in 100% yield.
The deprotected product (0.009g, 0.030mmol) was dissolved in methylene chloride (1.0mL) in an oven-dried 10 mL round bottom flask with magnetic stir bar. PDC (0.028g, 0.075mmol) and celite (0.028g) were added and reaction stirred overnight. TLC showed consumption of starting material. Reaction mixture charged directly onto a silica flash column (50% hexanes/50% ethyl acetate) for purification to afford 0.007g of (+)-7a as an oil in 78% yield.
Data for (+)-7a
[α]D23= +20.9 (c=0.25, CHCl3) 1H NMR (CDCl3, 300 MHz): δ 5.29 (m, 1H), 2.97 (t, 2H, J=8.00), 2.87(s, 3H), 2.47-2.40(m, 1H), 2.33-2.26(m, 2H), 2.13-1.92(m, 4H), 1.89-1.76(m, 4H), 1.72-1.55(m, 3H), 1.06 (d, 3H, J= 6.4 Hz), 0.79 (s, 3H). 13C NMR (CDCl3 400MHz): δ 210.71, 157.05, 120.99, 63.07, 54.86, 53.76, 40.60, 40.47, 35.03, 34.41, 32.56, 27.11, 23.97, 21.58, 20.58, 17.42. IR: 2929(s), 2833(m), 1697(s), 1527(m), 1508(m), 1451(w), 1423(w), 1366(m), 1231(s), 1203(m), 1120(s), 1072(w), 1034(m), 940(m), 902(w), 845(m), 769(m). HRMS: calculated for C16H26O3S: 299.1681; found: 299.1675.
(+)-AU-16-ene-25-SO2-Me (+)-3
Enantiomerically pure phosphine oxide (-)-8 (0.031g, 0.053 mmol) was dissolved in THF (1.5ml) in an oven-dried 10 mL round bottom flask with magnetic stir bar and purged with argon. Reaction vessel was cooled to -78°C and nBuLi (1.6M) (0.033mL, 0.053 mmol) was added dropwise resulting in a deep red color. After twenty minutes, ketone (+)-7a (0.007g, 0.023 mmol) was dissolved in THF (1 mL), cooled to -78°C, and added to the reaction via cannula. Reaction stirred for 6.5 hours at -78°C at which time it was quenched at -78°C by the addition of pH 7 buffer (2.00 mL) and allowed to come to room temperature. The reaction mixture was then rinsed into a separatory funnel and extracted with ethyl acetate (3×20 mL). The combined extracts were washed with H2O (1×20 mL), brine (1×20 mL), and dried over magnesium sulfate. The filtrate was concentrated in vacuo to give the crude product which was purified by gradient column chromatography (70% hexanes/30% ethyl acetate to 50% hexanes/50% ethyl acetate) affording 0.0035g of trifluoromethylsulfone TBS protected diol as an oil in 23% yield. After drying on the pump for 4 days, the ketone was yellow in color, which may indicate decomposition and could be the cause of the low yield.
The TBS protected product (0.0035 g, 0.005 mmol) was dissolved in acetonitrile (1.0mL) in an argon-purged 10 mL oven-dried round bottom flask equipped with a magnetic stir bar. After stirring for five minutes, HF (0.01g, 0.50 mmol, 49% aqueous solution) was added at rt via syringe. The reaction mixture was stirred in the dark at rt for 2 hours. TLC showed consumption of starting material. The reaction was diluted with ether (1.50 mL), and saturated NaHCO3 was added until CO2 liberation stopped. The reaction mixture was then rinsed into a separatory funnel and extracted with ethyl acetate (4×20 mL). The combined extracts were washed with H2O (1×20 mL), brine (1×20mL), and dried over magnesium sulfate. The filtrate was concentrated in vacuo to give the crude product which was purified by flash silica chromatography (100% EtOAc) to afford AU-16-ene-25-SO2-Me (+)-3 (0.001g, 0.002mmol) as an oil in 50% yield.
Data for (+)-3
1H NMR (CDCl3, 400 MHz): δ 6.37 (d, 1H, J=11.2 Hz), 6.10 (d, 1H, J=11.2Hz), 5.34(m,1H), 5.32(m, 1H), 5.01(m,1H), 4.44(m,1H), 4.27(m, 1H), 2.98(t, 2H, J=7.6Hz), 2.88(s, 3H), 2.83-2.80(m,1H), 2.61-258(m, 1H), 2.39-2.13(m, 4H), 2.02-1.97(m, 1H), 1.93-1.70(m, 6H), 1.69-1.60(m, 2H), 1.53-1.45(m, 3H), 1.05 (d, 3H, J=6.8 Hz), 0.69 (s, 3H).
**Insufficient material for 13C, but 1H nmr confirms structure.
AU-16-ene-25-SO2-i-Pr (+)-4
25-SO2-i-Pr C-8 TES-protected Alcohol (+)-6b
THF (1.5 ml) was added to isopropyl methylsulfone (0.054 g, 0.44 mmol) in an oven-dried 10 mL pear-shaped bottom flask. To this solution, nBuLi (1.6 M, 0.28 mL, 0.44 mmol) was added dropwise. The solution stirred at room temperature for one hour at which time 23-iodide (+)-5 (0.041 g, 0.089 mmol) was added via cannula in THF (1.50 mL). The reaction stirred 16 hours at which time TLC showed almost complete consumption of starting material. The reaction was quenched with H2O (2 mL) dropwise, then rinsed into a separatory funnel with ethyl acetate. The mixture was extracted with ethyl acetate (3×20 mL). The combined extracts were washed with ice water (1×20 mL) and dried over magnesium sulfate. The filtrate was concentrated in vacuo to give the crude product that was purified by column chromatography (90% hexanes/10% ethyl acetate), affording 0.027g of 25-isopropylsulfone (+)-6b as an oil in 69% yield and 10 mg recovered starting iodide.
Data for (+)-6b
[α]D21= +22.1 (c=0.50, CHCl3) 1H NMR (CDCl3, 400 MHz): δ 5.27 (m, 1H), 4.11(m, 1H), 3.11-3.05 (m, 1H), 2.89(t, 2H, J=8.0Hz), 2.28-2.20(m, 1H), 2.10-2.05(m,1H), 1.91-1.76(m, 4H), 1.71-1.57(m, 4H), 1.54-1.44(m, 3H), 1.37(d, 6H, J=6.8Hz), 1.00(s, 3H), 1.00 (d, 3H, J=6.8), 0.96-0.93 (m, 9H), 0.59-0.53 (m, 6H). 13C NMR (CDCl3 400MHz): δ 159.35, 20.28, 68.87, 55.05, 52.63, 49.40, 46.65, 35.74, 35.42, 34.85, 31.49, 30.70, 22.3, 19.65, 18.85, 18.01, 15.32, 15.25, 6.90, 4.87. IR: 2946(s), 2938(s), 2920(s), 2872(s), 1461(m), 1413(w), 1375(w), 1300(m), 1271(w), 1243(w), 1233(w), 1129(s), 1082(m), 1053(w), 1025(s), 1006(m), 958(m), 940(w), 921(m), 873(w), 845(m), 778(m), 741(m), 722(m). HRMS: calculated for C24H46O3SSi: 443.3015; found: 443.3026.
25-SO2-i-Pr C-8 Ketone (+)-7b
C-8 TES protected alcohol (+)-6b (0.034g, 0.070 mmol) was dissolved in acetonitrile (1.5 mL) in an argon-purged 10 ml oven-dried round bottom flask equipped with a magnetic stir bar. After stirring for five minutes, HF (0.054 g, 2.70 mmol, 49% aqueous solution) was added at rt via syringe. The reaction mixture was stirred in the dark at rt for 1 hour. TLC showed consumption of starting material. The reaction was diluted with ethyl acetate (1.50 mL), and saturated NaHCO3 was added until CO2 liberation stopped. The reaction mixture was then rinsed into a separatory funnel and extracted with ethyl acetate (3×10 mL). The combined extracts were washed with H2O (1×10 mL), brine (1×10mL), and dried over magnesium sulfate. The filtrate was concentrated in vacuo to give the crude product which was purified by column chromatography (50% hexanes/50% ethyl acetate) affording 0.016g of deprotected product as an oil in 80% yield.
The product (0.016g, 0.049mmol) was dissolved in methylene chloride (2.00mL) in an oven-dried 10 mL round bottom flask with magnetic stir bar. PDC (0.044g, 0.120mmol) and celite (0.044mg) were added and reaction stirred overnight. TLC showed consumption of starting material. Reaction mixture charged directly onto a silica flash column (50% hexanes/50% ethyl acetate) for purification to afford 0.010g of (+)-7b as an oil in 63% yield.
Data for (+)-7b
[α]d21= +12.7 (c=0.40, CHCl3) 1H NMR (CDCl3, 400 MHz): δ 5.28 (m,1H), 3.08-3.02 (m, 1H), 2.89-2.80 (m, 3H), 2.52-2.38(m, 1H), 2.27-2.25(m, 2H), 2.17-1.92(m, 5H), 1.87-1.76(m,3H), 1.67-1.60(m, 2H), 1.55-1.46(m, 1H), 1.35(d, 6H, J=6.8Hz), 1.05(d, 3H, J=7.2Hz), 0.78(s, 3H). 13C NMR (CDCl3 400MHz): δ 210.77, 157.15, 120.93, 63.00, 53.77, 52.95, 49.17, 40.49, 35.34, 34.42, 32.67, 27.11, 23.98, 21.56, 19.58, 17.43, 15.37. IR: 3046(m), 2865(m), 2882(m), 1716(s), 1546(w), 1489(w), 1461(m), 1423(w), 1356(m), 1300(s), 1262(m), 1205(w), 1120(s), 1053(m), 940(m), 873(m), 926(m), 750(m), 703(m), 674(m). HRMS: calculated for C18H30O3S: 327.1934; found: 327.1989.
(+)-AU-16-ene-25-SO2-i-Pr (+)-4
Enantiomerically pure phosphine oxide (-)-8 (0.028 g, 0.046 mmol) was dissolved in THF (1.5ml) in an oven-dried 10 mL round bottom flask with magnetic stir bar and purged with argon. Reaction vessel was cooled to -78°C and nBuLi (1.5 M) (0.031 mL, 0.046 mmol) was added dropwise resulting in a deep red color. After twenty minutes, ketone (+)-7b (0.006 g, 0.018 mmol) was dissolved in THF (1.00 mL), cooled to -78°C, and added to the reaction via cannula. Reaction stirred for 3 hours at -78°C at which time it was quenched at -78°C by the addition of pH 7 buffer (2.00 mL) and allowed to come to room temperature. The reaction mixture was then rinsed into a separatory funnel and extracted with ethyl acetate (3×20 mL). The combined extracts were washed with H2O (1×20 mL), brine (1×20 mL), and dried over magnesium sulfate. The filtrate was concentrated in vacuo to give the crude product which was purified by gradient column chromatography (70% hexanes/30% ethyl acetate to 50% hexanes/50% ethyl acetate) affording 0.010 g of isopropylsulfone TBS protected diol as an oil in 83% yield.
The TBS protected product (0.010 g, 0.015 mmol) was dissolved in acetonitrile (1.00 mL) in an argon-purged 10 mL oven-dried round bottom flask equipped with a magnetic stir bar. After stirring for five minutes, HF (0.030 g, 1.50 mmol, 49% aqueous solution) was added at rt via syringe. The reaction mixture was stirred in the dark at rt for 3 hours. TLC showed consumption of starting material. The reaction was diluted with ether (1.5 mL), and saturated NaHCO3 was added until CO2 liberation stopped. The reaction mixture was then rinsed into a separatory funnel and extracted with ethyl acetate (4×20 mL). The combined extracts were washed with H2O (1×20 mL), brine (1×20mL), and dried over magnesium sulfate. The filtrate was concentrated in vacuo to give the crude product which was purified by flash silica chromatography (100% EtOAc) to afford AU-16-ene-25-SO2-i-Pr (+)-4 (0.0043 g) as an oil in 62% yield.
Data for (+)-4
[α]D23= +1.60 (c=0.10, CHCl3) 1H NMR (CDCl3, 400 MHz): δ 6.33 (d, 1H, J=11.2 Hz), 6.08 (d, 1H, J=11.6Hz), 5.31(m,1H), 5.30(s, 1H), 4.98 (s, 1H), 4.41(m, 1H), 4.21 (m, 1H), 3.10-3.01(m, 1H), 2.87(t, 2H, J=7.6Hz), 2.81-2.78(m, 1H), 2.59-2.56(m, 1H), 2.37-2.27(m, 2H), 2.22-2.11(m, 2H), 2.02-1.98(m, 2H), 1.90-1.84(m, 2H), 1.82-1.71(m, 5H), 1.66-1.59(m, 3H), 1.53-1.44(m, 3H), 1.35(d, 6H, J=7.2Hz), 1.03 (d, 3H, J=6.8 Hz), 0.66 (s, 3H). 13C NMR (CDCl3 400MHz): δ158.80, 147.68, 142.25, 133.15, 124.85, 120.99, 117.00, 111.65, 74.22, 70.71, 66.88, 58.37, 52.80, 50.04, 49.33, 45.21, 42.90, 35.32, 32.61, 29.41, 28.75, 23.56, 21.51, 19.50, 17.05, 15.36, 15.31. IR: 3734(m, br), 2957(m), 2929(m), 2863(m), 1688(w), 1669(w), 1555(m), 1536(m), 1517(m), 1508(m), 1461(m), 1423(w), 1404(w), 1328(m), 1300(m), 1262(s), 1214(m), 1138(w), 1025(m), 968(w), 797(m), 769(s), 712(m), 693(m), 665(m). HRMS: calculated for C27H42O4S: 462.2804; found: 462.2818. UV (MeOH) λmax =262 nm (ε= 15,380).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Okamura WH, Zhu G. Vitamin D. Academic Press; San Diego: 1997. [Google Scholar]
- 2.Vieth R. Bone Miner. 1990;11:267–272. doi: 10.1016/0169-6009(90)90023-9. [DOI] [PubMed] [Google Scholar]
- 3.Holick MF. Vitamin D: Molecular Biology, Physiology, and Clinical Applications. Humana Press; Totowa: 1999. [Google Scholar]
- 4.Feldman D, Glorieux F, Pike JW. Vitamin D. Academic Press; San Diego: 1997. [Google Scholar]
- 5.Posner GH, Kahraman M. Overview: Rational Design of 1a,25-Dihydroxyvitamin D3 Analogs. In: Feldman D, Glorieux F, Pike JW, editors. Vitamin D. 2. Academic Press; San Diego: 2003. [Google Scholar]
- 6.Posner GH. J Nutr. 2002;132:3802S–3803S. doi: 10.1093/jn/132.12.3802S. [DOI] [PubMed] [Google Scholar]
- 7.Bouillon R, Okamura WH, Norman AW. Endocrine Reviews. 1995;16:200. doi: 10.1210/edrv-16-2-200. [DOI] [PubMed] [Google Scholar]
- 8.Usera AR, Dolan PM, Kensler TW, Posner GH. Bioorg Med Chem. 2007;15(16):5509–5518. doi: 10.1016/j.bmc.2007.05.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maienfisch P, Hall RG. Chimia. 2004;58:93–99. [Google Scholar]
- 10.Kirsch P. Modern Fluoroorganic Chemistry Synthesis, Reactivity, Applications. Wiley-VCH; Weinheim: 2004. [Google Scholar]
- 11.Chambers RD. Fluorine in Organic Chemistry. Blackwell Publishing; Oxford: 2004. pp. 1–7. [Google Scholar]
- 12.Uneyama K. Organofluorine Chemistry. Blackwell Publishing; Oxford: 2006. pp. 6–15.pp. 81–82.pp. 173–180.pp. 206–215. [Google Scholar]
- 13.Yamada S, Shimizu M, Yamamoto K. Medical Research Reviews. 2003;23(1):89–115. doi: 10.1002/med.10023. [DOI] [PubMed] [Google Scholar]
- 14.deGraaf C, Vermeulen MPE, Feenstra KA. J Med Chem. 2005;48(8):2725–2755. doi: 10.1021/jm040180d. [DOI] [PubMed] [Google Scholar]
- 15.Posner GH, Wang Q, Han G, Lee JK, Crawford K, Zand S, Brem H, Peleg S, Dolan P, Kensler TW. J Med Chem. 1999;42:3425–3435. doi: 10.1021/jm990267c. [DOI] [PubMed] [Google Scholar]
- 16.Dai H, Posner GH. Synthesis. 1994:1383–1398. [Google Scholar]
- 17.Posner GH, Lee JK, White MC, Hutchings RH, Dai H, Kachinski JL, Dolan P, Kensler TW. J Org Chem. 1997;62:3299–3314. doi: 10.1021/jo970049w. [DOI] [PubMed] [Google Scholar]