

Abstract
Local rates of cerebral protein synthesis (lCPSleu) were measured with the quantitative autoradiographic [1-14C]leucine method in a genetic mouse model (Pahenu2) of phenylketonuria. As in the human disease, Pahenu2 mice have a mutation in the gene for phenylalanine hydroxylase. We compared adult homozygous (HMZ) and heterozygous (HTZ) Pahenu2 mice with the background strain (BTBR). Arterial plasma concentrations of phenylalanine (Phe) were elevated in both HMZ and HTZ mutants by 21 times and 38%, respectively. In the total acid-soluble pool in brain concentrations of Phe were higher and other neutral amino acids lower in HMZ mice compared with either HTZ or BTBR mice indicating a partial saturation of the l-amino acid carrier at the blood brain barrier by the elevated plasma Phe concentrations. In a series of steady-state experiments, the contribution of leucine from the arterial plasma to the tRNA-bound pool in brain was found to be statistically significantly reduced in HMZ mice compared with the other groups, indicating that a greater fraction of leucine in the precursor pool for protein synthesis is derived from protein degradation. We found reductions in lCPSleu of about 20% throughout the brain in the HMZ mice compared with the other two groups, but no reductions in brain concentrations of tRNA-bound neutral amino acids. Our results in the mouse model suggest that in untreated phenylketonuria in adults, the partial saturation of the l-amino acid transporter at the blood–brain barrier may not underlie a reduction in cerebral protein synthesis.
Phenylketonuria (PKU) is an inherited disease of Phe metabolism that if not treated very early in life results in profound mental retardation (1). The classical form of PKU is characterized by mutations in the gene for Phe hydroxylase (PAH), negligible PAH activity in liver, elevated Phe concentrations in plasma, and metabolic products of Phe metabolism in urine. Despite extensive biochemical characterization of PKU, the mechanism by which hyperphenylalaninemia results in mental retardation is not understood. It has been hypothesized that an elevated plasma Phe concentration impedes entry of other neutral amino acids via the l-amino acid carrier at the blood–brain barrier, and that reduced influx may limit their availability for incorporation into tissue protein (2). There is ample evidence for the effective competitive inhibition of the transport system in hyperphenylalaninemic patients with PKU (3–5). It is only by inference that cerebral protein synthesis rates appear to be affected. Untreated patients with PKU have lower brain weights (6), changes in myelin structure (7), and reductions in dendritic arborization and numbers of synaptic spines (6). Selectively vulnerable regions of the brain are those that undergo development postnatally. During brain maturation when there is a net increase in brain protein, diminished rates of cerebral protein synthesis may not be adequate to support the growth and establishment of terminals and dendritic arbors so critical at this time.
To define the role of cerebral protein synthesis in PKU, animal models are essential. Before the recent development of a genetic model of PKU in mice (8), hyperphenylalaninemia was produced by systemic administration to rodents of large doses of Phe (2, 9) in conjunction with an inhibitor of PAH (10, 11) to prevent an increase in Tyr concentration. Animals were studied after acute or chronic treatment in adulthood and during maturation. These models, however, are imperfect homologues of PKU because of secondary effects of the PAH inhibitors.
Notwithstanding such imperfections, these were the only models available for neurochemical studies. Studies of developing mice administered high doses of Phe have shown disaggregation of brain polyribosomes and reduced rates of polypeptide chain elongation on polyribosomes prepared from brain homogenates (9, 12); both of these effects were reversed by treatment of the mice with a mixture of neutral amino acids (leucine, isoleucine, valine, threonine, tryptophan, tyrosine, methionine) (13). Direct effects of hyperphenylalaninemia on cerebral protein synthesis in vivo have, however, been more difficult to demonstrate because of the complications of making such measurements in intact animals. Several reports in the literature (2, 14–16) have led to conclusions that vary with the model studied and the method used to quantify cerebral protein synthesis rates.
Problems in the measurement of rates of cerebral protein synthesis in vivo with a radiolabeled tracer arise primarily from uncertainties about the specific activity (SA) of the tracer in the tissue precursor amino acid pool. Models for the behavior of the tracer in brain can be constructed and equations derived from which precursor SA can be estimated on the basis of measurements of the time course of SA in arterial plasma, but this presupposes that the sole source of amino acids in brain is the arterial plasma. We have determined that there is significant dilution of amino acids derived from plasma by amino acids released by breakdown of unlabeled protein in brain (17, 18); this “dilution” must be taken into account. In the present study, we have used a method that overcomes uncertainties about the precursor pool specific activity in the brain in vivo (17). The method was developed for use in the rat and adapted for the adult mouse. The radiolabeled tracer is [1-14C]leucine, which is used in conjunction with quantitative autoradiography to achieve spatial localization of the labeled protein in the brain for the determination of rates of protein synthesis in discrete brain regions (18). This method has been applied to a genetic mouse model of PKU (Pahenu2). The Pahenu2 mouse developed in the BTBR strain with germline mutagenesis (8, 19) has a mutation in the gene for PAH on chromosome 10. In mice homozygous for the mutation, the phenotype resembles human PKU. PAH activity is minimal in liver; concentrations of Phe are 10–20 times normal in plasma; animals are hypopigmented and exhibit impairments in performance of several behavioral tests of cognitive function (20). By these criteria, the Pahenu2 mouse appears to be a very good animal model of the human disease. Our results show that in the HMZ mouse, the fraction of leucine in the precursor pool coming from protein breakdown is increased, and lCPSleu is decreased compared with both BTBR and HTZ mice.
Materials and Methods
Chemicals.
Chemicals and materials were obtained from the following sources: l-[1-14C]leucine (SA, 60 mCi/mmol) and l-[3,4,5-3H]leucine (SA, 50 Ci/mmol), Du Pont/NEN; Escherichia coli tRNA, Sigma; vanadyl ribonucleoside complex and redistilled nucleic acid-grade phenol, Bethesda Research Laboratories; l-norleucine and 5-sulfosalicylic acid, Fluka; primers (upstream primer, 5′-ACCTTGTACTGGTTTCCGCCT-3′; and downstream primer, 5′-AGGTGTGTACATGGGCTTAG-3′), Cruachem, Herndon, VA; restriction endonuclease Alw26I, MBI Fermentas, Amherst, NY.
Animals.
All procedures were carried out in accordance with the National Institutes of Health Guidelines on the Care and Use of Animals and an animal study protocol approved by the National Institutes of Mental Health Animal Care and Use Committee. Pahenu2 breeding pairs (HTZ females and HMZ males) were obtained from The Jackson Laboratory. HTZ and HMZ pups produced by the breeding pairs were weaned between 21 and 23 days of age and crossbred in pairs of HTZ females and HTZ males to provide offspring in three experimental groups: HMZ, HTZ, and BTBR mice. All mice were housed in a central facility and maintained under controlled conditions of normal humidity and temperature with standard alternating 12-h periods of light and darkness. Food (NIH-31 rodent chow) and water were provided ad libitum. A total of 78 mature male mice between the ages of 10 and 18 weeks were studied.
Genotyping.
HMZ mice were easily distinguished from their HTZ and BTBR littermates by their lighter coat color. Phenotype was confirmed from concentrations of Phe in plasma elevated to about ×20 normal. To differentiate HTZ from BTBR littermates, genotyping was necessary. Genomic DNA was extracted (21) from a section of tail, and exon 7 of the PAH gene was amplified as described previously (19). The 132-bp PCR product was then subjected to restriction endonuclease digestion with Alw26I and incubated overnight at 37°C. The restriction fragments were separated by electrophoresis on a 20% polyacrylamide denaturing gel at 140 V for 16 h. The digestion fragments were: BTBR, 82 and 50 bp; HTZ, 82, 50, 48, and 34 bp; and HMZ, 50, 48, 34 bp.
Animal Preparation.
Mice were prepared for studies by insertion under light halothane anesthesia of polyethylene catheters (PE-10) into one femoral artery and one or two femoral veins. The catheters were tunneled under the skin to exit at the nape of the neck and then threaded through a flexible stainless steel swivel system attached to the skin with a button tether (Harvard Apparatus). Mice were allowed to recover from the surgery overnight in clear plastic cylinders 13 cm in diameter. This apparatus permitted mice to move freely throughout the recovery and experimental periods but not to gain access to the tubing. Food and water were available ad libitum.
Physiological Variables.
Mean arterial blood pressure, hematocrit, and arterial plasma glucose concentrations were measured to evaluate each animal's physiological state, as previously described (22). Body temperature was monitored with a Model BAT-12 thermometer (Sensortek, Clifton, NJ) or a microprobe implanted in the peritoneal cavity.
Method for the Measurement of lCPSleu.
The radiolabeled tracer is [1-14C]leucine, which is used in conjunction with quantitative autoradiography to achieve spatial localization of the labeled protein in the brain for the determination of rates of protein synthesis in discrete brain regions (17). The operational equation of the method is:
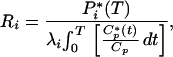 |
in which Ri is the rate of leucine incorporation into protein in tissue i; P*i(T) is the concentration of 14C fixed in tissue i at any time, T, after introduction of the tracer into the circulation; λi is equal to the fraction of leucine in the precursor pool for protein synthesis in tissue i that is derived from plasma; C*p(t) and Cp are the concentrations of labeled and unlabeled leucine in the arterial plasma, respectively, and t is the variable time. The evaluation of λi is carried out in a separate series of experiments by measuring the steady-state ratio of leucine SA in the tissue tRNA-bound pool to that of the arterial plasma.
Procedure for the Determination of lCPSleu.
Mice were surgically prepared and catheterized and their physiological states monitored as described above. The experimental period was initiated by an i.v pulse of 100 μCi/kg of l-[1-14C]leucine contained in ≈40 μl of physiological saline. Timed arterial samples were collected during the following 60 min for determination of the time courses of plasma concentrations of leucine and [14C]leucine. Blood sampling was more frequent during the period of most rapid change in the plasma concentration of [14C]leucine. The blood samples were immediately centrifuged to remove the red cells, and 5 μl of plasma from each sample was diluted in distilled water and deproteinized at 4°C by the addition of a solution of 16% (wt/vol) sulfosalicylic acid containing [3H]leucine (1 mCi/ml) and l-norleucine (0.04 mM) as internal standards for liquid scintillation counting and amino acid analysis, respectively. Labeled and unlabeled leucine concentrations in the acid-soluble fractions were assayed by liquid scintillation counting and by amino acid analysis, respectively. Concentrations of [14C]leucine and total leucine were calculated with correction for recovery based on the internal standard concentrations measured in each sample. At the end of the experimental interval, mice were killed by an i.v. injection of sodium pentobarbital, and brains were rapidly removed and frozen in isopentane cooled to −40°C with dry ice. Serial sections, 20 μm thick, were cut in a Leica 3050 cryostat (Leica, Deerfield, IL) at −18°C, thaw mounted on gelatin-coated slides, air dried, fixed, and washed in 10% formalin, and exposed to EMC-1 film (Kodak) along with calibrated [14C]methylmethacrylate standards as previously described (22). Autoradiograms were analyzed with an AIS image processing system (Imaging Research, St. Catherine's, ON, Canada) with a pixel size of 28 μm. The concentration of 14C in each region of interest was determined from the optical density vs. 14C concentration curve determined from the calibrated plastic standards, and lCPSleu was calculated by means of Eq. 1. Brain regions were identified by reference to a mouse brain atlas (23).
Determination of Steady-State Ratios of Leucine SA in Tissue Pools to that of Plasma.
λi for whole brain (λWB) was evaluated in a series of steady-state experiments as described previously (17). A constant arterial plasma SA for [3H]leucine was maintained for 90, 120, or 150 min by means of a programmed i.v. infusion of [3H]leucine designed to achieve and maintain a constant arterial plasma [3H]leucine concentration. The total infusion contained 3–8 mCi of [3H]leucine in a volume of about 0.4 ml. The SAs of [3H]leucine in the arterial plasma and acid-soluble and tRNA-bound pools in brain were determined as described below. Timed arterial blood samples (≈20 μl) were collected every 15 or 30 min during the infusion and centrifuged immediately to separate plasma, which was then deproteinized by addition of one-third of a volume of 16% (wt/vol) sulfosalicylic acid containing l-norleucine (0.04 mM) as an internal standard for amino acid analyses. The deproteinized samples were stored at −70°C until assayed for leucine and [3H]leucine concentrations. At the end of the infusions the mice were decapitated, and their brains were quickly removed and chilled to 0°C in ice-cold 0.25 M sucrose.
Extraction and Purification of Aminoacyl-tRNA.
Each brain was weighed and homogenized in 5 ml of 0.25 M sucrose (0°C) containing 10 mM vanadyl ribonucleoside complex to inhibit ribonuclease, 6 mg of uncharged tRNA as carrier, and l-norleucine (0.02 mM) added as an internal standard and centrifuged at 100,000 × g for 1 h. The tRNA-bound amino acids were purified as previously described (17). Briefly, the cytosolic protein and RNA in the supernatant fraction were precipitated in trichloroacetic acid, and the precipitates were washed repeatedly with HClO4. Protein was extracted with water-saturated phenol, and residual phenol was removed by extraction of the aqueous phase with anhydrous diethylether. The aminoacyl-tRNA was precipitated overnight at −20°C in ethanol containing 0.12 M potassium acetate, pH 5.5. The pure tRNA fraction was dissolved in 50 mM sodium carbonate (pH 10) and incubated at 37°C for 90 min to deacylate the aminoacyl-tRNA. Deacylated tRNA was precipitated overnight at −20°C in ethanol and removed by centrifugation (12,000 × g, 20 min) (17). The supernatant solution, which contained the previously tRNA-bound but now free amino acids, was dried in a stream of N2 and redissolved in 40 μl of 0.2 M sodium citrate (pH 2.2).
Extraction of Acid-Soluble Fraction in Brain Tissue.
A 100-μl volume of the supernatant solution derived from the 100,000 × g centrifugation of the whole-brain homogenate was deproteinized with sulfosalicylic acid and stored at −70°C until assayed for leucine and [3H]leucine concentrations (22).
Assay of SA of [3H]Leucine.
Deproteinized plasma samples and tissue fractions of acid-soluble and deacylated aminoacyl-tRNA pools were assayed for leucine concentration by postcolumn derivatization with o-phthaldehyde and fluorometric assay with a Beckman Model 7300 amino acid analyzer (Beckman, Fullerton, CA), and column eluates were collected every minute and assayed for 3H with a Model 2250CA Packard Liquid Scintillation Counter (Packard), as described previously (17). SA of each sample was calculated from total 3H in all fractions in the leucine peak and the total measured leucine content in the peak. The leucine concentration and the SA of [3H]leucine in the acid-soluble pool in the tissue was corrected for contamination by the leucine in the blood contained in the tissue. Values of 0.67 for the equilibrium distribution of free leucine between red cells and plasma measured in rats (22) and 30% for the hematocrit in rat brain (24) were used in these corrections; both corrections were less than 5%.
Calculation of the Value of λWB.
Values of λWB were calculated as the ratios of the measured steady-state SA of leucine in the tissue tRNA-bound pool to that of the free leucine in arterial plasma. The time course of the SA in arterial plasma and the SA of tRNA-bound leucine in whole brain at the end of the experimental interval was determined as described above. Only animals with relatively constant plasma leucine SAs were included in the series.
Determination of Statistical Analysis.
Measurements were analyzed for statistically significant differences among the three groups of animals by Bonferroni t tests for multiple comparisons (25).
Results
Physiological Measurements.
Mice from all three groups were well matched with respect to the physiological variables measured except for brain weight, which was statistically significantly lower (13%) in the HMZ than in BTBR or HTZ mice (Table 1).
Table 1.
Physiological variables
| BTBR (24) | HTZ (22) | HMZ (19) | |
|---|---|---|---|
| Age, days | 88 ± 3 | 84 ± 4 | 85 ± 3 |
| Body weight, g | 33 ± 2 | 34 ± 2 | 35 ± 2 |
| Brain weight, g | 0.42 ± 0.01 | 0.43 ± 0.004 | 0.37 ± 0.01*† |
| Hematocrit, % | 44 ± 2 | 43 ± 2 | 44 ± 2 |
| Arterial plasma glucose concentration, mM | 7.3 ± 0.3 | 7.8 ± 0.3 | 7.4 ± 0.3 |
| Mean arterial blood pressure, mmHg | 93 ± 3 | 96 ± 3 | 92 ± 2 |
Values are means ± SEM for the number of mice indicated in parentheses except for brain weight determined in 14, 12, and 10 BTBR, HTZ, and HMZ mice, respectively, and blood pressure measured in 18 of the 19 HMZ mice.
Statistically significantly different from BTBR mice, Bonferroni t-tests, P < 0.001.
Statistically significantly different from HTZ mice, Bonferroni t tests, P < 0.001.
Neutral Amino Acid Concentrations in Plasma and Brain.
The most striking change in the HMZ mice was the 21-fold increase in the Phe concentration in arterial plasma over that of BTBRs (Table 2). Even in HTZ mice, plasma Phe concentrations were statistically significantly increased compared with BTBR mice by 38%. There were some small differences in concentrations of the other neutral amino acids measured. Tyrosine was not quantified because in the hyperphenylalaninemic mice, tyrosine and Phe could not be completely separated from each other.
Table 2.
Neutral amino acid concentrations in arterial plasma and brain pools
| Arterial plasma
acid-soluble pool, μM
|
Tissue total acid-soluble pool,
nmol/g
|
Tissue tRNA-bound pool,
pmol/g
|
|||||||
|---|---|---|---|---|---|---|---|---|---|
| BTBR (24) | HTZ (22) | HMZ (19) | BTBR (14) | HTZ (12) | HMZ (10) | BTBR (14) | HTZ (12) | HMZ (10) | |
| Valine | 213 ± 11 | 260 ± 16* | 253 ± 11 | 61 ± 2 | 58 ± 3 | 32 ± 3§‡ | 104 ± 10 | 108 ± 8 | 122 ± 11 |
| Methionine | 49 ± 3 | 60 ± 4 | 46 ± 4† | ND | ND | ND | 56 ± 4 | 55 ± 5 | 60 ± 9 |
| Isoleucine | 99 ± 4 | 115 ± 6 | 113 ± 4 | 31 ± 1 | 30 ± 1 | 21 ± 2§‡ | 98 ± 4 | 105 ± 4 | 113 ± 6 |
| Leucine | 176 ± 8 | 209 ± 13 | 184 ± 8 | 58 ± 1 | 57 ± 2 | 37 ± 3§‡ | 128 ± 4 | 138 ± 4 | 146 ± 7 |
| Tyrosine | 44 ± 3 | 52 ± 4 | ND | 37 ± 3 | 32 ± 2 | 12 ± 2§‡ | 38 ± 4 | 37 ± 5 | 46 ± 5 |
| Phenylalanine | 71 ± 3 | 98 ± 6‡ | 1513 ± 70‡§ | 57 ± 3 | 62 ± 4 | 472 ± 86¶∥ | 63 ± 4 | 63 ± 4 | 74 ± 6 |
Values are means ± SEM for number of mice indicated in parentheses. Plasma phenylalanine was determined in 17 HMZ mice, and tRNA-bound valine was quantified in 13, 11, and 9 BTBR, HTZ, and HMZ mice, respectively. ND, not determined. Statistically significantly different from BTBR, *P < 0.05; ¶, P < 0.01; ‡, P < 0.001. Statistically significantly different from HTZ, †, P < 0.05; ∥, P < 0.0;, §, P < 0.001.
Brain acid-soluble neutral amino acid concentrations (Table 2) were similar in BTBR and HTZ mice. In HMZ mice, brain Phe concentration was eight times higher and other neutral amino acids determined were statistically significantly decreased compared with the other groups. In the tRNA-bound pool (Table 2), concentrations of neutral amino acids measured were similar in all three groups.
Steady-State Ratios of Whole Brain tRNA-Bound to Plasma [3H]Leucine SAs (λWB).
Only studies with fairly constant arterial plasma leucine SA (i.e., all samples taken during the last 20 min of the study within ±16% of the mean of those samples) were included in the series (Fig. 1). To establish that a steady state was reached, experiments of 90-, 120-, and 150-min duration were carried out for each genotype (Fig. 2), and the ratios of tRNA-bound to plasma leucine SAs determined. The ratio for the tRNA-bound pool was found to be unchanged between 120 and 150 min for all three groups. Steady-state SA ratios for the tRNA-bound pool were calculated from the mean of the 120- and 150-min infusions (Table 3). For the calculation of lCPSleu (Eq. 1), the value of λWB used for the HMZ mice was 0.34; for BTBR and HTZ mice, the pooled value of λWB was used (0.55 ± 0.02, mean ± SEM, n = 16).
Figure 1.

Typical steady-state experiments in HMZ (A), HTZ (B), and BTBR (C) mice illustrating the achievement and maintenance of a constant SA of [3H]leucine over 150 min and the relatively constant arterial plasma leucine concentration over this interval. The figure also shows the SA of the tRNA-bound leucine in brain at the end of the experiment.
Figure 2.

Relationship between duration of [3H]leucine square wave and ratio of whole-brain leucine SA in the tRNA-bound pool [Cpp*(T)/Cpp] to that of the arterial plasma in the three genotypes. Symbols are the means (±SEM) for the number of animals indicated in parentheses.
Table 3.
Steady-state ratios of tissue tRNA-bound to plasma [3H]leucine-specific activities
Values are means ± SEM for the number of animals indicated in parentheses.
Statistically significantly different from BTBR mice, Bonferroni t tests, P < 0.001.
Statistically significantly different from HTZ mice, Bonferroni t tests, P < 0.001.
Regional Rates of Cerebral Protein Synthesis.
lCPSleu was determined in 23 regions of brain (Fig. 3, Table 4). There were no statistically significant differences in lCPSleu between BTBR and HTZ mice in any of the regions examined. In comparison with either the BTBR or HTZ mice, lCPSleu in the HMZ mice was statistically significantly decreased in all regions. Decreases ranged from 9 to 32%; the largest effects were found in the dentate gyrus, ventral tegmental area, and cingulum.
Figure 3.

Digitized autoradiograms of coronal sections at three different levels in brain from mice representative of the three genotypes studied. Images have been color coded for lCPSleu. Sections illustrated were taken at the levels of the prelimbic cortex (Top), dorsal hippocampus (Middle), and cerebellum (Bottom). The color bar (Right) provides the calibration scale for the range of values of lCPSleu in nmol⋅g−1⋅min−1 for each color. Dorsal side is Top; right side is on Right. (Bar = 5 mm.)
Table 4.
Regional rates of cerebral leucine incorporation into protein, nmol/g of tissue per min
| BTBR (10) | HTZ (10) | HMZ (9) | |
|---|---|---|---|
| Sensory and motor areas | |||
| Primary motor cortex | 4.5 ± 0.2 | 4.8 ± 0.2 | 3.8 ± 0.2* |
| Somatosensory barrel cortex | 4.9 ± 0.2 | 5.2 ± 0.2 | 4.1 ± 0.3* |
| Primary auditory cortex | 4.9 ± 0.2 | 5.3 ± 0.2 | 4.2 ± 0.1*† |
| Primary visual cortex, binocular field | 4.9 ± 0.2 | 5.2 ± 0.2 | 4.1 ± 0.1*‡ |
| Primary visual cortex, monocular field | 5.0 ± 0.2 | 5.2 ± 0.1 | 3.9 ± 0.1*‡ |
| Caudate putamen | 3.4 ± 0.1 | 3.7 ± 0.2 | 2.9 ± 0.2† |
| Cerebellum, simple lobule, granule cell layer | 6.6 ± 0.2 | 7.0 ± 0.2 | 5.3 ± 0.2*‡ |
| Regions of the limbic system | |||
| Prefrontal cortex | 5.1 ± 0.2 | 5.5 ± 0.2 | 4.4 ± 0.2* |
| Prelimbic cortex | 5.4 ± 0.2 | 5.6 ± 0.2 | 4.6 ± 0.2§ |
| Anterior cingulate cortex | 5.3 ± 0.2 | 5.7 ± 0.2 | 4.7 ± 0.3§ |
| Hippocampus, pyramidal cell layer | |||
| CA1 | 6.4 ± 0.2 | 6.6 ± 0.1 | 4.8 ± 0.4*† |
| CA2 | 10.9 ± 0.3 | 10.9 ± 0.2 | 8.4 ± 0.6†§ |
| CA3 | 8.8 ± 0.3 | 9.3 ± 0.4 | 6.9 ± 0.4*† |
| Hippocampus, dentate gyrus | 6.8 ± 0.2 | 7.2 ± 0.2 | 5.0 ± 0.3†‡ |
| Hippocampus, lacunosum moleculare | 2.3 ± 0.1 | 2.6 ± 0.1 | 2.1 ± 0.2* |
| Subiculum | 4.9 ± 0.1 | 5.1 ± 0.2 | 4.0 ± 0.1*‡ |
| Ventral tegmental area | 5.2 ± 0.2 | 5.3 ± 0.1 | 3.6 ± 0.1*‡ |
| Cingulum | 2.4 ± 0.1 | 2.7 ± 0.1 | 1.9 ± 0.2* |
| Associative areas and habenula | |||
| Frontal cortex | 4.1 ± 0.2 | 4.4 ± 0.1 | 3.4 ± 0.1*† |
| Posterior parietal cortex | 5.0 ± 0.2 | 5.2 ± 0.1 | 4.0 ± 0.2*‡ |
| Perirhinal area | 4.8 ± 0.2 | 5.0 ± 0.2 | 3.8 ± 0.2*‡ |
| Habenula, medial nucleus | 8.1 ± 0.3 | 8.0 ± 0.2 | 6.4 ± 0.4*‡ |
| Habenula, lateral nucleus | 5.8 ± 0.2 | 5.9 ± 0.1 | 4.7 ± 0.3*‡ |
Values are the means ± SEM for the number of animals indicated in parentheses. Statistically significantly different from BTBR mice, Bonferroni t tests, †, P < 0.05; ‡, P < 0.01. Statistically significantly different from HTZ mice, Bonferroni t tests, §, P < 0.05; *, P < 0.01.
Discussion
The present study is, to our knowledge, the first study of amino acid compartmentation and protein synthesis in this genetic mouse model of PKU. The results show that in the adult HMZ mouse, lCPSleu is reduced throughout the brain below the levels in the HTZ and BTBR mice. In the brains of HMZ mice, concentrations of Phe are elevated, and those of the other neutral amino acids measured (including leucine) are decreased. The fraction of leucine in the precursor pool that is derived from protein breakdown is about 50% higher in the HMZ mice than in either HTZ or BTBR mice.
All three groups of mice showed similar regional variation in lCPSleu despite the generally 20% lower rates in the HMZ mice. Some of the highest rates are found in the pyramidal cell layer of the hippocampus, particularly in the CA2 region. Regions rich in myelin or synapses such as the cingulum or the stratum lacunosum moleculare of the hippocampus have the lowest lCPSleu.
The l-[1-14C]leucine method is based on a compartmental model for the behavior of leucine in brain that includes bidirectional carrier-mediated transport of leucine at the blood–brain barrier, an extracellular and multiple intracellular compartments in brain, and the possibility that unlabeled leucine from the breakdown of tissue protein can be reincorporated into tissue protein. The application of the model together with an appropriate experimental design, i.e., a pulse injection of a tracer dose of l-[1-14C]leucine followed by a 60-min interval for clearance of the tracer from the blood and the brain, gives rise to Eq. 1 (26). We have calculated that the correction for the lag between the integrated SAs of leucine in the arterial plasma and the precursor pool in the tissue is negligible. The calculations were based on our measurements of a 3.5-min average half life of leucine in gray matter of the rat (26). We have not determined the half life of leucine in gray matter of the Pahenu2 mouse, but it is reasonable to assume that it is similar in all three genotypes because of the comparable amount of time required for the tRNA-bound leucine pool to reach a steady state with leucine in the arterial plasma (Fig. 3).
The constant λi, the fraction of leucine in the precursor pool for protein synthesis that is derived from the arterial plasma, is determined in a separate series of experiments. The remainder (1-λi) must come from recycling of leucine from protein breakdown. In the present studies, values of λWB for each genotype were used in Eq. 1 to calculate the rates of protein synthesis in all of the local brain regions examined; this assumes that in each genotype, the fraction of recycled leucine in the precursor pool is constant from region to region. This is a reasonable assumption inasmuch as previous studies in normal conscious adult rats have shown that the value of λi in most gray matter regions falls within ±5% of the value of 0.58, the average measured for the brain as a whole (27), and it remains so even in conditions in which lCPSleu has been shown to be changed, such as the regenerating hypoglossal nucleus in which lCPSleu is increased by 20–30% (28). The values of λWB for the three genotypes were determined under the same conditions as those in which lCPSleu was determined, i.e., in conscious freely moving mice fed a diet with the full complement of protein and amino acids. Measurement of λWB requires a steady state for both labeled and unlabeled leucine. That a steady state was achieved was confirmed by the constant value for the ratio of SAs (tissue/plasma) over a prolonged period.
The finding that λWB is decreased in HMZ mice indicates that in these hyperphenylalaninemic animals with a reduced concentration of free leucine in the brain, a greater fraction of the tRNA-bound leucine pool comes from amino acids recycled from protein breakdown. Even with this increased channeling of leucine from protein breakdown into the precursor pool, the general reduction in lCPSleu in the HMZ mice suggests that the supply of amino acids is inadequate to maintain normal lCPSleu. The normal concentrations of the tRNA-bound amino acid pools in the HMZ mice, however, are difficult to reconcile with a decreased rate of protein synthesis because of a limited supply of amino acids. Moreover, it is unlikely that the effects we observed on lCPSleu in the HMZ mice are because of the hyperphenylalaninemia per se. It has been reported that there is no effect of acute hyperphenylalaninemia in adult rats on cerebral protein synthesis (16). Similarly, acute hypervalinemia (plasma concentrations of valine ×100 normal) also had no effect on rates of cerebral protein synthesis (29) despite significant reductions in brain concentrations of other neutral amino acids (18). The results of these two studies suggest that the size of the precursor amino acid pool (i.e., tRNA-bound amino acids) can be maintained by the increased contribution of amino acids derived from protein breakdown. In the adult brain in which there is no net change in protein content, amino acids from protein breakdown may serve as an adequate buffer against limitations in delivery of amino acids from the blood.
There are several possible explanations for the different effects on lCPSleu in the genetic mouse model of PKU and those with acute hyperaminoacidemias. One is a possible difference in the effects of acute and chronic hyperaminoacidemia. Buffering by amino acids derived from protein degradation may be possible in the short term, but eventually deficiencies of important metabolic intermediates may have far-reaching consequences. Another possibility is that in the Pahenu2 mice, a primary effect of hyperphenylalaninemia on brain protein synthesis may have occurred during brain development with consequential structural abnormalities. Decreased lCPSleu in adult HMZ mice may reflect those structural abnormalities. If this is the case, it should not be possible to restore lCPSleu to normal by placing these adult mice on a low Phe diet.
The advent of the Pahenu2 mouse model of PKU affords us the opportunity to further our understanding of the etiology of mental retardation in this disease and thereby to formulate and test new treatments. Ultimately a more thorough understanding of the role of protein synthesis in the ability of the brain to grow and develop normally and to undergo plasticity may give us insight into these processes so essential for normal brain function.
Acknowledgments
We thank T. Burlin for carrying out the amino acid analyses, M. Cook and J. Pietrosewicz for preparing tissue for autoradiography, R. Wang for overseeing the breeding colony, K. Schmidt for calculating infusion schedules, D. Ye and R. Cohen for counsel on genotyping, Brooke Miller for help with image processing, and S. Kaufman, K. Schmidt, and L. Sokoloff for helpful suggestions preparing the manuscript.
Abbreviations
- SA
specific activity
- PKU
phenylketonuria
- lCPSleu
local rate of leucine incorporation into cerebral protein
- HTZ
heterozygous
- HMZ
homozygous
References
- 1.Scriver C R, Kaufman S, Woo S L C. In: The Metabolic Basis of Inherited Disease. Scriver C R, Beaudet A L, Sly W S, Valle D, editors. New York: McGraw–Hill; 1989. pp. 495–546. [Google Scholar]
- 2.Agrawal H C, Bone A H, Davison A N. Biochem J. 1970;117:325–331. doi: 10.1042/bj1170325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Comar D, Saudubray J M, Duthilleul A, Delforge J, Maziere M, Berger G, Charpentier C, Todd-Pokropek J M. Eur J Pediatr. 1981;136:13–19. doi: 10.1007/BF00441705. [DOI] [PubMed] [Google Scholar]
- 4.Shulkin B L, Betz A L, Koeppe R A, Agranoff B W. J Neurochem. 1995;64:1252–1257. doi: 10.1046/j.1471-4159.1995.64031252.x. [DOI] [PubMed] [Google Scholar]
- 5.Knudsen G M, Hasselbalch S, Toft P B, Christensen E, Paulson O B, Lou H. J Inherit Metab Dis. 1995;18:653–664. doi: 10.1007/BF02436753. [DOI] [PubMed] [Google Scholar]
- 6.Bauman M L, Kemper Th L. Acta Neuropathol. 1982;58:55–63. doi: 10.1007/BF00692698. [DOI] [PubMed] [Google Scholar]
- 7.Poser C M, Van Bogaert L. Brain. 1959;82:1–9. doi: 10.1093/brain/82.1.1-a. [DOI] [PubMed] [Google Scholar]
- 8.Shedlovsky A, McDonald J D, Symula D, Dove W F. Genetics. 1993;134:1205–1210. doi: 10.1093/genetics/134.4.1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aoki F, Siegel F L. Science. 1970;168:129–130. doi: 10.1126/science.168.3927.129. [DOI] [PubMed] [Google Scholar]
- 10.MacInnes J W, Schlesinger K. Brain Res. 1971;29:101–110. doi: 10.1016/0006-8993(71)90420-3. [DOI] [PubMed] [Google Scholar]
- 11.Hughes J V, Johnson T C. J Neurochem. 1976;26:1105–1113. doi: 10.1111/j.1471-4159.1976.tb06993.x. [DOI] [PubMed] [Google Scholar]
- 12.Binek P A, Johnson T C, Kelly C J. J Neurochem. 1981;36:1476–1484. doi: 10.1111/j.1471-4159.1981.tb00589.x. [DOI] [PubMed] [Google Scholar]
- 13.Binek-Singer P, Johnson T C. Biochem J. 1982;206:407–414. doi: 10.1042/bj2060407. , 1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roberts S, Morelos B S. J Neurochem. 1976;26:387–400. doi: 10.1111/j.1471-4159.1976.tb04492.x. [DOI] [PubMed] [Google Scholar]
- 15.Wall K M, Pardridge W M. Biochem Biophys Res Commun. 1990;168:1177–1183. doi: 10.1016/0006-291x(90)91153-j. [DOI] [PubMed] [Google Scholar]
- 16.Dunlop D S, Yang X-R, Lajtha A. Biochem J. 1994;302:601–610. doi: 10.1042/bj3020601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smith C B, Deibler G E, Eng N, Schmidt K, Sokoloff L. Proc Natl Acad Sci USA. 1988;85:9341–9345. doi: 10.1073/pnas.85.23.9341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smith C B, Sun Y, Deibler G E, Sokoloff L. J Neurochem. 1991;57:1540–1547. doi: 10.1111/j.1471-4159.1991.tb06349.x. [DOI] [PubMed] [Google Scholar]
- 19.McDonald J D, Charlton C K. Genomics. 1997;39:402–405. doi: 10.1006/geno.1996.4508. [DOI] [PubMed] [Google Scholar]
- 20.Zagreda L, Goodman J, Druin D P, McDonald D, Diamond A. J Neurosci. 1999;19:6175–6182. doi: 10.1523/JNEUROSCI.19-14-06175.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.David Moore. Current Protocols in Molecular Biology. 1994;1:2.1.1. doi: 10.1002/j.1934-3647.1994.tb00220.x. [DOI] [PubMed] [Google Scholar]
- 22.Sun Y, Deibler G E, Jehle J, Macedonia J, Dumont I, Dang T, Smith C Beebe. Am J Physiol. 1995;268:R549–R561. doi: 10.1152/ajpregu.1995.268.2.R549. [DOI] [PubMed] [Google Scholar]
- 23.Franklin K B J, Paxinos G. The Mouse Brain in Stereotaxic Coordinates. San Diego: Academic; 1996. [Google Scholar]
- 24.Cremer J E, Seville M P. J Cereb Blood Flow Metab. 1983;3:254–256. doi: 10.1038/jcbfm.1983.35. [DOI] [PubMed] [Google Scholar]
- 25.Miller R G. Simultaneous Statistical Inference. New York: McGraw–Hill; 1966. pp. 67–70. [Google Scholar]
- 26.Smith C Beebe. Neurochem Res. 1991;16:1037–1045. doi: 10.1007/BF00965848. [DOI] [PubMed] [Google Scholar]
- 27.Sun Y, Deibler G E, Sokoloff L, Smith C B. J Neurochem. 1992;59:863–873. doi: 10.1111/j.1471-4159.1992.tb08324.x. [DOI] [PubMed] [Google Scholar]
- 28.Sun Y, Deibler G E, Smith C B. J Cereb Blood Flow Metab. 1993;13:1006–1012. doi: 10.1038/jcbfm.1993.126. [DOI] [PubMed] [Google Scholar]
- 29.Smith C Beebe, Sun Y. Am J Physiol. 1995;268:E735–E744. doi: 10.1152/ajpendo.1995.268.4.E735. [DOI] [PubMed] [Google Scholar]