

Abstract
Purpose
Inflammation caused by radiotherapy is a serious medical problem, and it is the main reason for dose restrictions during treatment of thorax-associated cancers. Here, we studied the role of interleukin-32 (IL-32), a novel protein only detected in human tissues, in Ionizing radiation (IR) induced vascular inflammation.
Methods and Materials
Irradiated (0 - 6 Gy) human umbilical vein endothelial cells (HUVECs) treated with or without various agents, a cytosolic phospholipase A2 (cPLA2) inhibitor, a Cox2 inhibitor, or lysophosphatidylcholines (LPCs), were used to assess IL-32 expression by Northern blot analysis and qRT-PCR. Expression of cell adhesion molecules as well as leukocyte adhesion to endothelial cells using THP-1 cells was analyzed as well.
Results
IR dramatically increased IL-32 expression in vascular endothelial cells through multiple pathways. IR induced IL-32 expression through NF-κB activation, through induction of cPLA2 and LPC, as well as induction of Cox2 and subsequent conversion of arachidonic acid to PGI2. Conversely, blocking NF-κB, cPLA2 and Cox2 activity impaired IR-induced IL32 expression. Importantly, IL-32 significantly enhanced IR induced expression of vascular cell adhesion molecules and leukocyte adhesion on endothelial cells.
Conclusion
This study identifies IL-32 as a positive regulator in IR-induced vascular inflammation, and neutralization of IL-32 may be beneficial in protecting from IR-induced inflammation.
Keywords: Endothelium, Angiogenesis, Ionizing radiation, Inflammation, IL-32
Introduction
Inflammation caused by radiotherapy is the main reason for dose restrictions for treating thorax-associated cancer, and radiation-induced pneumonitis can be fatal (1). Vascular endothelium is an active participant in inflammation. During inflammation initiation, circulating leukocytes must first be able to adhere selectively and efficiently to inflamed vascular endothelium. This process is facilitated by induction of vascular cell adhesion molecules on the inflamed endothelium, such as vascular cell adhesion molecule (VCAM)-1, intercellular adhesion molecule (ICAM)-1, and E-selectin. It is evident that the endothelium functions as a gatekeeper that controls the number and recruitment of distinct leukocyte subpopulations and, in doing so, determines the nature and extent of inflammation. It has been shown that IR increases vascular inflammation through induction of cell adhesion molecules such as, ICAM-1, VCAM-1, and E-selectin (2, 3).
IL-32 is a new cytokine that is detected in human hematopoietic cells. Genomic analysis indicates that IL-32 is only present in human tissues and absent in rodent (4). It induces expression of various cytokines, such as TNFα and IL-8, in monocytes (4) and activates the NF-κB and p38 mitogen-activated protein kinase in lymphocytes. Since IL-32 expression is regulated by inflammatory cytokines in human peripheral lymphocyte cells, it has been speculated that it may play a role in inflammatory/autoimmune diseases (4). Indeed, elevated expression of IL-32 was reported in rheumatoid arthritis (5-7), ulcerative colitis, and Crohn's disease (8, 9).
cPLA2s are key enzymes that catalyze the hydrolysis of membrane phospholipids to release bioactive lysophospholipids such as lysophosphatidylcholines (LPCs) and fatty acids (10). It specifically cleaves the sn-2 fatty acyl of phospholipid and has arachidonoyl preference. Therefore, activation cPLA2 generates arachidonic acid (AA) (11), which is a major substrate for inflammation and is further metabolized to prostaglandins by cyclooxygenase 1 and 2 (COX1/2). IR has been shown to increase prostacyclin (PGI2) production in endothelial cells (12-14), and this production was shown to be mediated by an increase in AA release and an activation of cyclooxygenase (COX) (15). Recently, we showed that IR treatment activated cPLA2 and increased production of LPCs in endothelial cells (16). Consistently, cPLA2α-deficient mice are resistant to various inflammatory stimulants (17-19).
Here, we studied the role of IL-32 in IR induced vascular inflammation. We found that IR treatment dramatically induces IL-32 expression in vascular endothelial cells via multiple ways, and IL-32 potentiates IR-induced vascular inflammation. Thus, neutralization of IL-32 function may offer a new means to control IR-induced inflammation.
Methods and Materials
Cell culture
Human umbilical vein endothelial cells (HUVECs) were purchased (Clonetics, San Diego, CA), and grown on 0.1 % gelatin-coated plates in endothelial growth medium, EGM (Clonetics). THP-1 cells were grown in RPMI1640 with 10% FBS. The adenoviral vectors directing the expression of IκB (AdIκBα), a mutated IκB as a NF-κB inhibitor, and GFP (AdGFP) and β-galactosidase (Adβ-gal) as viral vector controls were used (20). Palmitoyl LPC (16:0), stearoyl LPC (18:0), oleoyl LPC (18:1), and arachidoyl LPC (20:0) were purchased from Avanti Polar Lipids (Alabaster, AL). Wyeth cPLA2 inhibitor was synthesized by the Vanderbilt Chemistry Core.
Northern analysis
Cells were irradiated at 0, 3, or 6Gy. RNA was isolated 48h following IR using RNeasy kit (Qiagen, Valencia, CA) and subjected for Northern blot analysis. 32P labeled cDNA probes for IL-32 mRNA were hybridized using Express Hyb (BD Biosciences).
qRT-PCR
For the time course study, RNA was isolated from HUVECs following 0 or 3 Gy irradiation at specified time to measure IL-32 mRNA level. For the cPLA2 inhibitor study, HUVECs were irradiated with or without 3Gy following a treatment with a Wyeth cPLA2 inhibitor at 10 μM. For the LPC study, HUVECs were treated with 4 LPC species at 20 μM. For the IκB study, HUVECs were irradiated with 0 or 3Gy following infection with AdGFP or AdIκB at MOI=10. For the Cox2 inhibitor study, HUVECs were treated with celecoxib at 25 μM for 4h and then irradiated with 0 or 3Gy. Total RNA (1 μg) was used for the first-strand cDNA synthesis using iScript ™ cDNA synthesis kit (Bio Rad, Hercules, CA). Real-time PCR was done using IQ™ SYBR® Green supermix (Bio Rad) on iCycler (Bio Rad). IL-32 primers were 5′-CGACTTCAAAGAGGGCTACC (forward) and 5′-GAGTGAGCTCTGGGTGCTG (reverse). ICAM-1 primers were 5′-CCACAGTCACCTATGGCAAC (forward) and 5′-AGTGTCTCCTGGCTCTGGTT (reverse). VCAM-1 primers were 5′-GCTTCAGGAGCTGAATACCC (forward) and 5′-AAGGATCACGACCATCTTCC (reverse). E-selectin primers were 5′-TGAACCCAACAATAGGCAAA (forward) and 5′-CCTCTCATCATTCCACATGC (reverse). Each gene expression was normalized by β-actin.
Western blotting
Cell lysate was collected, and processed for Western blot and probed with an anti-Cox2 antibody (Santa Cruz). As a loading control, β-actin was used.
Cell Adhesion Assay
HUVECs were irradiated with 0 or 3Gy. THP-1 cells that were labeled using PKH26 Red Fluorescent Cell linker kit (Sigma-Aldrich, St. Louis, MO) were allowed to adhere to the monolayer of HUVECs for 30 min. Following extensive washing, adhered THP-1 cells were counted at randomly selected high power fields under fluorescent microscopy.
Statistics
Data were presented as mean ± SEM. Data were analyzed using Student's t-test and p-value less than 0.05 was considered statistically significant. p<0.05 is indicated as * and p<0.01 is indicated as ** in figures. All the experiments were repeated at least 3 times.
Results
IR induces expression of IL-32 via NF-κB in human endothelial cells
Inflammation induced by radiation therapy is a serious medical problem, and it is the main reason for dose restrictions for the treatment of thorax-associated cancers. Therefore, understanding the underline mechanism is important. Vascular endothelium separating tissues from circulating leukocytes plays critical roles in inflammation, through which it recruits leukocytes to injury sites via induction of cell adhesion molecules. In searching for the molecular mediators in IR induced vascular inflammation, we found that IL-32, a novel gene that is only detected in human cells, is expressed in human vascular endothelial cells. Interestingly, exposure of these cells to 3Gy, a dose commonly used in clinics, or 6Gy of IR dramatically induced IL-32 expression (Fig. 1a). Further analysis showed a time dependent gene induction with 3Gy treatment of IR (*p<0.05) (Fig. 1b). These findings support a new function of IL-32 in vascular inflammation in response to radiotherapy.
Fig. 1. IR induces IL-32 expression in human endothelial cells via NF-κB.
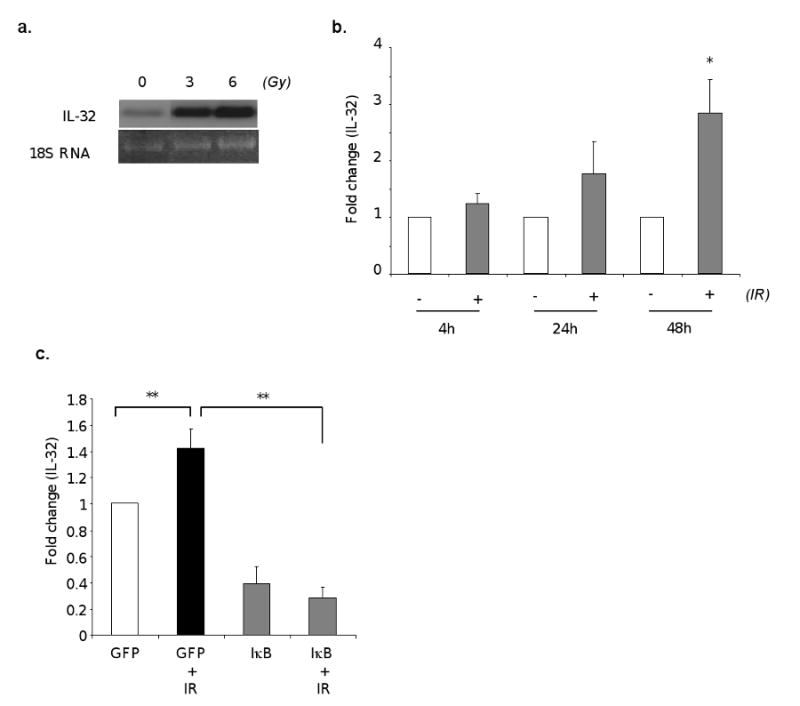
HUVECs were irradiated at the indicated dose. Total RNAs were isolated 48 hours after irradiation and analyzed for IL-32 expression by Northern blot (a). HUVECs were irradiated with 0 or 3 Gy. Total RNAs were isolated at indicated time, and IL-32 levels were measured by qRT-PCR (b). HUVECs were infected with AdGFP or AdIκB at MOI 10. Following overnight infection, cells were irradiated at 0 or 3Gy, harvested 48 hours later. IL-32 levels were measured by qRT-PCR (c). * p<0.05, **p<0.01.
NF-κB is a master regulator in gene expression associated with inflammation. To investigate the potential role of NF-κB in IR-induced IL-32 expression, we used adenoviral vector for efficient transgene expression in endothelial cells. The viral vector expressing GFP was used as a vector control as well as for evaluating infection efficiency. We achieved over than 85% infection efficiency in cultured human endothelial cells (data not shown). Using this approach, we found that expression of a mutant IκBα to inhibit NF-κB activation completely blocked IR induced IL-32 expression (p<0.01) (Fig. 1c). Furthermore, overexpression of the IκB mutant significantly reduced the basal levels of IL-32 when compared to vector controls (p<0.01) (Fig. 1c). This finding suggests NF-κB as a major regulator for IL-32 expression in endothelial cells.
IR induces IL-32 expression through induction of cPLA2 and Cox2
As we have previously demonstrated that exposure of endothelial cells to IR increases cPLA2 production (16), and cPLA2 is a key enzyme that produces bioactive lysophospholipids such as LPCs and fatty acids, important mediators in inflammation, we reasoned a role of cPLA2 in IL-32 gene induction upon the IR treatment. Using specific inhibitors, we found that blocking cPLA2 activation significantly inhibited IR-induced IL-32 expression (*p<0.05) (Fig. 2a). However, neutralization of cPLA2 has no effect on basal levels of IL-32, suggesting cPLA2 is likely only involved in IR mediated response. As activation of cPLA2 leads to release of LPCs from the phospholipids of the plasma membrane, we further determined whether LPCs could increase IL-32 expression. In a previous study, we found that exposure of endothelial cells to IR induced the production of four types of LPC species (unpublished data), which are palmitoyl-lysophophatidylcholine (16:0), stearoyl- lysophosphatidylcholine (18:0), oleyol-lysophosphatidylcholine (18:1) and arachidonoyl-lysophosphatidylcholine (20:0). Consistently, after exposure of human endothelial cells with either one of the four LPC species increased IL-32 expression (Fig. 2b). Furthermore, we found that neutralization of NF-κB using a mutant IκB construct significantly blocked LPC mediated IL-32 expression in these cells (Fig. 2c). These data suggest that IR treatment induces the production cPLA2 and LPC that upregulates IL-32 expression via NF-κB.
Fig. 2. IL-32 induction by IR partly depends on cPLA2 that produces LPCs.

HUVECs were treated with a Wyeth cPLA2 specific inhibitor at 10 μM or vehicle control for 30 min, followed by 3Gy IR. IL-32 levels were analyzed by qRT-PCR 48 hours after IR (a). *p<0.05. HUVECs were treated with indicated LPC species (20μM) at 0 and again at 24 hours. Total RNAs were isolated at 48 hours after IR and analyzed for IL-32 expression by qRT-PCR (b). HUVECs were infected with Adβ-gal or AdIκB at MOI=10. Following overnight infection, cells were treated with LPC for 48 hours. IL-32 levels were analyzed by qRT-PCR. Fold change of gene induction in the IκB group over the vector control was plotted (c). * p<0.05, **p<0.01.
In addition, activation of cPLA2 also leads to release of free fatty acid from the phospholipids of the plasma membrane, and the major free fatty acid produced by cPLA2 is arachidonic acid (AA). AA is a major substrate for inflammation and is further metabolized to prostaglandins by cyclooxygenase. Thus, we examined whether this pathway has a role in IR-induced IL-32 expression. Our data showed an increase production of Cox2 within an hour of low dose of IR exposure, which continued for 4 hours after IR exposure (Fig. 3a). Interestingly, blocking Cox2 activity using a specific inhibitor significantly inhibited IR-induced IL-32 expression in endothelial cells (p<0.05) (Fig. 3b). Further analysis suggests that it is the PGI2 but not PGE2, two major Cox2 metabolites, that is responsible for IR-induced IL-32 expression (p<0.05) (Fig. 3c). This finding is consistent with published results that IR increases PGI2 production in endothelial cells (12-14). Based on the observation of NF-κB is a key regulator in IR induced expression of IL-32, we further analyzed the role of NF-κB in PGI2-induced gene expression. As expected, we found that neutralization of NF-κB using a mutant IκB significantly impaired PGI2-mediated IL-32 expression (Fig. 3d). Taken together, our findings suggest that IR induces IL-32 expression in vascular endothelial cells through multiple mechanisms, via activation of cPLA2 and production of LPCs, induction of Cox2 and conversion of AA to PGI2, and subsequent activation of NF-κB.
Fig. 3. IR upregulates IL-32 expression partly through induction of Cox2 and PGI2.
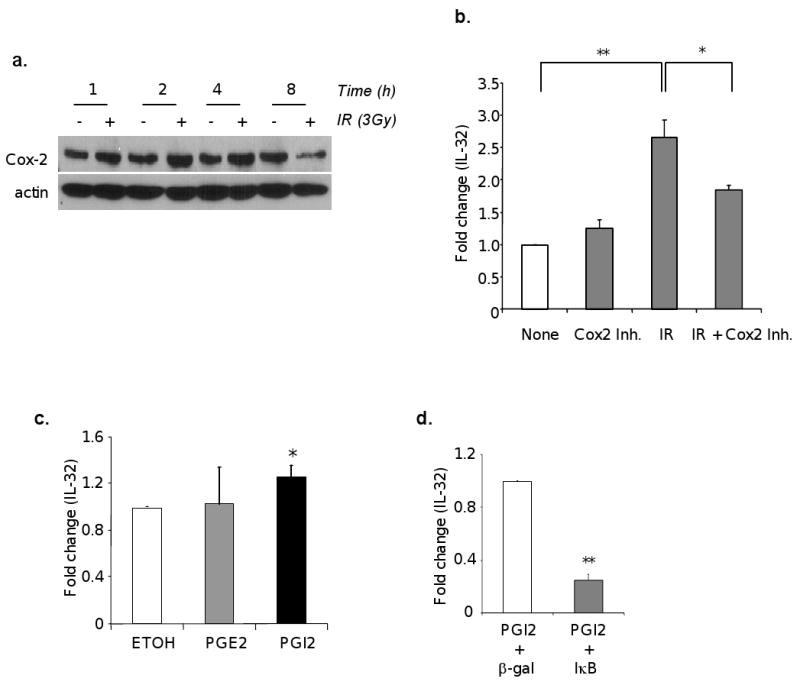
HUVECs were treated with or without 3Gy IR. The cells were harvested at the time indicated after IR. Protein extract was subjected to Western immunoblot for Cox-2 levels (a). HUVECs were treated with vehicle control or celecoxib (25 uM) for 4 hours, and followed by IR exposure at 3 Gy. Total RNAs were isolated and IL-32 mRNA was measured by qRT-PCR (b). HUVECs were treated with PGE2 (10μM), PGI2 (10μM), or ethanol (a vehicle control) for 24 hours, and IL-32 mRNA was measured using qRT-PCR (c). HUVECs were infected with Adβ-gal or AdIκB at MOI=10. Following overnight infection, cells were treated with PGI2 for 24 hours, and IL-32 mRNA was measured using qRT-PCR (d). *p<0.05, **p<0.01.
IL-32 potentiates IR-induced expression of cell adhesion molecules in endothelial cells and increases leukocyte adhesion
Next, we examined the function of IL-32 in vascular inflammation. Expression of cell adhesion molecules on inflamed endothelium is a paramount process in inflammation, and IR is known to upregulate these cell adhesion molecules. We used adenoviral vector for efficient IL-32 gene expression in endothelial cells, and GFP vector as a control. Consistent with published findings that IR increases the expression of cell adhesion molecules, we further showed that expression of IL-32 in endothelial cells potentiated cellular response to IR. There are higher levels of ICAM-1, VECAM-1 and E-selectin on vascular endothelial cells transfected with an IL-32 expression vector than the one in controls (Table 1). Accordingly, we observed a significant increase in leukocyte adhesion on vascular endothelial cells that were transfected with an IL-32 expression vector compared to controls (p<0.05). Expression of IL-32 alone in endothelial cells had no significant effects on leukocyte adhesion (Fig. 4). These data suggest that IL-32 alone is not sufficient to mediate the biological response, but it sensitizes vascular response to IR treatment. Taken together, our findings reveal a positive regulation, in which IR induces IL-32 expression that further enhances endothelial inflammatory response to IR exposure through production of cell adhesion molecules and leukocyte adhesion.
Table 1. IL-32 potentiates the expression of cell adhesion molecules after IR exposure.
HUVECs were transfected with a plasmid expressing IL-32 or control GFP, followed by exposure to 0 or 3Gy IR, respectively. Total RNAs were isolated 48 hours after IR. ICAM-1, VCAM-1, and E-selectin mRNA were measured by qRT-PCR. *p<0.05.
| ICAM-1 | VCAM-1 | E-SELECTIN | |
|---|---|---|---|
| GFP | 1 | 1 | 1 |
| GFP/IR | 1.4 | 1.3 | 1.7 |
| IL-32 | 1.3 | 1.7 | 2.2 |
| IL-32/IR | 2.2 * | 1.9 | 2.5 |
Fig. 4. Expression of IL-32 enhanced IR-induced leukocyte adhesion on vascular endothelium.

Leukocyte cell adhesion was performed on irradiated (3Gy) HUVECs transfetced with a plasmid vector expressing either IL-32 or GFP control 48 hours after IR, respectively. Following a 30 min of incubation with dye labeled THP-1 cells, adhered cells were visualized and counted in high power fields under fluorescent microscopy. The experiments were performed with 6 wells per group, and repeated 3 times. *p<0.05, **p<0.01.
Discussion
This study identifies IL-32 as a new mediator that potentiates IR induced vascular inflammation. We showed that IL-32 is present in vascular endothelial cells, and a clinical dose of IR significantly increased its expression via NF-κB. In addition, IR induces IL-32 expression through two interconnected mechanisms, in which IR increases LPC levels through activation of cPLA2, and through induction of Cox2 and subsequent production of PGI2. Most importantly, IL-32 enhanced endothelial response to IR exposure in the context of cell adhesion molecule expression and leukocyte adhesion (Fig. 5). These findings suggest that IL-32 positively regulates vascular inflammation. By doing so, it intensifies vascular inflammation upon IR exposure. Considering the significance of inflammation and IL-32 is only detected in human tissues, neutralization of IL-32 function could have important implication in controlling this serious side effect for human radiotherapy.
Fig. 5. Schematic diagram illustrating the gene regulation mechanisms of IR-induced IL-32 gene expression, and its role in vascular inflammation.

IL-32 was originally isolated from activated human natural killer cells. Recently, this gene was rediscovered in human lymphocytes, and renamed IL-32(4). Although IL-32 does not share sequence homology with any known cytokine families, IL-32 induces various cytokines, such as TNFα and IL-8 in lymphocytes and monocytic cells (4). Genomic search indicates that the highest homology to human IL-32 is in equine at 31.8%, and no homologue to this gene was found in mice. Since IL-32 expression is regulated by inflammatory cytokines in human peripheral lymphocyte cells, it has been speculated that it may play a role in inflammatory/autoimmune diseases (4). Further analysis indeed showed an elevation of IL-32 in human inflammatory diseases, such as in rheumatoid arthritis (5-7), ulcerative colitis and Crohn's disease (8, 9). In this study, we demonstrate a new role of IL-32 in vascular inflammation.
We recently showed that cPLA2 activity is increased in irradiated endothelial cells resulting in elevated production of LPC (16). Here, we demonstrate that cPLA2 is partly responsible for the induction of IR-induced IL-32 transcription in human endothelial cells. The hydrolysis of phospholipids at sn-2-acyl fatty acid position by cPLA2 results in two major products, LPCs and fatty acids (10). In mammalian cells, the sn-2-position of phospholipids is enriched with arachidonic acid (AA) (21), and the most abundant phospholipid in mammalian cell membranes is phosphatidylcholine. Therefore, in addition to the release of free AA, the activation of cPLA2 also increases the production of LPC. We studied the involvement of both products in the induction of IL-32. First, different fatty acid can be conjugated at acyl-1 position of phosholipids, and when fatty acids on acyl-2 position are liberated, it produces LPCs with different fatty acids at acyl-1 position. Using mass spectrometry, we found that exposure of endothelial cells with IR mainly induced four types of LPCs: palmitoyl-lysophophatidylcholine (16:0), stearoyl-lysophosphatidylcholine (18:0), oleyol-lysophosphatidylcholine (18:1) and arachidonoyl-lysophosphatidylcholine (20:0) (unpublished data). In this study, we show that all these four LPCs increase IL-32 expression in vascular endothelial cells.
Secondly, we tested the other product of cPLA2-dependent hydrolysis of phospholipids, AA, which is the major substrate for Cox1/2. Cox1/2 converts AA to prostaglandins, major mediators in inflammation. Our data showed that exposure of endothelial cells with IR induced Cox2 expression. Since both the substrate, AA, and the enzyme, Cox2, are elevated following IR, it is conceivable that the levels of prostaglandins would increase following IR. Indeed, several studies have shown that in irradiated endothelial cells, PGI2 is increased, while PGE2 is not altered (12-14). Consistently, we found that it is the PGI2 but nor PGE2 that increased IL-32 expression. Blocking Cox2 activation inhibited IR induced IL-32 expression. Together, these data reveal that IR induces IL-32 expression through two different but interconnected mechanisms, through activation of cPLA2 and production of LPCs as well as induction of Cox2 and production of PGI2. Importantly, blocking NF-κB activation significantly inhibited LPC and PGI2 mediated IL-32 expression, suggesting NF-κB as a downstream mediator in gene induction.
The vascular network, being the interface of the two blood and tissues, plays a key role in inflammation. Inflammatory cells are transported through blood vessel walls. Vascular endothelium is the first line of cells that interacting with circulating leukocytes. It regulates the recruitment and transmigration of leukocytes through the vessel wall to the target tissues to initiate inflammation. To some extent, vascular endothelium controls the intensity and the scope of inflammation. In this study, we found that radiation induces IL-32 expression in vascular endothelial cells, which further potentiates IR-induced vascular inflammation through enhanced expression of cell adhesion molecules and leukocyte adhesion.
In summary, this study has identified a new mediator involved in IR-induced vascular inflammation. IR induces IL-32 production through activation of cPLA2 and Cox2, as well as NF-κB. Elevated IL-32 further enhances IR induced vascular inflammation through upregulation of cell adhesion molecules and leukocyte adhesion, supporting a positive role of IL-32 in inflammation. Considering the significance of inflammation, targeting IL-32 may offer a new approach to control inflammation commonly associated with radiotherapy.
Acknowledgments
This work is supported in part by grants from NIH (CA108856, NS45888 and AR053718) to PCL and a training grant to HK (5T32CA093240).
Footnotes
Conflict of Interest Notification: None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Movsas B, Raffin TA, Epstein AH, et al. Pulmonary radiation injury. Chest. 1997;111:1061–1076. doi: 10.1378/chest.111.4.1061. [DOI] [PubMed] [Google Scholar]
- 2.Hallahan DE, Virudachalam S. Ionizing radiation mediates expression of cell adhesion molecules in distinct histological patterns within the lung. Cancer Res. 1997;57:2096–2099. [PubMed] [Google Scholar]
- 3.Hallahan DE, Virudachalam S, Kuchibhotla J. Nuclear factor kappaB dominant negative genetic constructs inhibit X-ray induction of cell adhesion molecules in the vascular endothelium. Cancer Res. 1998;58:5484–5488. [PubMed] [Google Scholar]
- 4.Kim SH, Han SY, Azam T, et al. Interleukin-32: a cytokine and inducer of TNFalpha. Immunity. 2005;22:131–142. doi: 10.1016/j.immuni.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 5.Shoda H, Fujio K, Yamaguchi Y, et al. Interactions between IL-32 and tumor necrosis factor alpha contribute to the exacerbation of immune-inflammatory diseases. Arthritis Res Ther. 2006;8:R166. doi: 10.1186/ar2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Joosten LA, Netea MG, Kim SH, et al. IL-32, a proinflammatory cytokine in rheumatoid arthritis. Proc Natl Acad Sci U S A. 2006;103:3298–3303. doi: 10.1073/pnas.0511233103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cagnard N, Letourneur F, Essabbani A, et al. Interleukin-32, CCL2, PF4F1 and GFD10 are the only cytokine/chemokine genes differentially expressed by in vitro cultured rheumatoid and osteoarthritis fibroblast-like synoviocytes. Eur Cytokine Netw. 2005;16:289–292. [PubMed] [Google Scholar]
- 8.Netea MG, Azam T, Ferwerda G, et al. IL-32 synergizes with nucleotide oligomerization domain (NOD) 1 and NOD2 ligands for IL-1beta and IL-6 production through a caspase 1-dependent mechanism. Proc Natl Acad Sci U S A. 2005;102:16309–16314. doi: 10.1073/pnas.0508237102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shioya M, Nishida A, Yagi Y, et al. Epithelial overexpression of interleukin-32alpha in inflammatory bowel disease. Clin Exp Immunol. 2007;149:480–486. doi: 10.1111/j.1365-2249.2007.03439.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nakanishi M, Rosenberg DW. Roles of cPLA2alpha and arachidonic acid in cancer. Biochim Biophys Acta. 2006;1761:1335–1343. doi: 10.1016/j.bbalip.2006.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clark JD, Milona N, Knopf JL. Purification of a 110-kilodalton cytosolic phospholipase A2 from the human monocytic cell line U937. Proc Natl Acad Sci U S A. 1990;87:7708–7712. doi: 10.1073/pnas.87.19.7708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eldor A, Vlodavsky I, Riklis E, et al. Recovery of prostacyclin capacity of irradiated endothelial cells and the protective effect of vitamin C. Prostaglandins. 1987;34:241–255. doi: 10.1016/0090-6980(87)90247-4. [DOI] [PubMed] [Google Scholar]
- 13.Hahn GL, Menconi MJ, Cahill M, et al. The influence of gamma radiation on arachidonic acid release and prostacyclin synthesis. Prostaglandins. 1983;25:783–791. doi: 10.1016/0090-6980(83)90003-5. [DOI] [PubMed] [Google Scholar]
- 14.Eldor A, Vlodavsky I, HyAm E, et al. The effect of radiation on prostacyclin (PGI2) production by cultured endothelial cells. Prostaglandins. 1983;25:263–279. doi: 10.1016/0090-6980(83)90109-0. [DOI] [PubMed] [Google Scholar]
- 15.Friedman M, Saunders DS, Madden MC, et al. The effects of ionizing radiation on the pulmonary endothelial cell uptake of alpha-aminoisobutyric acid and synthesis of prostacyclin. Radiat Res. 1986;106:171–181. [PubMed] [Google Scholar]
- 16.Yazlovitskaya EM, Linkous AG, Thotala DK, et al. Cytosolic phospholipase A(2) regulates viability of irradiated vascular endothelium. Cell Death Differ. 2008 doi: 10.1038/cdd.2008.93. [DOI] [PubMed] [Google Scholar]
- 17.Bonventre JV, Huang Z, Taheri MR, et al. Reduced fertility and postischaemic brain injury in mice deficient in cytosolic phospholipase A2. Nature. 1997;390:622–625. doi: 10.1038/37635. [DOI] [PubMed] [Google Scholar]
- 18.Hegen M, Sun L, Uozumi N, et al. Cytosolic phospholipase A2alpha-deficient mice are resistant to collagen-induced arthritis. J Exp Med. 2003;197:1297–1302. doi: 10.1084/jem.20030016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nagase T, Uozumi N, Ishii S, et al. Acute lung injury by sepsis and acid aspiration: a key role for cytosolic phospholipase A2. Nat Immunol. 2000;1:42–46. doi: 10.1038/76897. [DOI] [PubMed] [Google Scholar]
- 20.Jobin C, Panja A, Hellerbrand C, et al. Inhibition of proinflammatory molecule production by adenovirus- mediated expression of a nuclear factor kappaB super-repressor in human intestinal epithelial cells. J Immunol. 1998;160:410–418. [PubMed] [Google Scholar]
- 21.Bonventre JV. The 85-kD cytosolic phospholipase A2 knockout mouse: a new tool for physiology and cell biology. J Am Soc Nephrol. 1999;10:404–412. doi: 10.1681/ASN.V102404. [DOI] [PubMed] [Google Scholar]