

Abstract
Here we study conformational stabilization induced in a β-helical nanostructure by position-specific mutations. The nanostructure is constructed through the self-assembly of the β-helical building block excised from E. coli galactoside acetyltransferase (PDB code 1krr, chain A; residues 131-165). The mutations involve substitutions by cyclic, conformationally constrained amino acids. Specifically, a complete structural analysis of the Pro-Xaa-Val sequence [with Xaa being Gly, Ac3c (1-aminocyclopropane-1-carboxylic acid) and Ac5c (1-aminocyclopentane-1-carboxylic acid)], corresponding to the 148-150 loop region in the wild-type (Gly) and mutated (Ac3c and Ac5c) 1krr, has been performed using Molecular Dynamics simulations and X-ray crystallography. Simulations have been performed for the wild-type and mutants of three different systems, namely the building block, the nanoconstruct and the isolated Pro-Xaa-Val tripeptide. Furthermore, the crystalline structures of five peptides of Pro-Xaa-Val or Xaa-Val sequences have been solved by X-ray diffraction analysis and compared with theoretical predictions. Both the theoretical and crystallographic studies indicate that the Pro-Acnc-Val sequences exhibit a high propensity to adopt turn-like conformations, and this propensity is little affected by the chemical environment. Overall, the results indicate that replacement of Gly149 by Ac3c or Ac5c significantly reduce the conformational flexibility of the target site enhancing the structural specificity of the building block and the nanoconstruct derived from the 1krr β-helical motif.
Introduction
Currently, there is increasing interest in nanostructure design, which involves the ability to predictably manipulate the properties of the self-assembly of autonomous units with preferred conformational states.1-6 The units can be synthetic material science based or derived from functional biological macromolecules. Among the latter, peptides, proteins and nucleic acid molecules and their parts provide biological building blocks that may be assembled in a bottom-up approach into novel architectures, which may differ from those found in nature.5,6 Proteins are particularly suitable candidates for nanostructure design since they provide a broad range of molecular shapes (conformational motifs) that can be used to create diverse scaffolds. Successful design with proteins follows the physical principles of folding, stability and protein-protein interactions.7-9 In hierarchical design strategies, the assembled protein building blocks need to be in their preferred conformational states.10
Recently, we proposed the novel strategy of starting with the nanostructure shape created by making use of a library of protein building blocks with available structures.11-13 Such a strategy is attractive because the richness of the repertoire of the protein building block shapes and chemistries provides an immense array of potential nanoassemblies. The use of protein building blocks has additional advantages, since these structural elements have been perfected by evolution, are easily available, and are biocompatible. The challenge is to be able to predictably modulate them towards the desired goal. To successfully engineer nanostructures using protein building blocks, two criteria should be fulfilled. First, the desired conformation of the selected building block should be stable, and second, the association between the building blocks should be favorable, with an energy gap between the desired self-assembly and all other potential associations. To fulfill the first criterion, selection of a relatively stable structural unit such as a structural repeat taken from a commonly occurring protein architecture14-16 appears as a promising choice. For the second, attempting to preserve a native interface or construct also seems a reasonable approach.17
We have followed this strategy to design self-assembled nanostructures using building blocks extracted from β-helical proteins that contain a tubular or fibrillar motif in their folds.18 More specifically, we selected 17 building blocks from native left-handed β-helical proteins by slicing β-helices into two-turn repeat units. Four copies of each structural unit were stacked one atop the other with no covalent linkage between them. The stability of these self-assembled organizations was investigated through Molecular Dynamics (MD) simulations using atomistic models. We observed that a structural model based on the self-assembly of a two-turn repeat motif from E. coli galactoside acetyltransferase (PDB code 1krr, chain A; residues 131-165) produced a very stable nanoconstruct. Moreover, mesoscopic simulations using explicitly derived coarse-grained models19,20 indicated that the energetic stability of the nanostructure varies with the number of self-assembled building blocks, becoming disfavored after a threshold value (~ 45).
Although we are aware that no experimental evidence demonstrating that a building block consisting of two turns of β-helix remains intact in solution has been reported, the following considerations should be taken into account. Despite the classical vision that hierarchical three dimensional organization of globular proteins is the key event to stabilize secondary structure motifs, recent discoveries have shown that under the proper conditions short segments that present a high density of secondary structure motifs can become conformationally stable once excised from their original proteins.21 Hence, secondary structure repeats can be driven to show similar conformational tendencies when extracted from their native environment. For instance, if such motifs are associated to form longer polypeptide segments by small chemical modifications (such as capping of the terminal coils or the covalent linkage of the repeat segments) the main secondary structural features are preserved,21,22 and the most important non covalent interactions that characterized the structural motif remain mainly unaltered.23 Hence, the construction of nanostructures based on extracting polypeptide segments with high density of secondary structure has become for the last five years a standard strategy within the field of nanobiology. In order to preserve their native fold the building blocks require further manipulation, such as building structural repeats.24-26
In our case, the results previously obtained18 showed that if the proper protocol is found, the resulting nanoconstruct is stable. Our recent efforts have focused on proposing alternative paths to stabilize the native fold, combining in silico studies with the structural features of synthetic amino acids. The hypothetical constructs built in silico allow identification of those polypeptide segments are more flexible. In the context of the polypeptide dynamics, it means pointing to the parts that would most contribute to stabilize the unfolded states, presumed to be the most populated after the peptide is excised from the protein. This information is used to enhance the conformational specificity of β-helical nanoconstructs, by incorporating substitutions with non-proteinogenic amino acids in key positions of those mobile segments along the building blocks.27-30 The idea is to overpopulate the folded conformation by an extra stabilization of the flexible zones of the peptide. In silico this was accomplished by incorporating conformationally restricted amino acids in the most flexible regions of the β-helix, i.e the loops,27-29 even though in a few specific cases stabilization was achieved by replacing residues located in the β-sheet segments.30 It is worth noting that synthetic constrained amino acids were typically created to restrict their accessible conformational spaces to one of the conformations associated to the common secondary structural motifs. Furthermore, constrained amino acids promote such structural motifs in neigboring residues by altering their potential energy hypersurfaces,28,29 which produces a stabilization of the folded state with respect to the unfolded one. The most successful results27,28 were obtained by reducing the mobility of the loop regions by inserting 1-aminocycloalkane-1-carboxylic acids (Acnc, where n indicates the size of the cycloalkane ring), which are cyclic Cα-tetrasubstituted amino acids with a strong tendency to accommodate folded turn-like conformations.31-33 In particular, we observed that when the cyclopentane derivative (Ac5c, Figure 1) replaces Gly149 in the flexible loop defined by residues 148-150 of 1krr, the conformational specificity of both the building block and the self-assembled nanoconstruct increases significantly.28 The Gly149 residue seems therefore to be an appropriate target for position-specific mutations with constrained amino acids.
Figure 1.
Structure of two constrained amino acids of the Acnc family.
Accordingly, the role of Ac5c in the stabilization of the 1krr building block and the corresponding self-assembled nanoconstruct deserve further analysis for future nanobiological developments. Moreover, it is highly desirable to establish whether other residues of the Acnc series -in particular, the cyclopropane member (Ac3c, Figure 1)- produce a similar stabilizing effect when replacing Gly149. Previous studies on peptides incorporating Ac3c31-35 Ac5c31-33,36,37 showed that both residues share some conformational features inherent to their cyclic α-tetrasubstituted nature, while exhibiting distinctive structural properties which may be crucial in stabilizing the 1krr building block and nanoconstruct. It is also important to ascertain whether these conformationally restricted residues produce local stabilization or rather a global reduction of the conformational flexibility of the whole foldamer or/and the nanostructure. In this work, we report a complete structural analysis of the Pro-Xaa-Val sequence (with Xaa being Gly, Ac3c and Ac5c), which corresponds to the 148-150 loop region of the wild-type (Gly) and mutated (Ac3c, Ac5c) 1krr, using both computer simulation and experimental techniques. For this purpose, the influence of Ac3c and Ac5c on the conformation, flexibility and dynamics of the Pro-Xaa-Val sequence has been examined considering the wild-type and mutants of three different systems, which were simulated by MD: (i) the 1krr building block, (ii) the nanostructure constructed by stacking four replicas of the building block, i.e. four self-assembled repeat units, and (iii) the isolated tripeptide. The conformational preferences of the building blocks and the nanoconstructs were investigated following the evolution of the β-helical conformation through MD trajectories, while a conformational search procedure was applied to study the intrinsic conformational preferences of the tripeptides. Furthermore, for the latter system, theoretical predictions have been contrasted with experimental information provided by X-ray crystallography. Specifically, the molecular structure of five peptides of Pro-Xaa-Val or Xaa-Val sequences bearing different terminal groups have been solved by X-ray diffraction analysis. Combination of these experimental data with results derived from MD simulations provides a complete view of how the scenario of the examined loop region changes after the targeted replacements.
Methods
Molecular Dynamics Simulations
Systems Studied
Three different systems based on the building block corresponding to the segment 131-165 of the β-helical protein 1krr18 (Table 1) were simulated in this work. First, the potential stabilization of the loop region 148-150 (Pro-Gly-Val) when Gly149 is replaced by Ac3c and Ac5c was studied considering a single building block. These molecular systems were built using a cubic simulation box (initial dimensions 56.0×56.0×56.0 Å3) with 5656 water molecules. The second series of simulations were centered on the structural features of nanoconstructs built by placing 4 replicas of the β-helical building block assembled one on the top of the other and united via non-covalent interactions. These simulations were designed to examine whether Ac3c and Ac5c replacements are potential nuclei for the global stabilization of both the desired β-helical conformation and the nanoassembly. In these systems the simulation box was orthorhombic (initial dimensions 80.0×80.0×110.0 Å3) with 18041 water molecules. In both the building block and the nanoconstruct series, sodium ions were added to the system to reach electrical neutrality (1 and 4 cations, respectively). Finally, the intrinsic conformational preferences of the isolated tripeptide corresponding to the loop segment were studied in chloroform solution, the wild-type (Pro-Gly-Val) and mutated systems (Pro-Ac3c-Val and Pro-Ac5c-Val) being investigated. These simulations were performed considering a cubic simulation box (initial dimensions 44.5×44.5×44.5 Å3) with 772 chloroform molecules. All systems were conveniently blocked at the Nt and Ct termini by acetyl (Ac, MeCO-) and methylamide (-NHMe) groups, respectively.
Table 1.
Sequence of the 1krr building block used to construct the self-assembled nanostructure.
| PDB | Protein name | Residues | Sequence |
|---|---|---|---|
| 1krr | Galactoside acetyltransferase from E. coli | 131-165 | PITIGNNVWIGSHVVINP GVTIGDNSVIGAGSIVT |
General Simulation Conditions
All trajectories were generated using the NAMD program.38 Simulations of the building blocks and the nanoconstructs were performed in aqueous solution using the TIP3 model39 to represent explicit water molecules, while the OPLS440 model of chloroform was employed for the conformational search of the tripeptides. Energies were calculated using the AMBER force-field,41,42 the required parameters being taken from the AMBER libraries for all the residues with the exception of Ac3c and Ac5c, that were previously parametrized by our research group.36a,43,44
Atom pair distance cutoffs were applied at 14.0 Å to compute the van der Waals interactions. The electrostatic interactions were computed using the non-truncated electrostatic potential with Ewald Summations.45 The real space term was determined by the van der Waals cut off (14.0 Å), while the reciprocal term was estimated by interpolation of the effective charge into a charges mesh with a grid thickness of 5 points per volume unit, i.e. particle-mesh Ewald (PME) method.45 The lengths of bonds involving hydrogen atoms were constrained using the SHAKE algorithm.46 A numerical integration step of 2 fs was used in all simulations.
Before the corresponding MD production series, the thermodynamic variables of each system were equilibrated. For this purpose, the energy of each system was initially minimized to relax conformational and structural tensions using the conjugate gradient method for 5·103 steps. Next, different consecutive rounds of short MD runs were performed in order to equilibrate the density, temperature and pressure. Initially, solvent and charged sodium atoms were thermally relaxed by three consecutive runs, while the protein parts were kept frozen. First, 0.5 ns of NVT-MD at 500 K were used to homogeneously distribute the solvent and ions in the box. Second, 0.5 ns of isothermal and 0.5 ns isobaric relaxation were run. Finally, all the atoms of the system were subjected to 0.15 ns of steady heating until the target temperature was reached (298 K), and then 0.25 ns of NVT-MD at 298 K (thermal equilibration) followed by 0.5 ns of density relaxation (NPT-MD). Both temperature and pressure were controlled by the weak coupling method, the Berendsen thermo-barostat,47 using a time constant for heat bath coupling and a pressure relaxation time of 1 ps. The end of the density relaxation simulation was the starting point of each molecular simulation. All the systems based on 1krr were simulated at 298 K and constant pressure of 1 atm. The coordinates of all the production runs, which were 10 ns long, were saved every 1000 steps (2 ps intervals) for subsequent analysis.
Structural Analyses
The conformational specificity and conservation of the nanostructures was measured by calculating (i) the evolution of the backbone root-mean-square deviation (RMSD) through the simulation relative to the initial structure; and (ii) the root-mean-square fluctuation (RMSF) of individual residues averaged over the whole simulation. Both RMSD and RMSF were computed with respect to the backbone atoms (-N-Cα-C-). The temporal evolution of the backbone dihedral angles φ,ψ was used to show the mobility of the loop region under study. The conformation of a given residue is more clearly defined when the fluctuations of φ,ψ are smaller.
Conformational Search
The potential energy hypersurfaces of the Ac-Pro-Xaa-Val-NHMe tripeptides were explored by consecutive series of heating-cooling MD cycles following the principles of classic simulated annealing strategy.48,49 Thus, for each tripeptide, a randomly generated conformation was brought to 900 K and kept at this temperature for 25 ns, coordinates and velocities being stored every 200 ps. These 125 structures were cooled down to 298 K at a rate of 6 K per 25 ps and, subsequently, their conformational energies were relaxed by 3·103 steps energy minimization. Even though this procedure does not provide a complete exploration of the potential energy hypersurfaces, recent studies demonstrated that the lower energy regions of the conformational space are satisfactorily characterized by combining simulated annealing with molecular dynamics and energy minimization.50,51
Experimental Methods
General
Melting points were determined on a Gallenkamp apparatus and are uncorrected. IR spectra were registered on a Mattson Genesis FTIR spectrophotometer; νmax is given for the main absorption bands. 1H and 13C NMR spectra were recorded on a Bruker AV-400 or ARX-300 instrument at room temperature unless otherwise indicated, using the residual solvent signal as the internal standard; chemical shifts (δ) are expressed in ppm and coupling constants (J) in Hertz. Optical rotations were measured on a JASCO P-1020 polarimeter. High-resolution mass spectra were obtained on a Bruker Microtof-Q spectrometer.
Peptide Synthesis
The preparation of peptides of Pro-Xaa-Val sequence was carried out following standard procedures of peptide synthesis in solution52 starting from the corresponding valine derivative and using the tert-butoxycarbonyl group (Boc, tBuOCO-) as protection for the amino moieties (Figure 2). All coupling reactions were performed by the mixed anhydride method using isobutylchloroformate, while the Boc group was removed by treatment with a 3N solution of hydrogen chloride in anhydrous ethyl acetate. None of the three tripeptides thus obtained, Boc-Pro-Xaa-Val-NHMe (1a-3a), furnished single crystals suitable for X-ray diffraction analysis. For this reason, transformation of the Boc group into pivaloyl (Piv, tBuCO-) or acetyl (Ac, MeCO-) was considered. Pivaloylation and acetylation were carried out, respectively, by reaction with pivaloyl chloride or acetic anhydride in chloroform solution in the presence of N-methylmorpholine. The pivaloyl derivatives of both the Ac3c and Ac5c tripeptides (2b,3b) furnished single crystals of good quality, whereas all efforts to crystallize compounds of the Gly series (1a-c) were unsuccessful. Growing single crystals of the Boc-Xaa-Val-NHMe dipeptides, isolated as intermediates in the synthetic route (Figure 2), was also attempted. At this stage, only the Gly derivative (4a) crystallized. The Ac3c- and Ac5c-containing dipeptides provided single crystals after transformation into the corresponding acetyl (5c) and pivaloyl (6b) derivatives, respectively. The structure of all peptides synthesized is shown in Figure 2 together with their abbreviated codes. All compounds provided satisfactory physical and spectroscopical data. Those whose crystalline structure has been solved are given.
Figure 2.

Left: Synthetic route followed for the preparation of peptides (Xaa = Gly, Ac3c and Ac5c). Reagents: a) 3N HCl / EtOAc; b) iBuOCOCl, N-methylmorpholine (NMM); c) tBuCOCl, NMM; d) Ac2O, NMM. Right: Codes of the peptides synthesized (those whose crystalline structures have been solved by X-ray diffraction analysis are underlined).
Piv-Pro-Ac3c-Val-NHMe (2b)
White solid, mp 188°C (iPr2O/CH2Cl2). : -9.8 (c = 0.33, MeOH). IR (nujol) ν 3357, 3280, 3223, 1698, 1657, 1630, 1608 cm-1. 1H NMR (CDCl3, 400 MHz) δ 0.76-0.81 (m, 1H), 0.83 (d, 3H, J = 6.8 Hz), 0.87 (d, 3H, J = 6.8 Hz), 0.94-1.01 (m, 1H), 1.21 (s, 9H), 1.43-1.49 (m, 1H), 1.53-1.59 (m, 1H), 1.79-1.93 (m, 2H), 1.94-2.04 (m, 1H), 2.07-2.20 (m, 2H), 2.73 (d, 3H, J = 4.8 Hz), 3.67-3.76 (m, 2H), 4.14 (dd, 1H, J = 5.6 Hz, J = 8.0 Hz), 4.24 (dd, 1H, J = 7.2 Hz, J = 9.2 Hz), 6.57 (brs, 1H), 6.88 (m, 1H), 7.24 (d, 1H, J = 9.2 Hz). 13C NMR (CDCl3, 100 MHz) δ 16.44, 17.64, 18.34, 19.39, 26.22, 26.24, 27.23, 27.87, 30.65, 34.65, 38.86, 49.00, 60.31, 62.96, 171.67, 172.48, 174.35, 178.10. HRMS (ESI) C20H34N4O4Na [M+Na]+: calcd. 417.2472, found 417.2456.
Piv-Pro-Ac5c-Val-NHMe (3b)
White solid, mp 160°C (iPr2O/CH2Cl2). : -11.5 (c = 0.43, MeOH). IR (nujol) ν 3347, 1658, 1610 cm-1. 1H NMR (CDCl3, 300 MHz, 60°C) δ 0.93 (d, 3H, J = 6.9 Hz), 0.96 (d, 3H, J = 6.9 Hz), 1.31 (s, 9H), 1.57-1.75 (m, 2H), 1.76-1.96 (m, 4H), 1.97-2.20 (m, 5H), 2.37-2.48 (m, 1H), 2.49-2.60 (m, 1H), 2.79 (d, 1H, J = 4.8 Hz), 3.70-3.84 (m, 2H), 4.33 (dd, 1H, J = 5.1 Hz, J = 8.7 Hz), 4.43 (m, 1H), 6.50 (brs, 1H), 6.79 (m, 1H), 6.99 (d, 1H, J = 8.4 Hz). 13C NMR (CDCl3, 100 MHz) δ 17.39, 19.52, 24.14, 24.43, 26.22, 26.31, 27.30, 27.57, 29.58, 36.87, 38.05, 39.09, 48.86, 59.37, 63.01, 67.28, 172.23, 173.14, 173.83, 178.30. HRMS (ESI) C22H38N4O4Na [M+Na]+: calcd. 445.2785, found 445.2795.
Boc-Gly-Val-NHMe (4a)
White solid, mp 183°C (iPr2O/CH2Cl2). : -21.1 (c = 0.50, MeOH). IR (nujol) ν 3334, 3278, 1700, 1675, 1646 cm-1. 1H NMR (CDCl3, 400 MHz) δ 0.85 (d, 3H, J = 6.8 Hz), 0.88 (d, 3H, J = 6.8 Hz), 1.39 (s, 9H), 2.10 (m, 1H), 2.73 (d, 3H, J = 4.8 Hz), 3.76 (m, 2H), 4.18 (dd, 1H, J = 7.0 Hz, J = 8.6 Hz), 5.29 (m, 1H), 6.46 (m, 1H), 6.79 (m, 1H).13C NMR (CDCl3, 100 MHz) δ 18.01, 19.32, 26.16, 28.31, 30.76, 44.45, 58.65, 80.36, 156.27, 169.84, 171.66. HRMS (ESI) C13H25N3O4Na [M+Na]+: calcd. 310.1737, found 310.1733.
Ac-Ac3c-Val-NHMe (5c)
White solid, mp 193°C. : -14.0 (c = 0.45, MeOH). IR (nujol) ν 3381, 3325, 3301, 1665, 1644 cm-1. 1H NMR (CDCl3, 400 MHz) δ 0.83 (d, 3H, J = 6.8 Hz), 0.87 (d, 3H, J = 6.8 Hz), 0.88-0.93 (m, 1H), 0.98-1.03 (m, 1H), 1.41-1.52 (m, 2H), 1.97 (s, 3H), 2.10 (m, 1H), 2.75 (d, 3H, J = 4.7 Hz), 4.17 (dd, 1H, J = 6.0 Hz, J = 8.7 Hz), 6.78-6.87 (m, 3H). 13C NMR (CDCl3, 100 MHz) δ 16.95, 17.12, 17.78, 19.38, 23.18, 26.22, 30.60, 35.37, 59.03, 171.66, 172.02, 172.20. HRMS (ESI) C12H21N3O3Na [M+Na]+: calcd. 278.1475, found 278.1477.
Piv-Ac5c-Val-NHMe (6b)
White solid, mp 159°C (Et2O/CH2Cl2). : -38.1 (c = 0.27, AcOH). IR (nujol) ν 3344, 3306, 1667, 1641 cm-1. 1H NMR (CDCl3, 300 MHz, 60°C) δ 0.87 (d, 3H, J = 6.9 Hz), 0.91-0.99 (m, 4H), 1.23 (s, 9H), 1.66-1.99 (m, 5H), 2.11-2.30 (m, 1H), 2.39-2.54 (m, 2H), 2.79 (d, 3H, J = 4.8 Hz), 4.29 (dd, 1H, J = 4.8 Hz, J = 9.0 Hz), 5.92 (brs, 1H), 6.59 (m, 1H), 6.65 (brd, 1H, J = 9.0 Hz). 13C NMR (CDCl3, 100 MHz) δ 16.80, 19.06, 19.61, 23.94, 24.14, 26.24, 27.42, 29.14, 36.34, 37.69, 58.34, 67.41, 171.64, 173.41, 179.95. HRMS (ESI) C17H31N3O3Na [M+Na]+: calcd. 348.2258, found 348.2243.
X-Ray Diffraction
Single crystals of 2b, 3b, 4a, 5c and 6b were grown by slow evaporation from the solvents indicated in Table 2, where the main crystallographic data are also given. The X-ray diffraction data were collected at 100 K (6b), 115 K (4a) or room temperature (2b, 3b, 5c) on an Oxford Diffraction Xcalibur diffractometer provided with a Sapphire CCD detector, using graphite-monochromated Mo-Kαradiation (λ = 0.71073 Å). The structures were solved by direct methods using SHELXS-9753a and refinement was performed with SHELXL-9753b by the full-matrix least-squares technique with anisotropic thermal factors for heavy atoms. Hydrogen atoms were located by calculation (with the exception of the amide protons, which were found on the E-map) and affected by an isotropic thermal factor fixed to 1.2 times the Ueq of the carrier atom (1.5 for the methyl groups). Crystallographic data (excluding structure factors) for the structures in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication numbers CCDC 676501 to 676505. Copies of the data can be obtained, free of charge, on application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK [fax: +44(0)-1223-336033 or e-mail: deposit@ccdc.cam.ac.uk].
Table 2.
Crystallographic data.
| Piv-Pro-Ac3c-Val-NHMe | Piv-Pro-Ac5c-Val-NHMe | Boc-Gly-Val-NHMe | Ac-Ac3c-Val-NHMe | Piv-Ac5c-Val-NHMe | |
|---|---|---|---|---|---|
| 2ba | 3b | 4a | 5c | 6b | |
| Crystallization solvent | Et2O/CH2Cl2 | iPr2O/CH2Cl2 | iPr2O/CHCl3 | Et2O/CH2Cl2 | Et2O/CH2Cl2 |
| Crystal system | monoclinic | orthorhombic | orthorhombic | orthorhombic | orthorhombic |
| Space group | C2 | P212121 | P212121 | P212121 | P212121 |
| Unit cell dimensions a (Å) | 28.576(3) | 10.0590(6) | 9.0788(4) | 8.5773(2) | 9.4086(7) |
| b (Å) | 9.443(2) | 11.1909(5) | 9.4696(4) | 9.5980(3) | 11.0310(12) |
| c (Å) | 10.241(2) | 20.7908(9) | 18.1496(9) | 17.7198(4) | 18.4029(15) |
| β (deg.) | 106.566(17) | 90 | 90 | 90 | 90 |
| Z | 4 | 4 | 4 | 4 | 4 |
| Calculated density (g·cm-3) | 1.226 | 1.199 | 1.223 | 1.163 | 1.132 |
| Reflections collected / unique | 5386 / 3782 | 27691 / 4346 | 28118 / 2896 | 29374 / 2724 | 6929 / 3075 |
| (Rint = 0.061) | (Rint = 0.093) | (Rint = 0.055) | (Rint = 0.083) | (Rint = 0.064) | |
| Residual factors [I > 2σ(I)] R1 | 0.060 | 0.037 | 0.030 | 0.034 | 0.042 |
| wR2 | 0.065 | 0.066 | 0.060 | 0.067 | 0.070 |
| Residual factors [all data] R1 | 0.163 | 0.102 | 0.047 | 0.105 | 0.133 |
| wR2 | 0.079 | 0.076 | 0.062 | 0.077 | 0.081 |
| Largest diff. peak / hole (e· Å-3) | 0.15 / -0.17 | 0.22 / -0.12 | 0.15 / -0.14 | 0.10 / -0.09 | 0.14 / -0.18 |
Monohydrate, dichloromethane solvate.
Results and Discussion
The stabilization produced in both the building block and the self-assembled nanoconstruct when Gly149 is replaced by Ac5c in the excised 1krr motif was recently reported.28 In order to provide a reliable comparison of the stabilizing effects imparted by Ac3c and Ac5c when replacing this Gly residue (hereafter denoted as G149Ac3c and G149Ac5c mutants, respectively), in this work we have performed new MD simulations on the wild-type and the two mutants using identical simulation conditions. It should be remarked that, although the conformational specificity of the mutated building blocks and nanoconstructs is clearly evidenced in the next sections, the present work focuses on the structural aspects related to the 148-150 loop region, i.e. the Pro-Xaa-Val sequence. Accordingly, analysis and discussion of results mainly concentrate on the conformational description of the three residues involved in such peptide fragment.
Effects of the Mutation on the Building Block
The temporal evolution of the backbone RMSD of the building block derived from the 1krr β-helix for the wild-type sequence and the G149Ac3c and G149Ac5c mutants is depicted in Figure 3a. As can be seen, the RMSD values of both mutants are significantly smaller than those of the wild type. Furthermore, among the mutants, the structural deviations observed for G149Ac5c are slightly lower than those corresponding to the G149Ac3c system.
Figure 3.
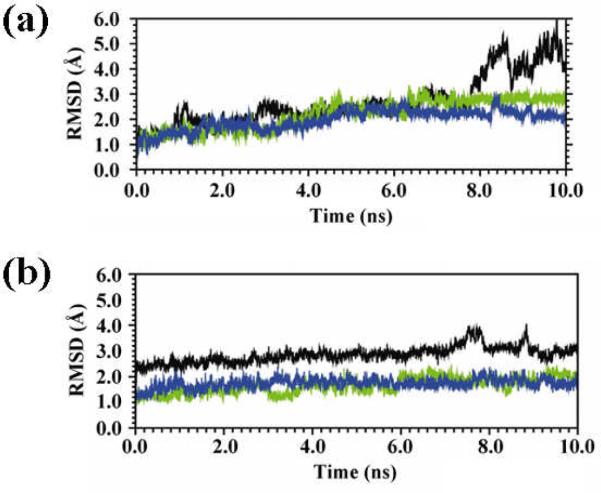
Evolution of the backbone RMSD of the building block (a) and the self-assembled nanoconstruct (b) derived from the 1krr β-helix for the wild-type sequence (black), the G149Ac3c mutant (green) and the G149Ac5c mutant (blue).
Figure 4 shows the temporal evolution of the backbone dihedral angles Φ and Ψ for the 148-150 Pro-Xaa-Val sequence (Xaa = Gly, Ac3c and Ac5c) as well as its neighboring residues derived from the simulations of the wild-type, G149Ac3c and G149Ac5c building blocks. As can be seen, Ac3c and Ac5c adopt a well-defined conformation, the variations of the Φ,Ψ dihedral angles being minimal in both cases. In contrast, large fluctuations are detected for the dihedral angle Φ of the Gly residue, which frequently exchanges from - 170° to -90°, i.e. the conformations adopted by Gly in the building block correspond to Φ,Ψ≈ -170°,0° and -90°,0° with half-life periods of ~ 0.8 and ~ 2.2 ns, respectively. The conformational rigidity introduced by the cyclic amino acids Ac3c and Ac5c also seems to affect the contiguous residues. Thus, the Val150 Φ and Pro148 Ψ angles are significantly different in the wild-type and in the two mutants. Specifically, Val150 Φ changes from ~ - 140° (wild-type) to ~ -60° (mutants), while Pro148 Ψ varies from ~ -30° (wild-type) to ~ 165° (mutants). Moreover, substitution of Gly149 also induces significant changes in the dihedral angle Ψ of both Asn147 (from ~ -30° to ~ 150°) and Thr151 (from ~ 170° to ~ - 40°), which are reflected in Figure 4.
Figure 4.

Temporal evolution of the backbone dihedral angles φ (a) and ψ (b) of the Asn147, Pro148, Xaa149, Val150 and Thr151 residues obtained from 10 ns MD simulations of the wild-type (Xaa = Gly, in black) and the G149Ac3c (Xaa = Ac3c, in green) and G149Ac5c (Xaa = Ac5c, in blue) mutants of the 1krr building block.
Interestingly, inspection of the RMSD (Figure 3a) and, especially, the RMSF (Figure 5a) values of the G149Ac3c and G149Ac5c building blocks reveals that the conformational specificity of the β-helix motif is enhanced not only by a decrease in the flexibility of the target site but also by the local conformational rearrangements undergone by the neighboring residues, which help to eliminate unfavorable energetic contributions. Indeed, the overall of the results indicate that such rearrangements extend to two residues at each side, and the RMSF also illustrates that the most significant ones affect those immediately adjacent.
Figure 5.

RMSF of the building block (a) and the self-assembled nanoconstruct (b) derived from the 1krr β-helix for the wild-type sequence (black), the G149Ac3c mutant (green) and the G149Ac5c mutant (blue). Red dashed lines indicate the position of the replacements.
A detailed analysis of the hydrogen bonds involving residues 147-151 for the wild-type and the mutated building blocks revealed the existence of up to eight different types of hydrogen-bonding interactions, which are listed and briefly described in Table 3. These interactions, which have been labeled using Roman numbers, can be classified into three groups: (a) backbone-backbone hydrogen bonds giving rise to turn-like conformations (interactions I and II); (b) side chain-backbone hydrogen bonds (interactions III-V); and (c) long-distance backbone-backbone hydrogen bonds involving residues 147-151 and those located in other turn repeat units, i.e. either below or above the segment under study (interactions VI-VIII). In all cases, hydrogen bonds were assigned using the following criteria: hydrogen⋯acceptor distance lower than 2.5 Å and donor-hydrogen⋯acceptor angle larger than 120°.
Table 3.
Hydrogen bonding interactions detected in the wild-type and/or the G149Xaa mutants (Xaa = Ac3c, Ac5c) of the 1krr building block and the corresponding self-assembled nanoconstructs.
| Label | Donor⋯ Acceptor | Comments |
|---|---|---|
| I | (Pro148) C=O⋯H-N (Val150) | Typically found in turn-like conformations |
| II | (Asn147) C=O⋯H-N (Val150) | Typically found in turn-like conformations |
| III | (Asn147) N-H⋯O=C (Pro148) | Asn interacts through the side chain |
| IV | (Xaa149) C=O⋯H-O (Thr151) | Thr interacts through the side chain |
| V | (Val1 50) C=O⋯H-O (Thr151)) | Thr interacts through the side chain |
| VI | (Asn147) N-H⋯O=C (Val164) | Asn interacts with a residue of the other turn repeat unit |
| VII | (Pro131) C=O⋯H-N (Xaa149) | Residue 149 interacts with a residue of the other turn repeat unit |
| VIII | (Ac) C=O⋯H-N (Xaa149) | Residue 149 interacts with the blocking group |
Figure 6 shows the temporal evolution of the I-VIII hydrogen bonds for the wild-type, G149Ac3c and G149Ac5c building blocks. Only two significant interactions are detected for the wild-type building block, which correspond to the formation of a turn-like conformation in the loop (I) and the hydrogen bond between Asn147 and the next turn repeat unit (VI). Surprisingly, when Ac3c replaces Gly149 such interactions disappear in about 4ns, the resulting mutant being dominated by the interaction between the hydroxylic side chain of Asn147 and the Pro148 carbonyl group (III). In contrast, the two interactions found for the wild-type are not only retained but even enhanced in the Ac5c-containing mutant, the most notable difference between the two systems being the coexistence of interactions I and II in the latter. Furthermore, the hydroxyl side group of Thr151 is hydrogen bonded to the Ac5c carbonyl (IV), thus enhancing the conformational rigidity of the loop.
Figure 6.
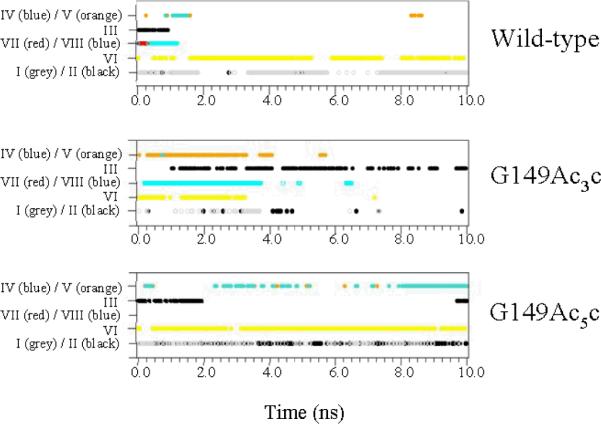
For the simulations of the wild-type, G149Ac3c and G149Ac5c building blocks, spatio-temporal evolution of the hydrogen bonds involving the segment comprised between residues 147 and 151. See Table 3 for definition of the hydrogen bond types.
Effects of the Mutation on the Nanoconstruct
The RMSD and RMSF profiles of the nanoconstructs built by stacking four replicas of the 1krr β-helical building block are displayed in Figures 3b and 5b, respectively. As can be seen, the nanostructures of the two mutants are clearly more stable than the wild-type one. Thus, although the latter is relatively stable on its own, replacement of Gly149 by Acnc residues reduces the flexibility of the system not only in this loop region but in the whole nanostructure (Figure 5b). This is visually evidenced for the G149Ac3c mutant by the selected snapshots depicted in Figure 7.
Figure 7.

Snapshots of the G149Ac3c self-assembled nanoconstruct taken at the beginning of the simulation and after 2.5, 5.0, 7.5 and 10.0 ns of MD.
In order to provide a representative description of the conformational effects produced by the synthetic residues on the G149Ac3c and G149Ac5c nanoconstructs, we concentrated on the analysis of the two central self-assembled structural units. The temporal evolution of the Φ,Ψ dihedral angles for the segment comprised between residues 147 and 151 in each of these two units is displayed in Figure 8. When Xaa is Gly, the results differ considerably from those discussed in the previous section for an isolated building block. Indeed, the backbone conformation of the wild-type nanoconstruct is similar to that obtained for the mutants of the isolated building block. Accordingly, self-assembling interactions seem to induce the conformational re-organization of the loop motif in the structural units of the nanoconstruct. Furthermore, the replacement of Gly149 by Ac3c or Ac5c in the nanoconstruct does not produce any significant conformational change in the backbone of the substituted position nor in its neighboring residues. The largest change occurs for the dihedral angle Φ of residue 149, which moved from around 120° (wild-type) to 90° (G149Ac3c) and 65° (G149Ac5c). However, a detailed inspection of the data provided in Figure 8 reveals that the fluctuations of the dihedral angles are smaller in the mutants than in the wild-type nanoconstruct, which is especially evident for the Φ dihedral of residue 149. Analyses of the RMSD (Figure 3b) and, especially, the RMSF (Figure 5b) values also evidence that the fluctuations are significantly smaller for the mutants than for the wild-type system. Thus, replacement of Gly by Ac3c or Ac5c produces an increase of the backbone rigidity without apparent structural rearrangements, which leads to the stabilization of the nanoconstruct.
Figure 8.

Temporal evolution of the backbone dihedral angles φ (a) and ψ (b) of the Asn147, Pro148, Xaa149, Val150 and Thr151 residues located in the two central structural units of the wild-type (Xaa = Gly, in black) and the G149Ac3c (Xaa = Ac3c, in green) and G149Ac5c (Xaa = Ac5c, in blue) mutants of the self-assembled nanoconstruct.
The temporal evolution of the hydrogen bonding interactions for the two central structural units of the three nanoconstructs is shown in Figure 9. Interestingly, the wild-type system is dominated by two interactions, VI and VIII, that occur between the residues of the segment investigated (Asn147 and Gly149, respectively) and residues located in the other turn repeat unit of the same building block (Val164 and the terminal acetyl group, respectively). Furthermore, a third type of interaction is detected in the wild-type nanoconstruct, although with lower frequency than VI and VIII. It corresponds to the hydrogen bond between the side chain of Thr151 and the backbone carbonyl group of Val150 (V). The hydrogen bond pattern of the G149Ac3c nanostructure is very similar to that discussed for the wild-type system. Indeed, the most remarkable difference is that the tendency to adopt turn-like conformations is higher for the mutant than for the wild-type system, as revealed by the intermittent formation of hydrogen bonds of type I along the last 7 ns of MD simulation.
Figure 9.

For the simulations of the wild-type, G149Ac3c and G149Ac5c self-assembled nanoconstructs, spatio-temporal evolution of the hydrogen bonds involving the segment comprised between residues 147 and 151 of the two central structural units. See Table 3 for definition of the hydrogen bond types.
Finally, the G149Ac5c nanoconstruct shows two differences in the hydrogen bonding interactions with respect to the wild-type and G149Ac3c. First, the turn-like conformation adopted by the Ac3c-containing mutant corresponds to a seven-membered hydrogen-bonded ring (C7 or γ-turn), while a ten-membered one (C10 or β-turn) is found in the nanostructure incorporating Ac5c. This is fully consistent with the intrinsic conformational properties exhibited by these synthetic residues.31-37 Thus, although both Ac3c and Ac5c show a high propensity to adopt turn-like conformations, the ability to accommodate a γ-turn motif has been observed experimentally for cyclopropane residues only.54-56 Second, no trace of interaction VIII was detected in the G149Ac5c nanoconstruct, hydrogen bonds between the segment under study and the adjacent turn repeat unit within the same building block being restricted to the VI type.
Comparison with the Crystal Structure of 1krr
Table 4 lists the backbone dihedral angles for residues 147-151 as found in the crystal structure of the 1krr β-helical protein, which has been extracted from the PDB.57 As can be seen, these values are significantly close to those obtained by simulating the self-assembled nanoconstruct (Figure 8) and, indeed, most of the backbone dihedral angles of residues 147-151 differ by less than 30°. However, comparison of the crystal structure with the dihedrals predicted for the building block reveals a higher discrepancy, particularly, for the wild-type system, where important variations are observed for the Asn147, Pro148 and Gly149 residues (Figure 4). This feature indicates that the loop formed by residues 147-151 adapts its conformation depending on the neighboring chemical environment. It should be noted that in the crystallized protein, different secondary and tertiary structures surround the 131-165 β-helical sequence, while in the self-assembled nanoconstruct this motif is flanked by identical repeating units, and water molecules surround the excised β-helical fragment when it is examined as an isolated building block. This conformational change appears to be more pronounced when the flexible Gly residue is present, whereas the Ac3cand Ac5c-containing sequences are less sensitive to environmental changes.
Table 4.
Backbone dihedral angles (in degrees) of residues 147-151 in the crystal structure of the β-helical protein1krr. Data extracted from the Protein Data Bank (PDB).
| Asn147 | Pro148 | Gly149 | Val1 50 | Thr151 | |
|---|---|---|---|---|---|
| φP | -70.8 | -51.9 | 95.7 | -69.0 | -152.8 |
| ψ | 135.4 | 140.5 | -21.6 | 159.0 | 148.9 |
The crystalline structure of the segment excised from 1krr to create the building block is displayed in Figure 10, which shows the hydrogen bonding interactions involving residues 147-151. The most remarkable difference between the crystallized protein and the simulated elements involves the backbone-backbone hydrogen bond of type II between the CO and NH groups of Asn147 and Val150, respectively, which is consistent with the formation of a β-turn (C10 conformation). Furthermore, the hydroxylic side chain of Thr151 is hydrogen bonded to the Val150 carbonyl group (interaction V in Table 3). Finally, some long-range interactions involving one residue of the loop, such as VI and VII, are also detected in the crystal structure of the β-helix (not shown in Figure 10). It should be noted that, in the simulated nanostructure (Figure 9), interactions V and VI were clearly detected, whereas VIII was favored over VII.
Figure 10.
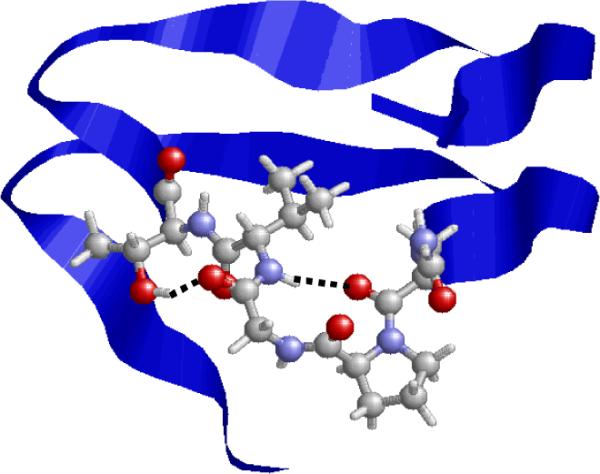
Fragment 131-165 in the X-ray crystal structure of the β-helical protein 1krr (galactoside acetyltransferase from E. Coli). Residues 147-151 are displayed using explicit atoms, while a blue stripe is used to represent the rest of the sequence. Hydrogen-bonding interactions found in the loop defined by residues 147-151 are indicated by dashed lines.
Conformational Preferences of the Pro-Xaa-Val Tripeptides
In order to ascertain how the intrinsic conformational properties of the residue occupying the targeted position (Xaa149) affect the structural behavior of the segment comprised between residues 147 and 151 of 1krr in both the isolated building block and the nanoconstruct, the potential energy surfaces of the Ac-Pro-Xaa-Val-NHMe (Xaa = Gly, Ac3c and Ac5c) tripeptides have been explored. Figure 11 represents the position of the minima found for the three compounds in the Φ,Ψ Ramachandran map of each residue. Table 5 lists the backbone dihedral angles of the minima with a relative energy lower than 2.5 kcal/mol, which are depicted in Figure 12. It should be noted that the terminal acetyl and methylamide groups in these tripeptides mimic, respectively, the Asn147 CO and Thr150 NH moieties in the protein fragment. However, interactions involving the functionalized side chains of Asn and Thr are not possible in these systems, as are not long-range interactions. Accordingly, these tripeptides are useful models to compare the conformational propensities of Gly, Ac3c and Ac5c within the sequence under investigation, but caution should be taken when comparing the results in this section with those described above for the isolated building blocks and the nanoconstructs.
Figure 11.

Comparison between the conformational preferences of the different residues in (a) Ac-Pro-Gly-Val-NHMe, (b) Ac-Pro-Ac3c-Val-NHMe and (c) Ac-Pro-Ac5c-Val-NHMe. The dihedral angles represented in the conformational maps correspond to the minima found by MD simulated annealing.
Table 5.
Relative energies (ΔE, in kcal/mol) and backbone dihedral angles (in degrees) of the Ac-Pro-Xaa-Val-NHMe (Xaa = Gly, Ac3c, Ac5c) conformers whose relative energy is within 2.5 kcal/mol above the global minimum.
| Conf. | ΔE | Pro |
Xaaa |
Val |
H-bonded conformation | |||
|---|---|---|---|---|---|---|---|---|
| φ | ψ | φ | ψ | φ | ψ | |||
| Ac-Pro-Gly-Val-NHMe | ||||||||
| #1 | 0.0 | -69.5 | 8.7 | -134.3 | 9.2 | -140.2 | -20.4 | C10, C13 |
| #2 | 1.0 | -51.3 | 65.8 | 150.2 | -18.2 | -137.7 | -6.5 | C7, C10, C13 |
| #3 | 1.8 | -50.1 | -18.7 | -102.1 | 0.7 | -133.9 | 7.9 | C10 |
| #4 | 2.5 | -73.7 | 41.6 | -64.6 | -9.3 | -137.4 | 27.7 | C7, C10 |
| Ac-Pro-Ac3c-Val-NHMe | ||||||||
| #1 | 0.0 | -42.5 | 102.0 | 81.9 | 2.2 | -136.2 | -17.7 | C10, C13 |
| #2 | 2.2 | -53.9 | -17.7 | -107.1 | 1.9 | -137.5 | -26.5 | C10, C13 |
| Ac-Pro-Ac5c-Val-NHMe | ||||||||
| #1 | 0.0 | -47.9 | 109.5 | 57.3 | 13.5 | -135.8 | -8.4 | C10, C13 |
| #2 | 2.5 | -50.2 | -26.6 | -54.8 | -14.4 | -127.4 | 18.3 | C10, C10 |
Xaa stands for Gly, Ac3c or Ac5c.
Figure 12.

Atomistic representation (balls and sticks) of the most relevant minima characterized for each tripeptide studied. The atom colors code is the CPK and the hydrogen bonds have been depicted.
The Φ,Ψ maps obtained for Ac-Pro-Gly-Val-NHMe indicate that, despite the presence of proline, whose Φ angle is confined to values around -60°, the conformational freedom of this tripeptide is relatively large. Thus, the flexible Gly residue adopts Φ,Ψ values typically associated with helical, turn-like and fully extended conformations, with equal preference for the left and right half of the Ramachandran map, corresponding to its achiral nature. However, all the Ac-Pro-Gly-Val-NHMe minima in Table 5 are characterized by a hydrogen bond between the acetyl CO group and the Val NH moiety, which forms the tenmembered cycle typical of a β-turn. This result is in line with the well known propensity of proline to occupy the i+1 position of β-turns58,59 and, therefore, of Pro-Xaa sequences to adopt this structural motif.58-61 Simultaneously, C13 and C7 hydrogen-bonded rings are present in some of these minima, where the acetyl carbonyl oxygen acts as a multiple hydrogen bond acceptor (Figure 12). On the other hand, Figure 13 shows the superposition of the segment Pro-Gly-Val in the 1krr crystal structure with all the minimum energy structures introduced in Table 5 for the Ac-Pro-Gly-Val-NHMe tripeptide. As can be seen, calculations on this small model compound reproduce very satisfactorily the conformational preferences experimentally found for the Pro-Gly-Val sequence in the 1krr protein.
Figure 13.
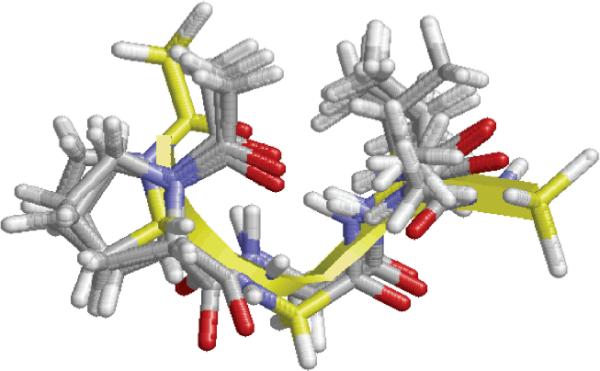
Superposition of the segment Pro-Gly-Val in the 1krr crystal structure (backbone highlighted in yellow) with all minimum energy structures introduced in Table 5 for the Ac-Pro-Gly-Val-NHMe tripeptide.
The behavior exhibited by Ac-Pro-Ac3c-Val-NHMe shows some important differences with respect to that discussed above for the Gly tripeptide. First, only two minima were characterized within an energy interval of 2.5 kcal/mol for the compound incorporating Ac3c, which reflects a notable restriction of the backbone flexibility. As shown in Figure 11, the φψ values preferred by the cyclopropane residue fall in the so-called bridge region of the Ramachandran map φψ ≈ ±90°,0°). This result is in agreement with the conformational propensities observed earlier for this amino acid using both theoretical34 and experimental35 methods. Interestingly, the two most stable minima characterized for Ac-Pro-Ac3c-Val-NHMe exhibit the same hydrogen-bonding scheme (Figure 12, Table 5), namely the acetyl CO interacts with both the Val NH and the terminal NHMe, thus leading to the coexistence of C10 and C13 intramolecularly hydrogen-bonded conformations. Indeed, the two minima differ only in the β-turn type accommodated by the Pro-Ac3c sequence, which corresponds to types I and II,60 respectively, in minima #2 and #1. Previous experimental studies on a Pro-Ac3 c dipeptide62 actually evidenced the presence of both β-turn types in chloroform and dichloromethane solutions.
The conformational space available to Ac-Pro-Ac5c-Val-NHMe was also found to be more restricted than that corresponding to the Gly tripeptide, with only two minima being located within 2.5 kcal/mol. The lowest energy minimum of Ac-Pro-Ac5c-Val-NHMe is characterized by intramolecularly hydrogen-bonded conformations of the C10 and C13 type and is very similar to the global minimum located for the Ac3c tripeptide not only in terms of hydrogen-bonded interactions but also regarding the dihedral angles. In contrast, the second minimum of Ac-Pro-Ac5c-Val-NHMe presents two consecutive β-turns (two C10 conformations), a structural motif not found for the Ac3c counterpart. This distinct behavior should be ascribed to the different conformational propensities inherent to Ac3c and Ac5c.31-37 Both are achiral cyclic Cα-tetrasubstituted α-amino acids and, as such, give rise to symmetrical Ramachandran maps (Figure 11), in which the conformational space available is clearly reduced with reference to Gly. However, remarkable differences are observed between them. Thus, the cyclopentane residue appears to exert a higher restriction on the values accessible to the φ angle, which are confined to two narrow regions near 60° and -60°. In comparison, the ψ dihedral is severely constrained in Ac3c and, indeed, this amino acid is known34,35 to fix the ψ value near 0° due to hyperconjugative effects between the cyclopropane ring and the adjacent carbonyl moiety.63
X-Ray Diffraction Studies
The conformational properties in solution predicted by MD simulations for the different systems considered and, in particular, for the Ac-Pro-Xaa-Val-NHMe tripeptides, have been compared with those observed in the crystalline state for related small peptides. The peptides synthesized in this work are shown in Figure 2 (see Methods section) and the molecular structures of those which yielded single crystals suitable for X-ray diffraction analysis are displayed in Figure 14. They correspond to two tripeptides of Pro-Ac3c-Val and Pro-Ac5c-Val sequences (2b and 3b, respectively) and three dipeptides of Gly-Val, Ac3c-Val and Ac5c-Val sequences (4a, 5c and 6b, respectively), bearing different terminal groups. Table 6 lists their backbone dihedral angles, while the intramolecular and intermolecular hydrogen bonds characterized in the crystalline structures are given in Tables 7 and 8, respectively. As in the previous section, the short length of the peptides crystallized does not allow the establishment of long-range hydrogen-bonding interactions. However, in the solid state, the CO and NH groups of neighboring peptide molecules may form hydrogen bonds, and these intermolecular interactions compete with intramolecular ones that stabilize turn-like conformations. The crystallographic studies in this section therefore allow us to determine whether the structural tendencies predicted for the Ac-Pro-Xaa-Val-NHMe tripeptides in a non-solvating medium are kept in an environment less propitious to intramolecular hydrogen bonding such as the crystal state.
Figure 14.

X-ray structure of the peptides crystallized. Heteroatoms (N,O) are shown as thermal ellipsoids and the intramolecular hydrogen bonds are represented as dashed lines. Most H atoms have been omitted for clarity.
Table 6.
Backbone dihedral angles (in degrees) in the X-ray structures solved in this work.
| Peptide | Pro |
Xaaa |
Val |
H-bonded conformation | |||
|---|---|---|---|---|---|---|---|
| φ | ψ | φ | ψ | φ | ψ | ||
| Piv-Pro-Ac3c-Val-NHMe (2b) | -65 | 143 | 74 | 2 | -130 | -49 | C10, C13 |
| Piv-Pro-Ac5c-Val-NHMe (3b) | -61 | 125 | 62 | 19 | -74 | 135 | C10 |
| Boc-Gly-Val-NHMe (4a) | -156 | 155 | -130 | 144 | - | ||
| Ac-Ac3c-Val-NHMe (5c) | -77 | -11 | -129 | 159 | - | ||
| Piv-Ac5c-Val-NHMe (6b) | -60 | -32 | -90 | 7 | C10 | ||
Xaa stands for Gly, Ac3c or Ac5c.
Table 7.
Parametersa of the intramolecular hydrogen bonds in the X-ray structures solved in this work.
| Peptide | Donor (D) | Acceptor (A) | d (H⋯A) | d (D⋯A) | < (D-H⋯A) | H-bonded conformation |
|---|---|---|---|---|---|---|
| Piv-Pro-Ac3c-Val-NHMe (2b) | (Val)NH | (Piv)CO | 2.35 | 3.034(5) | 129 | C10 |
| MeNH | (Piv)CO | 1.86 | 2.852(6) | 150 | C13 | |
| Piv-Pro-Ac5c-Val-NHMe (3b) | (Val)NH | (Piv)CO | 2.13 | 3.201(3) | 168 | C10 |
| Piv-Ac5c-Val-NHMe (6b) | MeNH | (Piv)CO | 1.92 | 2.934(3) | 163 | C10 |
Distances (d) in Å; angles in degrees.
Table 8.
Parametersa of the intermolecular hydrogen bonds in the X-ray structures solved in this work.
| Peptide | Donor (D) | Acceptor (A) | Symmetry operation | d (H⋯A) | d (D⋯A) | < (D-H⋯A) |
|---|---|---|---|---|---|---|
| Piv-Pro-Ac3c-Val-NHMe (2b) | (Ac3c)NH | (Ac3c)CO | -x+3/2, y-1/2, -z | 2.02 | 2.962(5) | 156 |
| (W1)Hb | (Val)CO | x, y, z | 1.84 | 2.847(5) | 168 | |
| (W2)Hb | (Val)CO | x, y, z | 1.94 | 2.897(5) | 172 | |
| Piv-Pro-Ac5c-Val-NHMe (3b) | (Ac5c)NH | (Pro)CO | x-1/2, -y+1/2, -z+2 | 2.21 | 3.100(3) | 160 |
| MeNH | (Val)CO | x+1/2, -y+3/2, -z+2 | 2.06 | 3.096(3) | 157 | |
| Boc-Gly-Val-NHMe (4a) | (Gly)NH | (Val)CO | -x+1, y+1/2, -z+1/2 | 2.00 | 2.9396(18) | 166 |
| (Val)NH | (Gly)CO | -x+1, y-1/2, -z+1/2 | 1.98 | 2.9090(17) | 160 | |
| MeNH | (Boc)CO | -x+1, y+1/2, -z+1/2 | 1.95 | 2.9119(18) | 175 | |
| Ac-Ac3c-Val-NHMe (5c) | (Ac3c)NH | (Ac)CO | x+1/2, -y+1/2, -z | 2.27 | 2.921(2) | 122 |
| (Ac3c)NH | (Val)CO | x+1/2, -y+1/2, -z | 2.51 | 3.263(2) | 132 | |
| (Val)NH | (Ac)CO | x+1/2, -y+1/2, -z | 2.65 | 3.428(2) | 131 | |
| MeNH | (Ac3c)CO | -x+1, y+1/2, -z+1/2 | 1.85 | 2.880(3) | 164 | |
| Piv-Ac5c-Val-NHMe (6b) | (Ac5c)NH | (Ac5c)CO | -x+1, y-1/2, -z+1/2 | 2.06 | 2.957(4) | 145 |
Distances (d) in Å; angles in degrees.
The peptide cocrystallized with two different water molecules (W1,W2).
As shown in Figure 14, the crystalline structure of Piv-Pro-Ac3c-Val-NHMe (2b) is characterized by the simultaneous presence of intramolecularly hydrogen-bonded conformations of the C10 and C13 types, with the pivaloyl CO group acting as the acceptor and the Val NH (C10) and the terminal NHMe (C13) being the donors (Table 7). In comparison, the Pro-Ac5c-Val derivative (3b) accommodates a β-turn (C10) conformation, while none of the Gly-containing tripeptides yielded single crystals of good quality probably due to the higher intrinsic flexibility of the Pro-Gly-Val sequence. Thus, although the global minimum predicted for all three Ac-Pro-Xaa-Val-NHMe tripeptides was characterized by the coexistence of C10 and C13 intramolecular hydrogen-bonded rings (Table 5), this double turn conformation is observed only in the crystalline Ac3c derivative. This could be indicative of a higher propensity of this amino acid to induce such a structural motif. Indeed, the two minima located for Ac-Pro-Ac3c-Val-NHMe within 2.5 kcal/mol exhibited this conformation. Moreover, the dihedral angles in the crystalline structure of 2b (Table 6) are in good agreement not only with those of the global minimum characterized for the Ac3c tripeptide (Table 5) but also, with exception of the ψ Val angle, with those detected for the Pro-Ac3c-Val sequence in both the building block and the nanoconstruct (Figures 4 and 8). Figure 15a shows the superposition of the crystal structure found for 2b with the minima listed in Table 5 for the model tripeptide, and snapshots separated by regular intervals of time of both the building block and the nanoconstruct. The excellent concordance indicates that the conformation induced by this restricted synthetic amino acid in the neighboring residues is scarcely influenced by the chemical environment.
Figure 15.

Superposition of the crystal structure found for 2b (a) and 3b (b) with the minima listed in Table 5 for the corresponding model tripeptide (left panel), and ten recorded snapshots separated by regular intervals of time of both the building block (central panel) and the nanoconstruct (right panel).
As mentioned, the Ac5c tripeptide (3b) accommodates in the crystal a C10 (β-turn) conformation centered at the Pro-Ac5c sequence and stabilized by a hydrogen bond between the pivaloyl CO and the Val NH moieties (Table 7). This interaction is present in the two most stable minima characterized for Ac-Pro-Ac5c-Val-NHMe (Table 5). However, in each of these minima, an additional intramolecular hydrogen bond was detected (of type C13 or C10 in minima #1 and #2, respectively), which is not found in the solid state. It this respect, it is interesting to note that the crystalline Ac5c-Val dipeptide (6b) does accommodate a β-turn (Figure 14), thus evidencing that both the Pro-Ac5c and the Ac5c-Val sequences are prone to adopt a C10 conformation and that, accordingly, a C10 + C10 structure (two consecutive β-turns) is accessible to the Pro-Ac5c-Val sequence. In comparison, the Pro-Ac3c-Val one has been shown to prefer a C10 + C13 conformation. These results are in perfect agreement with the structural propensities previously established for these cyclic residues.31-37 Thus, Ac5c is known to exhibit a marked preference for the 310/α-helical region of the Ramachandran map, corresponding to φ,ψ values around 60°,30° or -60°,-30°, which makes it a suitable candidate to occupy both position i+2 and i+1 of a β-turn (as observed in 3b and 6b, respectively). In contrast, Ac3c is usually found to accommodate φ,ψ values near 90°,0° or -90°,0° (the bridge region), and this is compatible with the i+2 position of a C10 conformation but not with the i+1 one. This feature explains why the Pro-Ac3c-Val sequence prefers a C10 + C13 structure rather than two consecutive C10 turns, as well as the lack of intramolecular hydrogen bonds in the crystalline structure of the Ac3c-Val dipeptide 5c (Figure 14). On the other hand, Figure 15b, which shows the superposition of the crystal structure of 3b with recorded snapshots of both the building block and the nanoconstruct, reflects an excellent agreement with the structural morphology found for the investigated turn region of the β-helix.
Among the Gly derivatives, we could only solve the X-ray diffraction structure of dipeptide 4a (Figure 14). Gly shows a much lower tendency to adopt folded conformations than the Acnc residues and, accordingly, it was not surprising to find that compound 4a accommodates an extended structure in the solid state, with both residues being located in the φ,ψ ≈ -150°,150° region of the conformational map (Table 6). This result seems to be in contradiction with the fact that all the low energy minima of Ac-Pro-Gly-Val-NHMe in Table 5 exhibit one or more intramolecular hydrogen bonds. However, it should be emphasized that the latter finding is not unexpected since these calculations were carried out in an organic solvent unable to interact with the polar amide groups in the molecule. Under such environmental conditions, the stability of conformers with intramolecular hydrogen bonds may be overestimated with respect to extended conformations, which seems to be the case for the Gly derivatives according to the results of the X-ray diffraction analysis. It is interesting to note that, in the crystal structure of 4a, all NH and CO groups in the molecule are involved in intermolecular hydrogen bonds (Table 8), which compete successfully with the intramolecular interactions detected in the calculations. Therefore, the conformational propensities of the Gly-containing sequences seem to be highly dependent on the environment, as mentioned above.
Conclusions
The results provided by MD simulations and X-ray crystallography on the systems investigated allow us to draw the following conclusions about the conformational impact of the position-specific mutations with Ac3c and Ac5c in the β-helical motif excised from 1krr:
Targeted replacements in the building block enhance the rigidity of the loop defined by the 148-150 residues, which is quite flexible in the wild-type system. The synthetic amino acid adopts a well-defined conformation and, simultaneously, reduces the conformational freedom of the neighboring residues. As a consequence, replacement of Gly149 by Ac3c or Ac5c results in the retention of the conformation found for this loop in the crystal structure of 1krr.
The influence of the mutations in the nanoconstruct is less pronounced than in the building block. This is because the self-assembly of β-helical motifs seems to induce a rearrangement in the loop of the wild-type system, which provides evidence that the conformation of the Gly-containing sequence is affected by the chemical environment. Results indicate that the native interface of the protein is similar to that achieved by self-assembling replicas of the building block one atop the other.
Simulations on the Ac-Pro-Xaa-Val-NHMe tripeptides (Xaa being Gly, Ac3c and Ac5c) in organic solution indicate that the conformational space of the Ac3c and Ac5c derivatives is more restricted than that of the Gly tripeptide. All three compounds are predicted to adopt turn-like conformations stabilized by intramolecular hydrogen bonds (C10 and C13, mainly). However, the ability to accommodate such structural motifs in the crystal state was confirmed only for the peptides incorporating the non-proteinogenic residues, whereas the crystallized Gly derivative was found to adopt an extended conformation.
A good concordance between the solid-state structures and the minimum energy conformations predicted for the Pro-Xaa-Val tripeptides was observed in the case of the Acnc residues. Moreover, both the crystallographic and theoretical studies highlighted the conformational features differentiating Ac3c from Ac5c, which are in line with the conformational preferences previously established for these residues.
The higher propensity to adopt turn-like conformations exhibited by the Pro-Acnc-Val sequences, evidenced by the X-ray studies, explains the ability of the Acnc residues to stabilize the flexible loop region in the β-helical motif excised from 1krr. In this sense, the agreement between the dihedral angles in the crystalline structure of the Pro-Acnc-Val tripeptides (in particular, the Ac5c derivative) and the Pro-Gly-Val sequence in the crystalline 1krr protein is remarkable.
Acknowledgements
Gratitude is expressed to the Centre de Supercomputació de Catalunya (CESCA) and to the Barcelona Supercomputing Center (BSC) for computational facilities. Financial support from the Gobierno de Aragón (research group E40) and Ministerio de Educación y Ciencia-FEDER (project CTQ2007-62245) is gratefully acknowledged. This project has been funded in whole or in part with Federal funds from the National Cancer Institute, National Institutes of Health, under contract number N01-CO-12400. The content of this publication does not necessarily reflect the view of the policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organization imply endorsement by the U.S. Government. This research was supported [in part] by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
References
- 1.Lomander A, Hwang WM, Zhang SG. Nano. Lett. 2005;5:1255. doi: 10.1021/nl050203r. [DOI] [PubMed] [Google Scholar]
- 2.Yokoi H, Kinoshita T, Zhang SG. Proc. Natl. Acad. Sci. 2005;102:8414. doi: 10.1073/pnas.0407843102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang SG. Biotechnol. Adv. 2002;20:231. [Google Scholar]
- 4.Zhang SG. Nat. Biotech. 2003;21:1171. [Google Scholar]
- 5.Nam KT, Kim DW, Yoo PJ, Chiang CY, Meethong N, Hammond PT, Chiang YM, Belcher AM. Science. 2006;312:885. doi: 10.1126/science.1122716. [DOI] [PubMed] [Google Scholar]
- 6.Huang Y;, Chiang CY, Lee SK, Gao Y, Hu EL, De Yoreo J, Belcher AM. Nano Lett. 2005;5:1429. doi: 10.1021/nl050795d. [DOI] [PubMed] [Google Scholar]
- 7.Rajagopal K, Ozbas B, Pochan DJ, Schneider JP. Eur. Biophys. 2006;35:162. doi: 10.1007/s00249-005-0017-7. [DOI] [PubMed] [Google Scholar]
- 8.Valery C, Artzner F, Robert B, Gulick T, Keller G, Gabrielle-Madelmont C, Torres ML, Cherif-Chejk R, Paternostre M. Biophys. J. 2004;86:2484. doi: 10.1016/S0006-3495(04)74304-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tang M, Waring AJ, Hong M. J. Am. Chem. Soc. 2005;127:13919. doi: 10.1021/ja0526665. [DOI] [PubMed] [Google Scholar]
- 10.Tsai HH, Tsai C-J, Ma BY, Nussinov R. Protein Sci. 2004;13:2753. doi: 10.1110/ps.04774004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tsai C-J, Zheng J, Alemán C, Nussinov R. Trends Biotech. 2006;24:449. doi: 10.1016/j.tibtech.2006.08.004. [DOI] [PubMed] [Google Scholar]
- 12.Alemán C, Zanuy D, Jiménez AI, Cativiela C, Haspel N, Zheng J, Casanovas J, Wolfson H, Nussinov R. Phys. Biol. 2006;3:S54. doi: 10.1088/1478-3975/3/1/S06. [DOI] [PubMed] [Google Scholar]
- 13.Tsai C-J, Zheng J, Haspel N, Wolfson H, Alemán C, Nussinov R. Proteins. 2007;68:1. doi: 10.1002/prot.21413. [DOI] [PubMed] [Google Scholar]
- 14.Kajander T, Cortajarena AL, Main ERG, Mochrie SG, Regan L. J. Am. Chem. Soc. 2005;127:10188. doi: 10.1021/ja0524494. [DOI] [PubMed] [Google Scholar]
- 15.Main ERG, Stott K, Jackson SE, Regan L. Proc. Natl. Acad. Sci. 2005;102:5721. doi: 10.1073/pnas.0404530102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kobe B, Kajava AV. Trends Biochem. Sci. 2000;25:509. doi: 10.1016/s0968-0004(00)01667-4. [DOI] [PubMed] [Google Scholar]
- 17.Padilla JE, Colovos C, Yeates TO. Proc. Natl. Acad. Sci. USA. 2001;98:2217. doi: 10.1073/pnas.041614998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haspel N, Zanuy D, Alemán C, Wolfson H, Nussinov R. Structure. 2006;14:1137. doi: 10.1016/j.str.2006.05.016. [DOI] [PubMed] [Google Scholar]
- 19.Curcó D, Nussinov R, Alemán C. J. Phys. Chem. B. 2007;111:10538. doi: 10.1021/jp072832q. [DOI] [PubMed] [Google Scholar]
- 20.Curcó D, Nussinov R, Alemán C. J. Phys. Chem. B. 2007;111:14006. doi: 10.1021/jp075386f. [DOI] [PubMed] [Google Scholar]
- 21.Main ERG, Stott K, Jackson SE, Regan L. Prod. Nat. Acad. Scien. USA. 2005;102:5721. doi: 10.1073/pnas.0404530102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lowe AR, Itzhaki LS. Prod. Nat. Acad. Scien. USA. 2007;104:2679. doi: 10.1073/pnas.0604653104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Binz HK, Kohl A, Plueckthun A, Gruetter MG. Proteins Struct. Funct. Bioinf. 2006;65:280. doi: 10.1002/prot.20930. [DOI] [PubMed] [Google Scholar]
- 24.Lee G, Abdi K, Jiang Y, Michaely P, Bennett V, Marszalek PE. Nature. 2006;440:246. doi: 10.1038/nature04437. [DOI] [PubMed] [Google Scholar]
- 25.Cortajarena AL, Yi F, Regan L. ACS Chem. Biol. 2008;3:161. doi: 10.1021/cb700260z. [DOI] [PubMed] [Google Scholar]
- 26.Cheng CY, Jarymowycz VA, Cortajarena AL, Regan L, Stone MJ. Biochemistry. 2006;45:12175. doi: 10.1021/bi060819a. [DOI] [PubMed] [Google Scholar]
- 27.Zheng J, Zanuy D, Haspel N, Tsai CJ, Alemán C, Nussinov R. Biochemistry. 2007;46:1205. doi: 10.1021/bi061674a. [DOI] [PubMed] [Google Scholar]
- 28.Zanuy D, Rodríguez-Ropero F, Haspel N, Zheng J, Nussinov R, Alemán C. Biomacromolecules. 2007;8:3135. doi: 10.1021/bm700561t. [DOI] [PubMed] [Google Scholar]
- 29.Zanuy D, Jiménez AI, Cativiela C, Nussinov R, Alemán C. J. Phys. Chem. B. 2007;111:3236. doi: 10.1021/jp065025k. [DOI] [PubMed] [Google Scholar]
- 30.Zanuy D, Rodríguez-Ropero F, Nussinov R, Alemán C. J. Struct. Biol. 2007;160:177. doi: 10.1016/j.jsb.2007.07.014. [DOI] [PubMed] [Google Scholar]
- 31.Venkatraman J, Shankaramma SC, Balaram P. Chem. Rev. 2001;101:3131. doi: 10.1021/cr000053z. [DOI] [PubMed] [Google Scholar]
- 32.Toniolo C, Crisma M, Formaggio F, Peggion C. Biopolymers (Pept. Sci.) 2001;60:396. doi: 10.1002/1097-0282(2001)60:6<396::AID-BIP10184>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 33.Benedetti E. Biopolymers (Pept. Sci.) 1996;40:3. doi: 10.1002/(sici)1097-0282(1996)40:1<3::aid-bip2>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 34.(a) Alemán C, Jiménez AI, Cativiela C, Pérez JJ, Casanovas J. J. Phys. Chem. B. 2002;106:11849. [Google Scholar]; (b) Alemán C. J. Phys. Chem. B. 1997;101:5046. [Google Scholar]; (c) Barone V, Fraternali F, Cristinziano PL, Lelj F, Rosa A. Biopolymers. 1988;27:1673. [Google Scholar]
- 35.(a) Valle G, Crisma M, Toniolo C, Holt EM, Tamura M, Bland J, Stammer CH. Int. J. Pept. Protein Res. 1989;34:56. doi: 10.1111/j.1399-3011.1989.tb01009.x. [DOI] [PubMed] [Google Scholar]; (b) Benedetti E, Di Blasio B, Pavone V, Pedone C, Santini A, Crisma M, Valle G, Toniolo C. Biopolymers. 1989;28:175. [Google Scholar]; (c) Crisma M, Bonora GM, Toniolo C, Barone V, Benedetti E, Di Blasio B, Pavone V, Pedone C, Santini A, Fraternali F, Bavoso A, Lelj F. Int. J. Biol. Macromol. 1989;11:345. doi: 10.1016/0141-8130(89)90006-8. [DOI] [PubMed] [Google Scholar]; (d) Benedetti E, Di Blasio B, Pavone V, Pedone C, Santini A, Barone V, Fraternalli F, Lelj F, Bavoso A, Crisma M, Toniolo C. Int. J. Biol. Macromol. 1989;11:353. doi: 10.1016/0141-8130(89)90007-x. [DOI] [PubMed] [Google Scholar]
- 36.(a) Alemán C, Zanuy D, Casanovas J, Cativiela C, Nussinov R. J. Phys. Chem. B. 2006;110:21264. doi: 10.1021/jp062804s. [DOI] [PubMed] [Google Scholar]; (b) Rao SN, Chan MF, Balaji VN. Bull. Chem. Soc. Jpn. 1997;70:293. [Google Scholar]
- 37.(a) Aschi M, Lucente G, Mazza F, Mollica A, Morera E, Nalli M, Paglialunga Paradisi M. Org. Biomol. Chem. 2003;1:1980. doi: 10.1039/b212247b. [DOI] [PubMed] [Google Scholar]; (b) Santini A, Barone V, Bavoso A, Benedetti E, Di Blasio B, Fraternalli F, Lelj F, Pavone V, Pedone C, Crisma M, Bonora GM, Toniolo C. Int. J. Biol. Macromol. 1988;10:292. doi: 10.1016/0141-8130(89)90007-x. [DOI] [PubMed] [Google Scholar]; (c) Crisma M, Bonora GM, Toniolo C, Benedetti E, Bavoso A, Di Blasio B, Pavone V, Pedone C. Int. J. Biol. Macromol. 1988;10:300. doi: 10.1016/0141-8130(89)90006-8. [DOI] [PubMed] [Google Scholar]; (d) Valle G, Crisma M, Toniolo C. Can. J. Chem. 1988;66:2575. [Google Scholar]; (e) Bardi R, Piazzesi AM, Toniolo C, Sukumar M, Balaram P. Biopolymers. 1986;25:1635. [Google Scholar]
- 38.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. J. Comput. Chem. 2005;26:1781. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. J. Chem. Phys. 1983;79:926. [Google Scholar]
- 40.Jorgensen WL, Brigss JM, Contreras ML. J. Phys. Chem. 1990;94:1683. [Google Scholar]
- 41.Wang J, Cieplak P, Kollman PA. J. Comput. Chem. 2000;21:1049. [Google Scholar]
- 42.Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA. J. Am. Chem. Soc. 1995;117:5179. [Google Scholar]
- 43.Alemán C, Casanovas J, Galembeck SE. J. Comput. Aided Mol. Design. 1998;12:259. doi: 10.1023/a:1007908630431. [DOI] [PubMed] [Google Scholar]
- 44.Gómez-Catalán J, Alemán C, Pérez JJ. Theor. Chem. Acc. 2000;103:180. [Google Scholar]
- 45.Darden T, York D, Pedersen L. J. Chem. Phys. 1993;98:10089. [Google Scholar]
- 46.Ryckaert JP, Ciccotti G, Berendsen HJC. J. Comput. Phys. 1977;23:327. [Google Scholar]
- 47.Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, Haak JR. J. Chem. Phys. 1984;81:3684. [Google Scholar]
- 48.Kirkpatrick S, Gelatt CD, Jr., Vecchi MP. Science. 1983;220:671. doi: 10.1126/science.220.4598.671. [DOI] [PubMed] [Google Scholar]
- 49.Steinbach PJ, Brooks BR. Chem. Phys. Lett. 1994;226:447. [Google Scholar]
- 50.Baysal C, Meirovitch H. J. Comput. Chem. 1999;20:1659. [Google Scholar]
- 51.Simmerling C, Elber R. J. Am. Chem. Soc. 1994;116:2534. [Google Scholar]
- 52.Bodanszky M, Bodanszky A. The Practice of Peptide Synthesis. Springer-Verlag; Berlin: 1994. [Google Scholar]
- 53.(a) Sheldrick GM. Program for the Solution of Crystal Structures. University of Göttingen; Göttingen: 1997. SHELXS-97. [Google Scholar]; (b) Sheldrick GM. Program for the Refinement of Crystal Structures. University of Göttingen; Göttingen: 1997. SHELXL-97. [Google Scholar]
- 54.Jiménez AI, Ballano G, Cativiela C. Angew. Chem. Int. Ed. 2005;44:396. doi: 10.1002/anie.200461230. [DOI] [PubMed] [Google Scholar]
- 55.Jiménez AI, Marraud M, Cativiela C. Tetrahedron Lett. 2003;44:3147. [Google Scholar]
- 56.Jiménez AI, Cativiela C, Marraud M. Tetrahedron Lett. 2000;41:5353. [Google Scholar]
- 57.Bernstein FC, Koetzle TF, Williams GJB, Meyer EF, Brice MD, Rodgers JR, Kennard O, Shimanouchi T, Tasumi M. J. Mol. Biol. 1977;112:535. doi: 10.1016/s0022-2836(77)80200-3. [DOI] [PubMed] [Google Scholar]
- 58.Chakrabarti P, Pal D. Prog. Biophys. Mol. Biol. 2001;76:1. doi: 10.1016/s0079-6107(01)00005-0. [DOI] [PubMed] [Google Scholar]
- 59.MacArthur MW, Thornton JM. J. Mol. Biol. 1991;218:397. doi: 10.1016/0022-2836(91)90721-h. [DOI] [PubMed] [Google Scholar]
- 60.Rose GD, Gierasch LM, Smith JA. Adv. Protein Chem. 1985;37:1. doi: 10.1016/s0065-3233(08)60063-7. [DOI] [PubMed] [Google Scholar]
- 61.Marraud M, Aubry A. Biopolymers. 1996;40:45. doi: 10.1002/(sici)1097-0282(1996)40:1<45::aid-bip3>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 62.Jiménez AI, Cativiela C, Aubry A, Marraud M. J. Am. Chem. Soc. 1998;120:9452. [Google Scholar]
- 63.Cramer CJ. J. Mol. Struct. (Theochem) 1996;370:135. [Google Scholar]