

Abstract
Historically, data for genetic studies are collected at one time point. However, for diseases with late onset or with complex phenotypes, such as Alzheimer disease (AD), restricting diagnosis to a single ascertainment contact may not be sufficient. Affection status may change over time, and some initial diagnoses may be inconclusive. Follow-up provides the opportunity to resolve these complications. However, to date, previous studies have not formally demonstrated that longitudinally re-contacting families is practical or productive.
To update data initially collected for linkage analysis of late-onset Alzheimer disease (LOAD), we successfully re-contacted 63 of 81 (78%) multiplex families (two to 17 years after ascertainment). Clinical status changed for 73 of the 230 (32%) non-affected participants. Additionally, expanded family history identified 20 additional affected individuals to supplement the data set. Furthermore, fostering ongoing relationships with participating families helped recruit 101 affected participants into an autopsy and tissue donation program. Despite similar presentations, discordance between clinical diagnosis and neuropathologic diagnosis was observed in 28% of those with tissue diagnoses.
Most of the families were successfully re-contacted, and significant refinement and supplementation of the data was achieved. We concluded that serial contact with longitudinal evaluation of families has significant implications for genetic analyses.
Introduction
Alzheimer disease (AD) is the most common neurodegenerative disorder, characterized by an insidious and inexorable decline in multiple cognitive domains, usually heralded by short-term memory loss (Small et al. 1997). Genetic factors are known to play a major role in the etiology of AD, and to date, variants in four genes have confirmed roles in AD pathogenesis. Rare causative autosomal dominant mutations in any of the three Mendelian genes (APP, PS1, or PS2) generally result in an early onset (before age 60) of disease (Goate et al. 1991; Levy-Lahad et al. 1995; Rogaev et al. 1995; Sherrington et al. 1995). In addition to these three genes, a common polymorphism of the APOE gene, ε4 (coding for apolipoprotein E, ApoE-ε4), is associated with increased risk and earlier age at onset (AAO) in both familial AD (at least two affected individuals in a family) and sporadic AD (no other identified affected individuals recognized in the family (Corder et al. 1993)). Combined, these confirmed genes account for only half of the overall genetic risk of AD (Farrer et al. 1997).
To facilitate identification of genes having large effects in late-onset AD (LOAD), the initial data sets consisted of families with two or more living affected individuals occuring in several generations (Pericak-Vance et al. 1991). These data sets were modest in size (10-32 LOAD multiplex families) (Pericak-Vance et al. 1988; Pericak-Vance et al. 1991; Schellenberg et al. 1988) However, having sufficient power to dissect the complex AD genetic etiology of more moderate effects requires larger data sets. To this end, recent genome screens in AD have utilized affected sibling pairs in addition to extended families (Myers et al. 2000; Lee et al. 2006; Blacker et al. 2003). Also, candidate gene association studies have used both case-control designs and family-based association methods based on discordant sibpairs (Bertram et al. 2007; Laird et al. 2000; Martin et al. 2003).
However, analysis of large, multigenerational families remains useful in mapping studies (Terwilliger et al. 2002), particularly those in complex heterogenous diseases (Ma et al. 2006). Additionally, it is critical that we continue to expand our knowledge of the natural history of AD. Characterizing phenotypic differences over time may play a critical role in delineating homogenous subsets of data aiding identification of AD genes (Ma et al. 2006; Shao et al. 2003; Scott et al. 2003). Today, many genetic studies of AD are in their third decade. Researchers have the opportunity to observe first-hand the transmission of a late-onset disorder over time, incorporate supplementary data, and clarify the relationship between clinical and histopathological findings. Longitudinal follow-up emerges as a powerful tool to help dissect of the underlying genetic etiology of AD.
Methods
Family ascertainment and follow-up
Eighty-one Caucasian multiplex late-onset AD families (average AAO greater than 60) were ascertained for genetic studies of AD. To meet inclusion criteria, a family must have had at least two living affected individuals (usually siblings) available for clinical evaluation and DNA sampling. Additionally, each family had either: a) two affected individuals, each of whom had a clinical diagnosis of probable AD and/or b) one affected individual with a clinical diagnosis of possible AD, and one affected individual later confirmed to have definite AD by autopsy (Mirra et al. 1991).
Families were ascertained through the Joseph and Kathleen Bryan Alzheimer Disease Research Center (ADRC) and the Center for Human Genetics (CHG) at Duke University Medical Center (DUMC). Families were referred for potential study inclusion by neurologists participating in AD clinical research, community neurologists specializing in the treatment of dementia, research nurse clinicians from the autopsy and tissue donation program, or family members.
Ascertainment included assembling a pedigree, gathering family medical history (from family interview, clinical evaluation, and/or medical records), and collecting a blood sample for DNA extraction. At study initiation, affection status was determined by clinical diagnosis of a referring research neurologist. As the study expanded, more rigorous criteria were incorporated. Structured clinical interviews, including psychometric evaluation utilizing the Clinical Dementia Rating scale (Hughes et al. 1982) and Mini-Mental State Exam (Folstein et al. 1975), or Modified Mini-Mental State Exam (Teng and Chui 1987), were administered. Wherever possible, formal psychometric tests were applied to previously ascertained individuals as well. Affection status was determined by consensus of physicians and physician assistants experienced in clinical dementia research. Probands and other sampled affected subjects all met NINCDS-ADRDA criteria for the diagnosis of AD (McKhann et al. 1984). In concert with clinical ascertainment, an autopsy and brain tissue donation program was established to provide tissue for research and neuropathologic diagnoses.
Families were contacted annually through research newsletters and brief questionnaires. The research newsletters highlighted ongoing genetic research, underscored the importance of follow-up, and explained the tissue donation program. The brief questionnaires contained open-ended queries regarding dementia progression and any new onset of cognitive problems in previously unaffected participants, as well as unsampled family members. Reply to questionnaires was voluntary; individuals who did not reply were not re-contacted. In addition to annual newsletters, regular attempts were made to gather additional information on any individual who was reported by the family to have developed cognitive difficulties.
Autopsy and tissue donation services were offered to all families enrolled in the research study. Families who expressed interest in autopsy and tissue donation were contacted, and we established a tissue donation plan when appropriate. Because participation in the autopsy and tissue donation program was voluntary, not all families enrolled in the genetic research component of the study were also enrolled in the autopsy component of the study.
Fifteen years after enrollment began, a systematic follow-up was initiated. A letter was sent to each family member of the 81 families that previously enrolled or expressed interest in receiving newsletters. Subsequently, each individual or their representative was contacted by telephone. Whenever possible, individuals were then seen in person. Otherwise, contact was by telephone, and medical records were obtained as appropriate. During follow-up contact, extant vital and cognitive status of every family member was solicited, and arrangements were made for the collection of additional clinical information and blood samples.
Clinical Evaluation
Informed consent for clinical evaluations, blood samples, autopsy and tissue donation, and telephone follow-up was obtained for all participating individuals under protocols approved by the DUMC Institutional Review Board (IRB).
Clinical diagnosis of AD was made using NINCDS-ADRDA guidelines (McKhann et al. 1984). Clinical information was obtained from several sources. Individuals were identified either from an historical report of dementia of insidious onset and gradual progression (according to at least two family observers), a review of medical records, a clinical examination, or a combination of the above. With regard to family-based historical reports of dementia, no disagreements among family members were experienced. Cases were categorized as affected (diagnosis of probable, or possible AD), unclear (usually dementia of unknown cause or mild cognitive impairment), or unaffected (no history of, or no evidence on screening evaluation of cognitive impairment). If diagnosis was based on observer history and/or limited medical record information, then data appropriate to rule out the presence of atypical neurological features were deemed not available. AAO was defined as the age at which a family historian reported onset of significant cognitive problems that interfered with normal activity, or the age at onset of problems as documented in the medical record. Family history data was updated and expanded to capture any individuals in the family with newly diagnosed AD.
Pathological Evaluation
All neuropathologic diagnoses were made by neuropathologists (C.M.H. and others) with experience in the diagnosis of neurodegenerative diseases. Pathological diagnosis of AD was based on senile plaque counts using CERAD criteria (Mirra et al. 1991). Neurofibrillary tangle (NFT) counts were also recorded using Braak and Braak (B&B) (Braak and Braak 1991) staging, defined as: stage 1-2: mesial temporal lobe NFTs; stage 3-4: some neocortical NFTs; and stage 5-6: frequent neocortical NFTs. Lewy bodies were identified with α-synuclein immunostaining techniques, and the McKeith diagnostic rating protocol for their presence (brainstem, limbic/transitional, neocortical) (McKeith et al. 1996).
A pathologic diagnosis of dementia with Lewy bodies (DLB) was assigned to brain tissue with pathologic findings of diffuse Lewy bodies and B&B staging limited to stage 0-2 plaques (Braak and Braak 1991). If pathologic findings were B&B stage 3 or greater and Lewy bodies were present, the assigned pathologic diagnosis of Alzheimer disease and Lewy bodies (AD-LB) was assigned (Heyman et al. 1999; McKeith et al. 1996).
Gross pathologic findings of severe symmetric temporal and/or frontal lobe atrophy, in conjunction with pathognomonic silver-staining cytoplasmic inclusions, resulted in the neuropathologic diagnosis of Pick’s disease (Uchihara et al. 2003). Progressive supranuclear palsy (PSP) was diagnosed by findings of tau-positive diffuse NFTs in both the neocortex and multiple subcortical structures, as well as neuronal loss and gliosis in specific subcortical areas (Rampello et al. 2005; Josephs et al. 2006).
Statistical Analysis
We compared the distributions of demographical features across different subgroups of neuropathological diagnoses by autopsy. The two-sided t-test was used to examine the difference in age at onset (AAO) between the group of individuals with a sole neuropathological diagnosis of AD (AD-only), and the group of individuals with other diagnoses (either pathologic findings in addition to those of AD, or findings consistent with a non-AD etiology for the dementia). The chi-square test was performed to compare the sex ratio between the AD-only group and other-diagnoses group. To evaluate the association between the APOE e4 allele and autopsy neuropathological diagnoses, we performed the Fisher’s exact test, due to small numbers in some subgroups. Individuals were included in these analyses regardless of family relationships, and thus, the variance may be underestimated. The computer program FASTLINK was used to calculate parametric linkage to APOE (affecteds only, both dominant and recessive models) on the 63 families in their original and updated forms. A disease allele frequency of 0.001 with penetrance of 0.0001 was used for the dominant model, while the recessive model had a disease allele frequency of 0.2 with penetrance of 0.0001.
Results
Initial Data and Clinical Diagnoses
Of the 81 families ascertained, we were able to re-contact 63 (78%). Among the 63 families, 148 individuals were initially determined to be affected with AD, 19 individuals had unclear cognitive status, and 211 individuals were unaffected. The average AAO of the 148 affected individuals was 69.9 years, with a gender distribution of 99 females (67%) and 49 males (33%).
Follow-up Data and Clinical Diagnoses
After updating family history data for the available 63 families, a supplement of 20 individuals (primarily first cousins of the probands) were identified and categorized as affected. In addition, many previously ascertained individuals whose original cognitive statuses were unclear or unaffected, converted to affected. Of the 19 individuals who originally had an unclear cognitive status, 10 (53%) converted to affected status, eight died without change in cognitive status, and the remaining individual had no available follow-up data. Of the 211 individuals originally considered unaffected, 38 (18%) converted to affected status and 25 (12%) converted to cognitive status unclear.
The follow-up data set comprises 216 affected individuals, with 135 females (62.5%) and 81 males (37.5%). Mean AAO was 71.3 years (SD 7.0), and was not significantly different from the original data set (p=0.08). These results are summarized in Table 1.
Table 1.
Autopsy neuropathologic diagnoses and demographics for 101 individuals with a clinical diagnosis limited to late-onset AD alone
| Group # | Neuropathologic Diagnosis | n | n-female (%) | sex p-value | Mean AAO +/- (SD) | AAO p-value |
|---|---|---|---|---|---|---|
| 1 | AD only | 73 | 55 (75) | n/a | 71.5 (7.0) | n/a |
| 2 | AD-LB | 18 | 8 (42) | 0.011 | 67.6 (5.2) | 0.029 |
| 3 | DLB | 7 | 2 (29) | 0.009 | 67.4 (8.3) | 0.149 |
| 4 | Picks | 2 | 1 (50) | -------- | 60.0 (1.4) | 0.024 |
| 5 | AD-PSP | 1 | 0 (0) | -------- | 68 ---- | ------- |
| 6 | 2+3 (any-LB) | 25 | 10 (40) | 0.001 | 67.6 (6.0) | 0.015 |
| 7 | 2+3+4+5 (any co-morbidity) | 28 | 11 (39) | 0.001 | 67.0 (6.0) | 0.003 |
| total | 1+2+3+4+5 | 10 1 |
67 (66) | ------- | 70.4 (6.9) | ------- |
n/a is not applicable (i.e. comparator group)
Sex p-value: the chi-square nominal p-value for the difference in sex ratio between the clinical diagnosis group and the AD only pathological group
AAO p-value: the t-test nominal p-value for the difference in age-at-onset between the clinical diagnosis group and the AD only pathological group
APOE gene frequencies in the initial data set of 148 individuals were APOE-2 = 0.007; APOE-3 = 0.496; and APOE-4 = 0.50, while the frequencies in the 48 individuals converting to affected in the follow-up analysis were 0.043, 0.5, and 0.457 respectively (none differed significantly, p>0.05). Two-point LOD scores at the APOE-4 locus for the initial data set were 1.279 under.a dominant model and 1.505 under a recessive model, while the follow-up data set LOD scores were 2.920 and 2.856 respectively, representing a more than tenfold increase in likelihood of linkage between APOE and AD in the updated dataset.
Neuropathologic Characteristics and Clinical Correlations
Individuals
Neuropathologic diagnoses were completed for 101 individuals with clinically diagnosed AD (see figure A). Autopsy confirmed that 73 of the 101 (72%) clinically diagnosed AD participants also had the pathologic diagnosis of definite AD. This group, with concordant neuropathologic diagnosis, was predominantly female (75%).
Figure A.
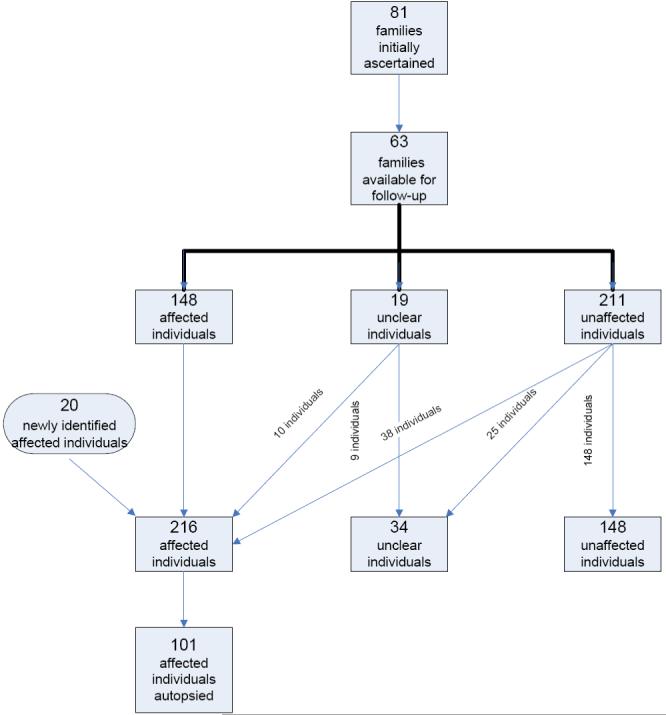
Flowchart showing original dataset and clinical follow-up.
However, the remaining 28 autopsies revealed diagnoses that were not consistent with the assigned clinical diagnosis of AD alone. Nineteen autopsies identified significant unrecognized co-morbid neurodegenerative disorders in addition to AD pathology, while nine other autopsies demonstrated evidence of an etiology of dementia unrelated to AD. The group of patients with neuropathologic diagnoses other than AD alone was significantly less female (38% female; p=0.001), and was significantly younger (by 4.5 years) than the group diagnosed with AD alone (p = 0.003) (Table 1). APOE e4 allele status was not significantly different between these 28 individuals (84.6% of whom had at least one e4 allele; Table 2) and the 73 with definite AD by neuropathology (79.3% with at least one e4 allele; data not shown).
Table 2.
Autopsy neuropathologic diagnoses, demographics, and APOE alleles for 28 individuals with a clinical diagnosis limited to late-onset AD alone, and findings on autopsy.discordant with, or in additional to, those consistent with AD
| Family | Individual | AAO | AOD | APOE allele 1 | APOE allele 2 | Neuropathological diagnosis |
|---|---|---|---|---|---|---|
| A | 1 | 63 | 70 | 4 | 4 | AD-LB |
| B | 1 | 70 | 77 | 3 | 4 | AD-LB |
| B | 108 | 68 | 83 | 3 | 4 | AD-LB |
| C | 136 | 64 | 79 | 3 | 4 | AD-LB |
| D | 1 | 75 | 86 | 3 | 4 | AD-LB |
| E | 1 | 69 | 77 | 3 | 4 | AD-LB |
| F | 113 | 68 | 72 | 3 | 4 | AD-LB |
| G | 1 | 59 | 71 | 4 | 4 | AD-LB |
| H | 1 | 74 | 85 | 3 | 3 | AD-LB |
| I | 105 | 70 | 87 | 3 | 4 | AD-LB |
| J | 9000 | 73 | 84 | 3 | 4 | AD-LB |
| K | 1 | 55 | 72 | 3 | 3 | AD-LB |
| L | 1 | 66 | 71 | 4 | 4 | AD-LB |
| M | 1 | 70 | 91 | 3 | 4 | AD-LB |
| N | 106 | 70 | 77 | 4 | 4 | AD-LB |
| O | 1 | 70 | 76 | 3 | 4 | AD-LB |
| P | 1 | 63 | 72 | 3 | 4 | AD-LB |
| Q | 125 | 70 | 77 | 3 | 4 | AD-LB |
| R | 107 | 68 | 71 | 3 | 4 | AD-PSP |
| D | 105 | 59 | 74 | 3 | 3 | DLB |
| R | 1 | 61 | 69 | 3 | 4 | DLB |
| R | 1001 | 82 | 90 | 3 | 3 | DLB |
| E | 105 | 70 | 75 | 3 | 4 | DLB |
| G | 105 | 67 | 74 | n/a | n/a | DLB |
| S | 1 | 60 | 73 | 3 | 4 | DLB |
| T | 1 | 73 | 81 | 3 | 4 | DLB |
| U | 1 | n/a | 81 | 3 | 3 | PICK |
| V | 1 | 61 | 81 | 3 | 4 | PICK |
n/a is data not available
Of the 19 autopsies with co-morbid neurodegenerative disorders, characteristic neuropathology necessary for diagnosis of AD (B&B stage 3 or greater) was accompanied by significant Lewy bodies (AD-LB) in 18. One individual had dual neuropathologic diagnoses of AD and progressive supranuclear palsy (AD-PSP).
Clinically, six of the 18 individuals with AD-LB manifested some parkinsonian features (bradykinesia, wide-based and unsteady gate, increased muscle tone or masked facies) and one of those six had hallucinations, while another had a fluctuating sensorium. However, none met sufficient clinical criteria for a diagnosis of Parkinson disease or DLB. Complicating the clinical differentiation were those individuals who had only AD pathology and no evidence of Lewy bodies, but did have some parkinsonian features. The 12 other individuals diagnosed with AD-LB upon autopsy manifested clinical findings consistent with AD alone, with no clinical parkinsonian features.
The individual with AD-PSP had a dementia AAO of 68, and clinical characteristics typical of PSP. On exam, impaired upward and downward gaze were noted, as well as questionably increased asymmetric muscle tone. However, the clinical characteristics of PSP were only recognized post-mortem during a thorough medical chart review.
Nine individuals had dementias without neuropathologic evidence of AD. Seven had Lewy body pathology consistent with DLB. Picks disease accounted for the remaining two non-AD cases of dementia.
Clinical diagnosis of dementia with Lewy bodies (DLB) was based on consensus criteria (McKeith et al. 1996). However, no individual met sufficient clinical criteria to receive a clinical diagnosis of DLB. Several cases did manifest a subsyndromal range of clinical findings characteristic of DLB; three of the seven had significant motor features (bradykinesia, wide-based and unstable gait, and increased tone), including one who also had masked facies.. One other individual had hallucinations, and fluctuating sensorium. The remaining three individuals with DLB had clinical features consistent with AD alone.
Families
Fifty-two of the 63 families (83%) included in the follow-up had family members come to autopsy. Twenty-one of these families had only one individual autopsied. Fifteen of these 21 individuals received a matching tissue diagnosis of definite AD. However, four others had a pathologic diagnosis of both AD and LB pathology, and only one of these four had any identified motor features. Pathologic diagnosis of DLB was rendered from autopsy for each of the two remaining individuals. Both of these expressed subsyndromal motor features.
Thirty-one families had two or more individuals come to autopsy. A pathologic diagnosis of definite AD (without evidence of another dementing process) was made in all autopsied individuals in only 15 (48%) of these families. Ten families (32%) had at least one individual with AD pathology, and at least one other individual with both AD and LB pathology. However, only four of the eleven individuals in the ten families with this combination of anatomic pathologies (AD-LB) had clinical features suggestive of DLB: mild motor features (1), gait change (1), motor features (1), and hallucinations (1). The other six had no clinical DLB features. Another two families, with the clinical diagnosis of AD alone, had one individual with co-morbid AD and LB pathology, and one with DLB, upon autopsy. There were no individuals in either family with clinical evidence of DLB or parkinsonism, despite autopsy evidence of significant Lewy bodies in all autopsied individuals.
Two families had disparate pathologic reports for AD and Picks disease. One family had an individual with definite AD and another individual with Picks disease, while the other family had two individuals with definite AD and one individual with Picks disease. In one of these individuals, questionable frontal release signs (snout and palmomental reflexes) were noted.
One family had three individuals with disparate pathologic diagnoses including AD, AD-LB pathology (syncope, mildly increased tone), and DLB (hallucinations, parkinsonian response to certain neuroleptic medications). A final extraordinary family had four individuals come to autopsy, and the pathologic diagnoses were definite AD (ataxia, slightly increased muscle tone in the upper extremities, and visual hallucinations), AD-PSP (see individual results), and DLB (2 individuals without clinical DLB symptoms).
Discussion
For diseases with a complex phenotype or a late onset, stability of initial diagnoses may be problematic. For probands, clinical diagnosis is limited by the sensitivity of the diagnostic criteria. Critical co-morbid disorders and alternative diagnoses may be overlooked. For example, in this study, all 101 individuals who came to autopsy met clinical criteria of AD alone, however, only 73 of those individuals had evidence of AD alone upon autopsy. Additionally, the discrepancies between clinical diagnosis and histopathologic diagnosis increased as the number of autopsies in the family increased. While all families in the study were offered autopsy and tissue diagnoses, it is possible that this trend is a result of sampling bias. Families containing greater numbers of affected individuals, or individuals with symptoms outside of the typical range for AD, may have preferentially elected to participate in the autopsy and tissue donation program.
We also found that the group with neuropathologic diagnoses other than AD alone were significantly less female (38% vs. 75%) and were significantly younger (67.0 years vs. 71.5 years) than those with pathology limited to AD. These differences may represent important clues to underlying heterogeneity that could not be determined from clinical assessment alone. Furthermore, for non-proband individuals in a pedigree, diagnosis is limited to those disorders that are significantly expressed at the time of ascertainment. Any disease manifesting after ascertainment is therefore missed. Longitudinal follow-up of individuals participating in research provides the opportunity to resolve much of the concomitant ambiguity. With follow-up, initial diagnostic impressions may be clarified. Additionally, diagnoses may be confirmed through alternative sources, such as autopsy.
Fortunately, members of multiplex families with LOAD are receptive to repeated contacts by research staff, and these repeat contacts have been fruitful. Previously ascertained unaffected individuals in the families develop dementia in significant numbers, confirming the familial nature of the diagnosis. When the families where first ascertained and examined, 211 individuals were classified as unaffected or unclear. Upon follow-up analysis in this study, 48 individuals out of the 211 (22.7%) converted to affected status. The prevalence of AD for those 65 years of age or greater is 10.3% (Evans et al., 1989). Also, in the course of follow-up and expansion of family history data 20 new affected individuals were identified who were previously unknown to the study.
Awareness of change in affection status is critical for genetic analyses. For instance, previously believed discordant sibling pairs may obscure interpretation of results due to lack of power (Martin et al. 2003). Follow-up provides a means to reevaluate these sibling pairs, possibly identifying newly affected individuals in the analyses. The need for follow-up in studies of age-related disorders is critical to reliably assign affection status and avoid misclassifications. Additionally, follow-up provides an opportunity to identify and ascertain supplemental individuals in families, furtherincreasing statistical power for genetic analyses.
Studies such as this one focusing on the genetics of AD came to prominence in the 1980s and early 1990s. In 1997, the National Institute on Aging and the Reagan Institute proposed consensus criteria for pathologic diagnosis incorporating the staging of neurofibrillary changes of Braak and Braak (NIA-Reagan Working Group 1997). The NIA-Reagan criteria establish probabilistic categories in an attempt to assess whether clinical dementia is caused by AD pathology. Both CERAD and NIA-Reagan recognized that Lewy bodies, the hallmark lesions in Parkinson disease, are present in variable number and location in a significant number of brains of patients with AD.
The biological significance of plaques, tangles, and Lewy bodies in any single patient, as well as in patient groups is yet to be determined. Tissue donation and autopsies in those with a clinical diagnosis of AD are an important additional source of information. Pathologic diagnosis may confirm, clarify, or expand the clinical diagnosis of the etiology of dementia. Banked tissue also facilitates more comprehensive investigation, such as gene expression profiling. By discussing the utility of autopsy and tissue donation in our center’s periodic newsletters, participants from the extended families have been successfully recruited. Family members participated in the autopsy and tissue donation program in significant numbers. One hundred and one affected individuals came to autopsy, out of a total of 196 affected individuals included the study (148 affected individuals at initial ascertainment and 48 who converted to affected during the follow-up years). Two unaffected individuals participated in the autopsy program as controls (data not shown). Maintaining relationships with the families encouraged generous participation.
Both the utility and the confounding nature of anatomic pathologic diagnosis are highlighted by our results. Studies of unrelated individuals clinically diagnosed with AD have demonstrated neuropathological features either not consistent with-AD, or additional to those of AD, in 21 to 38% of autopsied cases (Gearing et al. 1995; Jicha et al.2006; Lim et al. 1999). In this study, the pathologic diagnosis confirmed the clinical diagnosis of AD in all autopsied cases in 31 of the families in which at least one autopsy was performed. However, in the remaining 21 families (40%), additional or different pathology was recognized. Fourteen families exhibited AD pathology, plus Lewy bodies in 15 of their 32 autopsied cases. There were no consistent or uniform clinical distinctions between those individuals with AD pathology versus those with AD and Lewy body pathology. It is unclear whether the pathogenesis (genetic or non-genetic) of disease in these families is comparable to the process in families with AD pathology alone. Additionally, at least one individual in each of six families had a pathologic diagnosis of DLB. Three of these individuals had motor features, one had hallucinations and fluctuations in cognition, but the other three had clinical evidence of AD alone. Individuals with any clinical features of DLB and their families may not be appropriate for inclusion in a genetic analysis of AD. Nevertheless, none of the individuals with DLB met sufficient clinical criteria for a diagnosis consistent with DLB.
Alternatively, clinical heterogeneity could be reflective of genetic heterogeneity. Stratification on clinical features could identify a more homogenous subset for genetic analysis. Six families had disparate pathological diagnoses. Gene-environment interactions may cause pathologic lesions characteristic of AD to occur in one sibling, and those of DLB or Picks disease to occur in a second sibling. However, an intriguing second possibility is suggested by recent findings of the LRRK2 mutation in autosomal dominant PD (Zimprich et al. 2004). In the LRRK2 case, genetic homogeneity (identical mutations in different individuals) results in phenotypic heterogeneity in terms of neuropathologic features. Thus the pathologic changes may not be related to the primary risk factor, but instead be due to modifying genes or secondary effects.
The two families with affected individuals that were clinically diagnosed with AD, but who had autopsy evidence of DLB alone, serve as a reminder of the complexity of clinical diagnosis. The pathologic findings in these two families are of particular interest, since one family included a sibling of an affected individual with a clinical diagnosis of Parkinson’s disease (but no dementia), and the other family included three siblings of an affected individual, all of whom had a clinical diagnosis of AD without motor features. Families with disparate pathological diagnoses could represent underlying genetic heterogeneity. Including them in genetic analyses with families whose members have only AD pathology potentially confounds analyses.
The apparently discordant clinical characteristics and pathologic findings also suggest intriguing questions concerning the biology of AD and its relationship to other neurodegenerative disorders. Given a standard set of clinical characteristics, clinicians categorize patients into diagnostic groups. Genetic analyses assume the pathobiological mechanisms that result in the clinical disease are similar, at least in the majority of cases (i.e., there is a low phenocopy rate). Given what is now understood about the complex nature of “genetic” disorders, an alternative hypothesis asserts that the consequences of an allele of “moderate effect” are modified by additional factors. These factors, genetic and/or environmental, impact the pathobiology, producing significant clinically observed signs and symptoms. For two siblings, each with the same allele of moderate effect, one may have clinical and pathological AD, while the second may have clinical and even pathological DLB. Analysis of data in complex genetic disorders is made more difficult by such findings.
Follow-up of large multiplex families in which multiple members have late onset Alzheimer disease is productive and important. Through follow-up, affection status may be clarified, additional family members may be ascertained, and the natural history of the disease may be more fully understood. Furthermore, enrolling individuals in autopsy and tissue donation programs allows pathological diagnoses, and facilitates additional biologic investigation. These refined data may be critical to increasing the power of genetic analyses both by enlarging the data set and by clarifying diagnostic categories. Periodic follow-up with family members provides additional material useful in the continuing effort to elucidate the biological structures that are the bases of what is clinically recognized as the dementia of Alzheimer disease(s), as well as other disorders causing degenerative dementia.
Acknowledgments
This study is funded by grants from the National Institutes of Health/National Institute of Aging (R01 AG019757, R01 AG021547, P50 AG005128), the Alzheimer’s Association, Inc., the Claude D. Pepper Older Americans Independence Center (P50 AG11268), the McKnight Endowment Fund for Neuroscience, the Louis D. Scientific Award of the Institut de France, and from donor contributors to the Duke Center for Human Genetics research program.
References
- Bertram L, McQueen MB, Mullin K, Blacker E, Tanzi RE. Systematic meta-analyses of Alzheimer Disease genetic association studies: the AlzGene database. Nat Genet. 2007;39:17–32. doi: 10.1038/ng1934. [DOI] [PubMed] [Google Scholar]
- Blacker D, Bertram L, Saunders AJ, Moscarillo TJ, Albert MS, Wiener H, Perry RT, Collins JS, Harrell LE, Go RC, Mahoney A, Beaty T, Fallin MD, Avramopoulos D, Chase GA, Folstein MF, McInnis MG, Bassett SS, Doheny KJ, Pugh EW, Tanzi RE. Results of a high-resolution genome screen of 437 Alzheimer’s Disease families. Hum Mol Genet. 2003;12:23–32. doi: 10.1093/hmg/ddg007. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E. Demonstration of amyloid deposits and neurofibrillary changes in whole brain sections. Brain Pathol. 1991;1:213–216. doi: 10.1111/j.1750-3639.1991.tb00661.x. [DOI] [PubMed] [Google Scholar]
- Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261:921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- Evans DA, Funkenstein DH, Albert MS, Scherr PA, Cook NR, Chown MJ, Hebert LE, Hennekens CH, Taylor JO. Prevalence of Alzheimer’s Disease in a community population of older persons. Higher than previously reported. JAMA. 1989;262:2551–2556. [PubMed] [Google Scholar]
- Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, Myers RH, Pericak-Vance MA, Risch N, Van Duijn CM, APOE and Alzheimer Disease Meta Analysis Consortium Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. JAMA. 1997;278:1349–1356. [PubMed] [Google Scholar]
- Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”: a practical method for grading the cognitive state of patients for the clinician. J Psychiat Res. 1975;12:189–198. doi: 10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- Gearing M, Mirra SS, Hedreen JC, Sumi SM, Hansen LA, Heyman A. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part X. Neuropathology confirmation of the clinical diagnosis of Alzheimer’s Disease. Neurology. 1995;45:461–466. doi: 10.1212/wnl.45.3.461. [DOI] [PubMed] [Google Scholar]
- Goate A, Chartier-Harlin MC, Mullan M, Brown J, Crawford F, Fidani L, Giuffra L, Haynes A, Irving N, James L, Mant R, Newton P, Rooke K, Roques P, Talbot C, Pericak-Vance MA, Roses A, Williamson R, Rossor M, Owen M, Hardy J. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature. 1991;349:704–706. doi: 10.1038/349704a0. [DOI] [PubMed] [Google Scholar]
- Heyman A, Fillenbaum GG, Gearing M, Mirra SS, Welsh-Bohmer KA, Peterson B, Pieper C. Comparison of Lewy body varient of Alzheimer’s Disease with pure Alzheimer’s Disease. Neurology. 1999;52:1839–1844. doi: 10.1212/wnl.52.9.1839. [DOI] [PubMed] [Google Scholar]
- Hughes CP, Berg L, Danziger WL, Cohen LA, Martin RL. A new clinical scale for the staging of dementia. British Journal of Psychiatry. 1982;140:566–572. doi: 10.1192/bjp.140.6.566. [DOI] [PubMed] [Google Scholar]
- Jicha GA, Parisi JE, Dickson DW, Johnson K, Cha R, Ivnik RJ, Tangalos EG, Boeve BF, Knopman DS, Braak H, Peterson RC. Neuropathologic outcome of mild cognitive impairment following progression to clinical dementia. Arch Neurol. 2006;63:674–681. doi: 10.1001/archneur.63.5.674. [DOI] [PubMed] [Google Scholar]
- Josephs KA, Petersen RC, Knopman DS, Boeve BF, Whitwell JL, Duffy JR, Parisi JE, Dickson DW. Clinicopathologic analysis of frontotemporal and corticobasal degenerations and PSP. Neurology. 2006;66:41–48. doi: 10.1212/01.wnl.0000191307.69661.c3. [DOI] [PubMed] [Google Scholar]
- Laird NM, Horvath S, Xu X. Implementing a unified approach to family-based tests of association. Genet Epidemiol. 2000;19(Suppl 1):S36–S42. doi: 10.1002/1098-2272(2000)19:1+<::AID-GEPI6>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Lee D, Lockey R, Mohapatra S. Folate receptor-mediated cancer cell specific gene delivery using folic acid-conjugated oligochitosans. J Nanosci Nanotechnol. 2006;6:2860–2866. doi: 10.1166/jnn.2006.465. [DOI] [PubMed] [Google Scholar]
- Levy-Lahad E, Wasco W, Poorkaj P, Romano DM, Oshima J, Pettingell WH, Yu CE, Jondro PD, Schmidt SD, Wang K, Crowley AC, Fu YH, Guenette SY, Galas D, Nemens E, Wijsman EM, Bird TD, Schellenberg GD, Tanzi RE. Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science. 1995;269:973–977. doi: 10.1126/science.7638622. [DOI] [PubMed] [Google Scholar]
- Lim A, Tsuang D, Kukull W, Nochlin D, Leverenz J, McCormick W, Bowen J, Teri L, Thompson J, Peskind ER, Raskind M, Larson EB. Clinico-neuropathological correlation of Alzheimer’s Disease in a community-base case series. J Am Geriatr Soc. 1999;47:564–569. doi: 10.1111/j.1532-5415.1999.tb02571.x. [DOI] [PubMed] [Google Scholar]
- Ma DQ, Cuccaro ML, Jaworski J, Haynes C, Abramson RK, Wright HHGJ, Haines JL, Pericak-Vance MA. Dissecting the Locus Heterogeneity of Autism: Significant Linkage to Chromosome 12q14. Molecular Psychiatry. doi: 10.1038/sj.mp.4001927. in press. [DOI] [PubMed] [Google Scholar]
- Martin ER, Bass MP, Hauser ER, Kaplan NL. Accounting for linkage in family-based tests of association with missing parental genotypes. Am J Hum Genet. 2003;73:1016–1026. doi: 10.1086/378779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeith IG, Galasko D, Kosaka K, Perry EK, Dickson DW, Hansen LA, Salmon DP, Lowe J, Mirra SS, Byrne EJ, Lennox G, Quinn NP, Edwardson JA, Ince PG, Bergeron C, Burns A, Miller B, Lovestone S, Collerton D, Jansen ENH, Ballard C, de Vos RAI, Wilcock GK, Jellinger KA, Perry RH. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): Report of the consortium on DLB International Workshop. Neurology. 1996;47:1113–1124. doi: 10.1212/wnl.47.5.1113. [DOI] [PubMed] [Google Scholar]
- McKhann G, Drachman D, Folstein M. Clinical diagnosis of Alzheimer’s disease: Report of the NINCDS-ADRDA Work Group under the auspices of the Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology. 1984;34:939–944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, Van Belle G, Berg L. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology. 1991;41:479–486. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- Myers A, Holmans P, Marshall H, Kwon J, Meyer D, Ramic D, Shears S, Booth J, DeVrieze FW, Crook R, Hamshere M, Abraham R, Tunstall N, Rice F, Carty S, Lillystone S, Kehoe P, Rudrasingham V, Jones L, Lovestone S, Perez-Tur J, Williams J, Owen MJ, Hardy J, Goate AM. Susceptibility locus for Alzheimer’s disease on chromosome 10. Science. 2000;290:2304–2305. doi: 10.1126/science.290.5500.2304. [DOI] [PubMed] [Google Scholar]
- NIA-Reagan Working Group Consensus recommendations for the postmortem diagnosis of Alzheimer’s disease. The National Institute on Aging and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer’s Disease. Neurobiol Aging. 1997;18:S1–S2. [PubMed] [Google Scholar]
- Pericak-Vance MA, Bebout JL, Gaskell PC, Yamaoka LH, Hung WY, Alberts MJ, Walker AP, Bartlett RJ, Haynes CA, Welsh KA, Earl NL, Heyman A, Clark CM, Roses AD. Linkage studies in familial Alzheimer disease: evidence for chromosome 19 linkage. Am J Hum Genet. 1991;48:1034–1050. [PMC free article] [PubMed] [Google Scholar]
- Pericak-Vance MA, Yamaoka LH, Haynes CS, Speer MC, Haines JL, Gaskell PC, Hung WY, Clark CM, Heyman AL, Trofatter JA. Genetic linkage studies in Alzheimer’s disease families. Exp Neurol. 1988;102:271–279. doi: 10.1016/0014-4886(88)90220-8. [DOI] [PubMed] [Google Scholar]
- Rampello L, Butta V, Raffaele R, Vecchio I, Battaglia G, Cormaci G, Alvano A. Progressive supranuclear palsy: a systematic review. Neurobiol Dis. 2005;20:179–186. doi: 10.1016/j.nbd.2005.03.013. [DOI] [PubMed] [Google Scholar]
- Rogaev EI, Sherrington R, Rogaeva EA, Levesque G, Ikeda M, Liang Y, Chi H, Lin C, Holman K, Tsuda T. Familial Alzheimer’s disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer’s disease type 3 gene. Nature. 1995;376:775–778. doi: 10.1038/376775a0. [DOI] [PubMed] [Google Scholar]
- Schellenberg GD, Bird TD, Wijsman EM, Moore DK, Boehnke M, Bryant EM, Lampe TH, Nochlin D, Sumi SM, Deeb SS, Beyreuther K, Martin GM. Absence of linkage of chromosome 21q21 markers of familial Alzheimer’s disease. Science. 1988;241:1507–1510. doi: 10.1126/science.3420406. [DOI] [PubMed] [Google Scholar]
- Scott WK, Hauser ER, Schmechel DE, Welsh-Bohmer KA, Small GW, Roses AD, Saunders AM, Gilbert JR, Vance JM, Haines JL, Pericak-Vance MA. Ordered-subsets linkage analysis detects novel Alzheimer disease Loci on chromosomes 2q34 and 15q22. Am J Hum Genet. 2003;73:1041–1051. doi: 10.1086/379083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Y, Cuccaro ML, Hauser ER, Raiford KL, Menold MM, Wolpert CM, Ravan SA, Elston L, Decena K, Donnelly SL, Abramson RK, Wright HH, DeLong GR, Gilbert JR, Pericak-Vance MA. Fine mapping of autistic disorder to chromosome 15q11-q13 by use of phenotypic subtypes. Am J Hum Genet. 2003;72:539–548. doi: 10.1086/367846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, Chi H, Lin C, Li G, Holman K, Tsuda T, Mar L, Foncin JF, Bruni AC, Montesi MP, Sorbi S, Rainero I, Pinessi L, Nee L, Chumakov I, Pollen DA, Brookes A, Sanseau P, Polinsky RJ, Wasco W, Da Silva HAR, Haines JL, Pericak-Vance MA, Tanzi RE, Roses AD, Fraser PE, Rommens JM, St George-Hyslop PH. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature. 1995;375:754–760. doi: 10.1038/375754a0. [DOI] [PubMed] [Google Scholar]
- Small GW, Rabins PV, Barry PP, Buckholtz NS, DeKosky ST, Ferris SH, Finkel SI, Gwyther LP, Khachaturian ZS, Lebowitz BD, McRae TD, Morris JC, Oakley F, Schneider LS, Streim JE, Sunderland T, Teri LA, Tune LE. Diagnosis and treatment of Alzheimer disease and related disorders. Consensus statement of the American Association for Geriatric Psychiatry, the Alzheimer’s Association, and the American Geriatrics Society. JAMA. 1997;278:1363–1371. [PubMed] [Google Scholar]
- Teng EL, Chui HC. The modified Mini-Mental State (3MS) examination. Journal of Clinical Psychiatry. 1987;48:314–318. [PubMed] [Google Scholar]
- Terwilliger JD, Haghighi F, Hiekkalinna TS, Goring HH. A bias-ed assessment of the use of SNPs in human complex traits. Curr Opin Genet Dev. 2002;12:726–734. doi: 10.1016/s0959-437x(02)00357-x. [DOI] [PubMed] [Google Scholar]
- Uchihara T, Ikeda K, Tsuchiya K. Pick body disease and Pick syndrome. Neuropathology. 2003;23:318–326. doi: 10.1046/j.1440-1789.2003.00523.x. [DOI] [PubMed] [Google Scholar]
- Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, Kachergus J, Hulihan M, Uitti RJ, Calne DB, Stoessl AJ, Pfeiffer RF, Patenge N, Carbajal IC, Vieregge P, Asmus F, Muller-Myhsok B, Dickson DW, Meitinger T, Strom TM, Wszolek ZK, Gasser T. Mutations in LRRK2 Cause Autosomal-Dominant Parkinsonism with Pleomorphic Pathology. Neuron. 2004;44:601–607. doi: 10.1016/j.neuron.2004.11.005. [DOI] [PubMed] [Google Scholar]