

Abstract
Previous studies in pulmonary arterial smooth muscle cells (PASMCs) showed that the TRPC1 channel mediates capacitative Ca2+ entry (CCE), but the molecular signal(s) that activate TRPC1 in PASMCs remains unknown. The aim of the present study was to determine if TRPC1 mediates CCE through activation of STIM1 protein in mouse PASMCs. In primary cultured mouse PASMCs loaded with fura-2, cyclopiazonic acid (CPA) caused a transient followed by a sustained rise in intracellular Ca2+ concentration ([Ca2+]i). The transient but not the sustained rise in [Ca2+]i was partially inhibited by nifedipine. In addition, CPA increased the rate of Mn2+ quench of fura-2 fluorescence that was inhibited by SKF 96365, Ni2+, La3+ and Gd3+, exhibiting pharmacological properties characteristic of CCE. The nifedipine-insensitive sustained rise in [Ca2+]i and the increase in Mn2+ quench of fura-2 fluorescence caused by CPA were both inhibited in cells pretreated with antibody raised against an extracellular epitope of TRPC1. Moreover, STIM1 siRNA reduced the rise in [Ca2+]i and Mn2+ quench of fura-2 fluorescence caused by CPA, whereas overexpression of STIM1 resulted in a marked increase in these responses. RT-PCR revealed TRPC1 and STIM1 mRNAs, and Western blot analysis identified TRPC1 and STIM1 proteins in mouse PASMCs. Furthermore, TRPC1 was found to co-immunoprecipitate with STIM1, and the precipitation level of TRPC1 was increased in cells subjected to store depletion. Taken together, store depletion causes activation of voltage-operated Ca2+ entry and CCE. These data provide direct evidence that CCE is mediated by TRPC1 channel through activation of STIM1 in mouse PASMCs.
Intracellular calcium plays an important role in regulating vascular smooth muscle tone. An increase in intracellular Ca2+ concentration ([Ca2+]i) activates contractile proteins and results in contraction. [Ca2+]i can be increased through the release of Ca2+ from the sarcoplasmic reticulum (SR) and Ca2+ entry from extracellular space through voltage-operated Ca2+ channels (VOCCs), receptor-operated channels (ROCs) or store-operated channels (SOCs) (Barritt, 1999; Parekh & Putney, 2005). Recently, Ca2+ entry through SOCs (so-called capacitative Ca2+ entry, CCE) has gained considerable attention in vascular smooth muscle research (Ng & Gurney, 2001; Trepakova et al. 2001; Albert & Large, 2002; Flemming et al. 2002; Wilson et al. 2002; Weirich et al. 2005; McElroy et al. 2008; Ng et al. 2008). CCE is activated in response to Ca2+ release induced by agonists activating receptors coupled to the inositol 1,4,5-trisphosphate (IP3) signalling pathway, or by agents that inhibit the SR Ca2+-ATPase (SERCA), such as cyclopiazonic acid (CPA) or thapsigargin (Albert & Large 2003; Parekh & Putney, 2005; Leung et al. 2007). However, the molecular composition of SOCs and the signal(s) that activate these channels in vascular smooth muscle remain unclear.
Over the past decade, there is increasing evidence that members of canonical subgroup of transient receptor potential non-selective cation channel (TRPC) constitute tetramers of both ROCs and SOCs (Parekh & Putney, 2005; Pedersen et al. 2005; Albert et al. 2007). In general, TRPC1, 4 and 5 are sensitive to store depletion and function as SOCs, whereas TRPC3, 6 and 7 function as ROCs that are gated by G-protein-phospholipase C and diacylglycerol (Pedersen et al. 2005). Recently, several studies have confirmed the existence of TRPC channels in various vascular preparations (Leung et al. 2007; Albert et al. 2007), including pulmonary artery smooth muscle cells (PASMCs) (Ng & Gurney, 2001; Walker et al. 2001; Wang et al. 2003; Lu et al. 2008; McElroy et al. 2008). Using inhibitory antibodies, antisense and siRNA methods, several studies have presented evidence for TRPC1 being an essential component for SOCs in vascular smooth muscle cells, including aortic smooth muscle cells (Xu & Beech, 2001; Brueggemann et al. 2006), cerebral artery cells (Bergdahl et al. 2005), mesenteric artery cells (Saleh et al. 2006, 2008), portal vein cells (Saleh et al. 2008); coronary artery cells (Takahashi et al. 2007a; Saleh et al. 2008) and PASMCs (Sweeney et al. 2002). Interestingly, TRPC1 and TRPC5 have been shown to colocalize and associate with one another in rabbit pial arteriole (Xu et al. 2006), suggesting that TRPC1/TRPC5 may form heterotetramers in vascular smooth muscle. Thus, it is possible that TRPC1 may be an important candidate to form SOCs in PASMCs, either as a homotetramer or a heterotetramer with other TRPC channels.
A recent advance in the understanding of the potential molecular composition of SOCs has been the discovery of a transmembrane protein STIM1 (stromal-interacting molecule 1), which has been shown to mediate a well characterized store-operated current, the so-called calcium release activated calcium current (Icrac) in non-excitable cells (Smyth et al. 2006; Lewis, 2007). STIM1 was found to act as a sensor within the stores (Roos et al. 2005; Zhang et al. 2005) and also may play a role in the plasma membrane (Zhang et al. 2005; Spassova et al. 2006) to activate Icrac. To date, there is very little information on the role of STIM1 in smooth muscle cells. STIM1 mRNA was shown to be expressed in human airway smooth muscle cells (Peel et al. 2006), cultured human coronary artery smooth muscle cells (Takahashi et al. 2007b), mouse aorta smooth muscle cells (Dietrich et al. 2007) and human saphenous vein cells (Li et al. 2008), and siRNA targeting STIM1 resulted in reduction of Ca2+ entry and whole cell current activated by CPA or thapsigargin (Peel et al. 2006; Takahashi et al. 2007b; Li et al. 2008). More recently, STIM1 mRNA and protein were found to express in rat PASMCs (Lu et al. 2008). However, the role of STIM1 in the activation of CCE in PASMCs remains unknown.
Recently, over-expression of STIM1 increased TRPC1 activity and both proteins were shown to associate with one another (Huang et al. 2006; López et al. 2006; Takahashi et al. 2007a). Interestingly, TRPC1 has been shown to form a complex with STIM1 to activate SOCs in human salivary gland cells (Ong et al. 2007; Cheng et al. 2008) and saphenous vein cells (Li et al. 2008). However, there is no evidence for the functional interaction between STIM1 and TRPC1 in the activation of SOCs in PASMCs. The aims of the present study were to investigate if store depletion activates CCE in mouse PASMCs and to determine whether CCE is mediated by TRPC1 through activation of STIM1 in these cells.
Methods
PASMCs isolation and cell culture
Male C57BL/6 mice were killed with pentobarbital sodium (50 mg kg−1i.p.) followed by cervical dislocation, as approved by the university of Nevada Reno Institutional Care and Use Committee. The heart and lungs were removed and second and third branches of the intrapulmonary artery were dissected in a low-Ca2+ physiological salt solution (PSS) composed of the following (mm): 125 NaCl, 5.36 KCl, 0.34 Na2HPO4, 0.44 K2HPO4, 1.2 MgCl2, 11 Hepes, 10 glucose and 0.05 CaCl2 (pH 7.4 adjusted with Tris). To disperse cells, pulmonary arterial tissue was incubated with the low-Ca2+ PSS containing (in mg ml−1): 1 collagenase type XI, 2 trypsin inhibitor, 0.45 protease, 1.3 taurine, 2 bovine serum albumin (fat free) for 30 min at 5°C followed by 8 min at 33°C. The tissue was then transferred to an enzyme-free, low-Ca2+ PSS and triturated with a fire-polished Pasteur pipette. The resulting dispersed PASMCs were subjected to cell culture as previously described (Dai et al. 2005; Ng et al. 2008). Freshly dispersed PASMCs were plated onto a 60 mm cell cultured dish and incubated with Dulbecco's modified Eagle medium (DMEM) containing 10% newborn calf serum (NCS), penicillin (100 units ml−1) and streptomycin (100 μg ml−1). Cells were incubated in a humidified atmosphere of 5% CO2 in air at 37°C and grown to 90–95% confluence. These primary cultured cells were then trypsinized and passaged onto a coverslip and grown to 70–80% confluence. Confluent cells were then growth arrested in 0.1% NCS medium for 24 h prior to experimental use.
Measurement of intracellular Ca2+
The cytosolic Ca2+ concentration was estimated in PASMCs loaded with fura-2 acetoxymethyl ester (fura-2 AM) (Molecular Probes, Eugene, OR, USA) using a dual excitation digital Ca2+ imaging system (IonOptix Inc., Milton, MA, USA) equipped with an intensified CCD camera as previously described (Wilson et al. 2002; Ng et al. 2008). PASMCs were loaded with 10 μm fura-2 AM for 1 h in the dark at room temperature and placed on the coverslip in a 0.2 ml perfusion chamber mounted on an inverted epifluorescence microscope (Nikon) outfitted with a 40× oil immersion objective (NA 1.3, Nikon). Cells were washed several times at 1 ml min−1 to remove extracellular fura-2 AM with 2 mm Ca2+-PSS composed of the following (mm): 126 NaCl, 5 KCl, 0.3 NaH2PO4, 10 Hepes, 1 MgCl2, 2 CaCl2, 10 glucose (pH 7.4 with NaOH). Cells were illuminated with xenon arc lamp at 340 ± 15 and 380 ± 12 nm (Omega Optical, Brattleboro, VT, USA) and emitted light was collected from regions that encompassed single cells with a CCD camera at 510 nm (Nikon). Images were acquired at 1 Hz and stored on the compact disk for later analysis. Background fluorescence was collected automatically and subtracted from the acquired fluorescence video images during each experiment. The ratio of fluorescence (R) excited at the two excitation wavelengths was used to estimate intracellular Ca2+ concentration ([Ca2+]i) as described by Grynkiewicz et al. 1985:
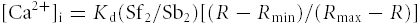
The values for Sf2 (fluorescence measured at 380 nm in Ca2+-free solution), Sb2 (fluorescence measured at 380 nm in Ca2+-saturating conditions), Rmin (minimum ratio) and Rmax (maximum ratio) were determined from in situ calibrations of fura-2 for each cell. The dissociation constant for Ca2+ binding, Kd, was assumed to be 224 nm (Grynkiewicz et al. 1985). To determine Rmin, cells were dialysed with 4 μm ionomycin in Ca2+-free PSS containing 10 mm EGTA at the end of each experiment. Rmax was determined from cells dialysed with 4 μm ionomycin in PSS containing 10 mm CaCl2. ΔR is the change in fluorescence ratio by subtracting the fluorescence ratio from the basal fluorescence ratio. Δ[Ca2+]i is the change in [Ca2+]i by subtracting the estimated [Ca2+]i from the basal [Ca2+]i.
In experiments where the effects of store depletion were investigated, CPA was used to deplete the SR Ca2+ stores in Ca2+-free PSS followed by re-exposure of cells with 2 mm Ca2+-PSS as previously described (Wilson et al. 2002; Ng et al. 2008). An elevation in [Ca2+]i above basal levels during 2 mm Ca2+ re-addition was used as a marker of CCE mediated extracellular Ca2+ entry. In experiments where the Ca2+ influx through SOCs were studied, the rate of Mn2+-induced quenching of fura-2 fluorescence was recorded during excitation at 360 nm in nominally Ca2+-free PSS containing nifedipine (Ng et al. 2008). In experiments where the effects of LaCl3 and GdCl3 were studied, an EGTA- and phosphate-free Hepes-PSS was used to avoid precipitation and chelation of La3+ and Gd3+ (Wang et al. 2003; Snetkov et al. 2003; Ng et al. 2008). In experiments where the effect of TRPC1 antibody was studied, cultured PASMCs were pre-incubated with TRPC1 (1 : 100, Alomone Laboratories, Jerusalem, Israel) at 37°C for 24 h before the experiments started. For negative control, TRPC1 antibody was pre-adsorbed with TRPC1 antigen peptide (1 μg peptide per 1 μg antibody) at room temperature for 2 h and then incubated with PASMCs at 37°C for 24 h before experimental recording.
Total RNA isolation and RT-PCR
Total RNA was isolated from cultured mouse PASMCs using TRIZOL reagent (Invitrogen, CA, USA) as per the manufacturer's instructions. First strand cDNA was prepared from the RNA preparations by using Superscript III reverse transcriptase (Invitrogen). The resulting cDNA was then amplified by PCR with primers specific for mouse TRPC1 (sense, 5′-CCTTCTCATACTGTGGATTATTG-3′; antisense, 5′-GTACCAGAACAGAGCAAAGCA-3′) and STIM1 (sense, 5′-CAATGGTGATGTGGATGTGGAAGA-3′; antisense, 5′-AGTAACGGTTCTGGATATAGGCAAACC-3′). Primers for mouse β-actin (sense, 5′-TGGCTACAGCTTCACCACC-3′; antisense, 5′-ACTCCTGCTTGCTGATCCAC-3′) were used as an internal control. The amplification cycle parameters were 95°C for 10 min, 35 cycles at 95°C for 30 s (denaturation), 58°C for 30 s (annealing), and 72°C for 45 s (extension). Sample was then heated at 72°C for 7 min to ensure complete product extension. For reverse transcription (RT) controls, reverse transcriptase was omitted from cDNA reaction. Amplified products were resolved by gel electrophoresis, purified and verified by sequencing.
Transfection of PASMCs with STIM1 siRNA
PASMCs were transiently transfected with STIM1 siRNA (ID: s74488, Silencer Select Pre-designed siRNA, Ambion, Austin, TX, USA) using siPORT Amine transfection reagent (Ambion) according to the manufacturer's instruction. For every 35 mm cell culture dish of cells, 10 μl of STIM1 siRNA (50 μm) was diluted in 90 μl of OPTIMEM I (Invitrogen). Then, 10 μl of siPORT Amine was diluted in 90 μl of OPTIMEM I and mixed with the diluted siRNA. The mixture (200 μl) was incubated at room temperature for 20 min to allow formation of transfection complexes. Primary cultured PASMCs were then trypsinized and incubated in DMEM containing 10% NCS and antibiotics, and the cells were subsequently passaged onto three 35 mm cell culture dishes. To each culture dish, the transfection complexes were added onto the cells to give a final volume of 2.5 ml in growth medium and a final concentration of 200 nm STIM1 siRNA. The plate was swirled gently to ensure uniform distribution of the transfection complexes. The cells were incubated with the transfection complexes at 37°C for 24 h and grown to 70–80% confluence. The cells were then incubated in the growth arrested medium containing 0.1% NCS at 37°C for 24 h prior to experimental use. For negative control, the cells were transfected with a scrambled siRNA (Silencer Negative Control no. 1 siRNA, Ambion) using the same transfection method.
Generation of recombinant STIM1 adenovirus
STIM1 cDNA was isolated from mouse brain and cloned into pcDNA3.1 according to the manufacturer's instructions (Invitrogen) and the STIM1 construct was confirmed using terminator cycle sequencing. Recombinant adenoviruses for STIM1 were then produced in a pAdTrack-CMV/pAdEasy recombinant containing green fluorescent protein (Ad-GFP-STIM1), purified and amplified by using the AdEasy adenoviral vector system (Stratagene, La Jolla, CA, USA). To produce adenoviruses, the STIM1 adenovirus recombinants were transfected into viral packaging cell line using the MBS mammalian transfection kit (Stratagene). Adenoviruses were then harvested, plaque-purified and titred by an agarose overlay plaque assay as previously described (Graham & Prevec, 1995). The same procedure was used to generate a control adenovirus containing GFP (Ad-GFP) with no insertion of STIM1 gene. For infection, cultured PASMCs were incubated with adenovirus in DMEM containing 0.1% NCS for 24 h. The cells were then washed with fresh 0.1% NCS medium for another 24 h. Infected cells were monitored by observing the number of green cells under fluorescence miscroscope and were subsequently used for calcium imaging study or Western blot analysis.
Western blot analysis and co-immunoprecipitation
Total protein was obtained from cultured mouse PASMCs by using RIPA extraction buffer containing protease and phosphatase inhibitors as previously described (Dai et al. 2005). For western blot analysis, equal amounts of total protein (50 μg) were resolved by 10% sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis and transferred onto nitrocellulose membranes for 90 min at 24 V (Genie blotter, Idea Scientific Company, Minneapolis, MN, USA). The membranes were then blocked for 1 h with LI-COR blocking solution (LI-COR, Lincoln, NE, USA) and probed with a rabbit polyclonal TRPC1 antibody (1 : 100, Alomone Laboratories) or mouse monoclonal STIM1 antibody (1 : 100, BD Biosciences Pharmingen, San Diego, CA, USA). The membranes were simultaneously probed with goat polyclonal GAPDH antibody (1 : 20 000, Santa Cruz Biotechnology, Santa Cruz, CA, USA) as an internal control. The primary antibodies were incubated overnight at 4°C and after washout membranes were incubated with two secondary antibodies in LI-COR solution for 45 min at room temperature: one coupled to an infrared fluorescence marker with emission wavelength of 800 nm (1 : 100 000; IR 800, Rockland Immunochemicals, Gilbertsville, PA, USA), and the other coupled to an infrared fluorescence marker with emission wavelength of 680 nm (1 : 100 000; Alexa Fluor 680, Molecular Probes). Immunoblots were then scanned to obtain double-colour fluorescent images with an Odyssey scanner (LI-COR).
For co-immunoprecipitation of STIM1 and TRPC1, 0.5 mg of total protein was first diluted with an equal volume of PSS (with protease inhibitors) and mixed with 10 μg of Stim1 antibody (EXBIO, Czech Republic), and incubated with agitation at 4°C for 2 h. Then, 100 μl of slurry of agarose beads conjugated to goat anti-mouse antibodies (Sigma) was washed with 1 ml PSS and incubated overnight with the protein–antibody complex at 4°C on an end-over-end mixer. The beads–protein–antibody complex was then washed three times with 1 ml of PSS. The protein was released from the beads by adding 35 μl of 4× SDS loading buffer and incubated for 20 min at room temperature prior to loading on a 10% SDS gel. After gel electrophoresis, the separated protein was transferred onto nitrocellulose membrane. To demonstrate immunoprecipitation of STIM1, the blot was probed with Stim1 antibody (1 : 100; BD Biosciences). To demonstrate co-immunopreciptation of STIM1 and TRPC1, the blot was subsequently probed with TRPC1 antibody (1 : 100, Alomone).
Drug solutions and data analysis
CPA, nifedipine, MnCl2, NiCl2, LaCl3 and GdCl3 were obtained from Sigma-Aldrich Co. (St Louis, MO, USA). SKF 96365 and ionomycin were obtained from Calbiochem (San Diego, CA, USA). TRPC1 antibody was obtained from Alomone Laboratories. STIM1 antibodies were obtained from BD Biosciences and EXBIO. STIM1 siRNA and negative control siRNA were obtained from Ambion. CPA, nifedipine and ionomycin were dissolved in dimethylsulphoxide. Other drugs were dissolved in deionized water. Data are expressed as means ±s.e.m. of n cells from at least five cell culture dishes passaged from three primary cultured dishes of separate seedings. Statistical comparisons employed Student's unpaired t tests or one-way analysis of variance (ANOVA) with Tukey's pairwise comparison as appropriate. A value of P < 0.05 was considered significant.
Results
Store depletion causes activation of VOCC and CCE in mouse PASMCs
To study Ca2+ entry pathway(s) activated by store depletion in mouse PASMCs, control experiments were first performed in Ca2+-free solutions followed by re-addition of 2 mm Ca2+ (Fig. 1A). Removal of extracellular Ca2+ caused a decrease in [Ca2+]i. Subsequent addition of 2 mm Ca2+ elicited a very small transient rise in [Ca2+]i, 42 ± 14 nm (ΔR= 0.11 ± 0.03) above basal levels (Fig. 1A and C; n= 22, P < 0.01), which decayed slowly to the baseline. Figure 1B shows that when cells were superfused with Ca2+-free PSS containing 10 μm CPA, an early transient increase in [Ca2+]i was observed indicative of Ca2+ release from the intracellular stores (arrow). This early transient rise in [Ca2+]i decayed slowly to a mean level below baseline. Subsequent addition of 2 mm Ca2+ in the presence of CPA elicited a significant transient rise in [Ca2+]i of 209 ± 31 nm (ΔR= 0.49 ± 0.03) followed by a sustained rise in [Ca2+]i of 67 ± 6 nm (ΔR= 0.21 ± 0.02) above basal levels (Fig. 1B and C; n= 117, P < 0.01). Part of the transient increase in [Ca2+]i was mediated by Ca2+ influx through VOCCs because nifedipine significantly reduced the increase in [Ca2+]i to 137 ± 10 nm (Fig. 1B and D; ΔR= 0.35 ± 0.03, n= 296, P < 0.05), at a concentration (10 μm) causing maximal inhibition of these channels in PASMCs (Ng et al. 2008). However, nifedipine did not affect the sustained increase in [Ca2+]i (Fig. 1B and D).
Figure 1. Store depletion increases [Ca2+]i in mouse PASMCs.

Store depletion increases [Ca2+]i in cultured mouse PASMCs. A, in control experiments, removal of external Ca2+ caused a decrease in fluorescence ratio below basal level. Subsequent addition of 2 mm Ca2+ caused a very small transient increase in fluorescence ratio, which slowly returned to basal levels (dotted line). B, when applied in Ca2+-free solution, depletion of intracellular stores with 10 μm CPA transiently elevated fura-2 fluorescence ratio, indicating Ca2+ release from the intracellular stores (arrow). Re-addition of 2 mm Ca2+ in the continued presence of CPA caused a transient followed by a sustained increase in fluorescence ratio. The transient but not the sustained component was reduced by 10 μm nifedipine (Nif). C, mean changes in [Ca2+]i compared to the resting [Ca2+]i in control (n= 22) and store depletion (n= 117) experiments. Filled bars indicate mean decrease in [Ca2+]i. Shaded and open bars indicate mean transient and sustained rise in [Ca2+]i, respectively. **P < 0.01 and ++P < 0.01, compared to the resting [Ca2+]i (ANOVA). D, bar graph showing the mean changes in transient and sustained rise in [Ca2+]i caused by store depletion after re-addition of 2 mm Ca2+, in the absence (n= 117) and presence (n= 296) of 10 μm nifedipine. *P < 0.05 (unpaired t test).
We previously found in canine PASMCs that store depletion caused a dihydropyridine-insensitive increase in [Ca2+]i through activation of CCE (Wilson et al. 2002; Ng et al. 2008). To determine if the dihydropyridine-insensitive components activated by CPA in mouse PASMCs recruit a Ca2+ influx pathway similar to CCE, the effect of store depletion on Mn2+ quench of fura-2 fluorescence was tested in the presence of 10 μm nifedipine. In control experiments, the effect of store depletion on Mn2+ quench of fura-2 was examined as described previously (Ng et al. 2008). Figure 2A shows the fluorescence intensity recorded at an excitation wavelength of 360 nm in a single PASMC. Removal of extracellular Ca2+ did not cause any decline in fluorescence intensity. The addition of 30 μm MnCl2 in the presence of nifedipine caused the fluorescence to decline slightly. Subsequent depletion of the SR Ca2+ stores by 10 μm CPA resulted in a 120 ± 7% (Fig. 2F, n= 212) increase in the rate of decline of fluorescence, corresponding to enhanced Mn2+ quench of fura-2 indicative of store depletion activated Ca2+ entry (Ng et al. 2008). The pharmacology of the store depletion activated Ca2+ entry was further studied by testing the effects of known blockers of store-operated channels, SKF 96365, Ni2+, La3+ and Gd3+ (Parekh & Putney, 2005) in the presence of nifedipine. Figure 2B–F shows that 50 μm SKF96365 (Fig. 2B), 500 μm Ni2+ (Fig. 2C), 100 μm La3+ (Fig. 2D) and Gd3+ (Fig. 2E) inhibited CPA-activated Mn2+ quench of fura-2 fluorescence, from 120 ± 7% (n= 212) to 4 ± 10% (n= 92), 20 ± 19% (n= 51), −15 ± 6% (n= 67) and −38 ± 6% (n= 47), respectively (Fig. 2F, P < 0.01).
Figure 2. Store depletion increases the rate of Mn2+ quench of fura-2 fluorescence in mouse PASMCs.

A, depletion of intracellular Ca2+ stores with 10 μm CPA increased the rate of Mn2+ quench of fura-2 fluorescence in the presence of 10 μm nifedipine. B–E, the increase in Mn2+-quench of fura-2 activated by store depletion was inhibited by 50 μm SKF 96365 (B), 500 μm Ni2+ (C), 100 μm La3+ (D) and 100 μm Gd3+ (E). F, bar graph showing percentage change in fura-2 quench rate after store depletion, in the absence (n= 212) and presence of SKF96365 (n= 92), Ni2+ (n= 51), La3+ (n= 67) and Gd3+ (n= 47). **P < 0.01 (ANOVA).
TRPC1 and STIM1 expression in mouse PASMCs
To determine if TRPC1 channel and STIM1 protein are expressed in mouse PASMCs, mRNA and protein expression were detected using RT-PCR and Western blot analysis, respectively. Figure 3A (upper panel) shows that TRPC1 mRNA is expressed in cultured mouse PASMCs with a predicted size of 516 bp and was confirmed to have the correct nucleotide sequence. TRPC1 protein was also found in cultured mouse PASMCs (lower panel). It is noteworthy that cross-reactivity of TRPC1 antibody with other TRPC channels is negligible because there is only one significant band detected by the TRPC1 antibody in the molecular weight range of 80–200 kDa. Thus, it is unlikely that the TRPC1 antibody used in our present study binds to other TRPC proteins in mouse PASMCs because the predicted molecular weights of other TRPC proteins in the TRPC family also fall in this molecular weight range. As shown in Fig. 3B, STIM1 mRNA (473 bp, upper panel) and protein (lower panel) were also detected in cultured mouse PASMCs.
Figure 3. TRPC1 and STIM1 expression in mouse PASMCs.

A, upper panel, RT-PCR products from cultured mouse PASMCs amplified using primers for mouse TRPC1 (516 bp) and β-actin (498 bp). Three separate RT-PCR reactions were performed in the presence (+) and absence (–) of reverse transcriptase (RT). Lower panel, TRPC1 protein and GAPDH were detected in cultured mouse PASMCs using Western blot analysis. A negative control was performed by pre-incubating TRPC1 antibody with the antigen peptide. Experiments were performed in 5 separate Western blot analyses. B, upper panel, RT-PCR products from cultured mouse PASMCs amplified using primers for mouse STIM1 (473 bp) and β-actin (498 bp). Data shown for β-actin and STIM1 expressions are from two separate part of a same gel. Three separate RT-PCR reactions were performed in the presence (+) and absence (–) of reverse transcriptase (RT). Lower panel, STIM1 protein and GAPDH were detected in cultured mouse PASMCs using Western blot analysis. Experiments were performed in 6 separate Western blot analyses.
TRPC1 and STIM1 mediate CCE in mouse PASMCs
To determine if TRPC1 channels are responsible for CCE in mouse PASMCs, the effects of TRPC1 antibody were investigated in cells subjected to store depletion in the presence of 10 μm nifedipine. The anti-TRPC1 antibody from Alomone is raised against the extracellular amino acid sequence 557–571, which is predicted to lie in the pore-forming region of the protein (Sage et al. 2002; Ahmmed et al. 2004). This antibody is widely used to study CCE in many various cell types, including pulmonary artery cells (Kunichika et al. 2004), endothelial cells (Ahmmed et al. 2004; Jho et al. 2005) and glomerular mesangial cells (Du et al. 2007). In control experiments, TRPC1 antibody was pre-adsorbed with TRPC1 antigen peptide and incubated with the cells for 24 h before recording. Figure 4A shows that the nifedipine-insensitive transient increase in [Ca2+]i caused by 10 μm CPA in control cells was not different from that in TRPC1 antibody-treated cells (Fig. 4B). However, the nifedipine-insensitive sustained increase in [Ca2+]i was abolished in TRPC1 antibody-treated cells as compared to the control cells, from 55 ± 5 nm (ΔR= 0.18 ± 0.01, n= 156) above baseline to 6 ± 21 nm (ΔR= 0.09 ± 0.06, n= 139) below baseline (Fig. 4A and B; P < 0.01). To further confirm that TRPC1 mediates CCE in mouse PASMCs, we compared the effects of 10 μm CPA on Mn2+ quench of fura-2 fluorescence in control cells to cells treated with TRPC1 antibody. Figure 4C shows that CPA caused a marked 112 ± 5% (n= 117) increase in Mn2+ quench of fura-2 in the presence of 10 μm nifedipine in control cells. This increase in Mn2+ quench rate was significantly reduced to 36 ± 16% (Fig. 4C and D; n= 48, P < 0.01) in cells treated with TRPC1 antibody.
Figure 4. TRPC1 mediates CCE in mouse PASMCs.

A, TRPC1 antibody (1 : 100) inhibited the CPA-induced sustained but not transient increase in fura-2 fluorescence ratio in the presence of 10 μm nifedipine. B, bar graph showing mean changes in transient and sustained increase in [Ca2+]i caused by 10 μm CPA after re-addition of 2 mm Ca2+ in the presence of 10 μm nifedipine, in control cells (filled bars, TRPC1 Ab+peptide, n= 156) and in cells treated with TRPC1 antibody (open bars, n= 139). **P < 0.01 (unpaired t test). C, TRPC1 antibody (1 : 100) inhibited the increase in Mn2+ quench of fura-2 fluorescence caused by 10 μm CPA in the presence of 10 μm nifedipine. D, bar graph showing percentage change in fura-2 quench rate after store depletion in the presence of 10 μm nifedipine, in control cells (filled bar, TRPC1 Ab+peptide, n= 117) and in cells treated with TRPC1 antibody (open bar, n= 48). **P < 0.01 (unpaired t test).
To determine if STIM1 protein mediates CCE in mouse PASMCs during store depletion, we first verified if siRNA knockdown of STIM1 mRNA reduced the expression level of STIM1 protein in cultured mouse PASMCs. Figure 5A shows that endogenous STIM1 protein was detected at similar levels in non-transfected cells and in cells transfected with 200 nm scrambled siRNA (negative control). The protein level was significantly reduced in cells transfected with 200 nm STIM1 siRNA compared to non-transfected cells and cells transfected with scrambled siRNA. We then examined the effect of STIM1 siRNA on CPA-induced rise in [Ca2+]i in the presence of 10 μm nifedipine. Figure 5B shows that 10 μm CPA caused an increase in nifedipine-insensitive transient and sustained rise in [Ca2+]i in cells transfected with 200 nm scrambled siRNA (negative control). Both transient and sustained increase in [Ca2+]i were significantly reduced in STIM1 siRNA-transfected cells from 139 ± 17 nm (ΔR= 0.27 ± 0.02, n= 51) to 74 ± 8 nm (ΔR= 0.19 ± 0.03, n= 84, P < 0.01) and 33 ± 3 nm (ΔR= 0.08 ± 0.01, n= 51) to 14 ± 4 nm (ΔR= 0.04 ± 0.01, n= 84, P < 0.01), respectively (Fig. 5B and C). To confirm that endogenous STIM1 mediates CCE in mouse PASMCs, we compared the effects of CPA on Mn2+ quench of fura-2 fluorescence between control cells transfected with scrambled siRNA and cells transfected with STIM1 siRNA. Figure 5D shows that 10 μm CPA caused a 109 ± 8% (n= 119) increase in Mn2+ quench of fura-2 in the presence of 10 μm nifedipine in negative control cells. This increase in Mn2+ quench rate was significantly reduced to 46 ± 8% (Fig. 5D and E, n= 107, P < 0.01) in cells transfected with 200 nm STIM1 siRNA.
Figure 5. siRNA knockdown of STIM1 reduces CCE in mouse PASMCs.

A, STIM1 protein and GAPDH were detected in non-transfected mouse PASMCs and in PASMCs transfected with 200 nm scrambled siRNA (negative control). The expression of STIM1 but not GAPDH reduced significantly in cells transfected with 200 nm STIM1 siRNA. Experiments were performed in 3 separate Western blot analyses. B, siRNA knockdown of STIM1 reduced the CPA-induced transient and sustained increase in fura-2 fluorescence ratio in the presence of 10 μm nifedipine. C, bar graph showing mean changes in transient and sustained increase in [Ca2+]i caused by 10 μm CPA after re-addition of 2 mm Ca2+ in the presence of 10 μm nifedipine, in negative control cells (filled bars, n= 51) and in STIM1 siRNA-transfected cells (open bars, n= 84). **P < 0.01 (unpaired t test). D, siRNA knockdown of STIM1 reduced the increase in Mn2+ quench of fura-2 fluorescence caused by 10 μm CPA in the presence of 10 μm nifedipine. E, bar graph showing percentage change in fura-2 quench rate after store depletion in the presence of 10 μm nifedipine, in negative control cells (filled bar, n= 119) and in STIM1 siRNA-transfected cells (open bar, n= 107). **P < 0.01 (unpaired t test).
To further examine if STIM1 protein contributes to CCE, we overexpressed STIM1 protein in cultured mouse PASMCs and then compared the effects of 10 μm CPA on these cells to the control cells in the presence of 10 μm nifedipine. Figure 6A shows that endogenous STIM1 protein was detected at similar levels in non-infected cells and in cells infected with adenovirus containing GFP (Ad-GFP). The protein level was significantly increased in cells infected with STIM1-GFP adenovirus (Ad-GFP-STIM1) as compared to non-infected cells and Ad-GFP cells. Figure 6B shows that 10 μm CPA caused an increase in nifedipine-insensitive transient (108 ± 18 nm, ΔR= 0.27 ± 0.05, n= 62) and sustained rise in [Ca2+]i (40 ± 18 nm, ΔR= 0.11 ± 0.04, n= 62) in Ad-GFP cells. This increase in [Ca2+]i was slightly smaller but not significantly different from that in non-infected cells (transient, 151 ± 16 nm, ΔR= 0.31 ± 0.03, n= 61; sustained, 50 ± 9 nm, ΔR= 0.12 ± 0.02, n= 61, P > 0.05). It is likely that the smaller increase in [Ca2+]i observed in Ad-GFP cells was due to the perturbation of cell function caused by the adenoviruses. Thus, Ad-GFP cells were used as a control in subsequent experiments for comparison of CCE in Ad-GFP-STIM1 cells because they were both treated under the same infection conditions. Figure 7A shows that 10 μm CPA caused an increase in nifedipine-insensitive transient and sustained rise in [Ca2+]i in Ad-GFP cells. Both the transient and sustained increase in [Ca2+]i were significantly enhanced in STIM1-overexpressed cells from 91 ± 23 nm (ΔR= 0.21 ± 0.04, n= 56) to 269 ± 19 nm (ΔR= 0.63 ± 0.06, n= 75, P < 0.01) and 36 ± 23 nm (ΔR= 0.07 ± 0.04, n= 56) to 93 ± 10 nm (ΔR= 0.20 ± 0.02, n= 75, P < 0.05), respectively (Fig. 7A and B). To further confirm that STIM1 mediates CCE in mouse PASMCs, we compared the effects of 10 μm CPA on Mn2+ quench of fura-2 fluorescence in Ad-GFP cells to STIM1-overexpressed cells. Figure 7C shows that CPA caused a 52 ± 6% (n= 88) increase in Mn2+ quench of fura-2 in the presence of 10 μm nifedipine in Ad-GFP cells. This increase in Mn2+ quench rate was significantly increased to 157 ± 12% (Fig. 7C and D; n= 103, P < 0.01) in STIM1-overexpressed cells.
Figure 6. Overexpression of STIM1 in mouse PASMCs.

A, STIM1 protein and GAPDH were detected in non-infected mouse PASMCs and in PASMCs infected with adenovirus containing GFP (Ad-GFP). The expression of STIM1 but not GAPDH increased markedly in cells infected with STIM1-GFP-adenovirus (Ad-GFP-STIM1). Experiments were performed in 3 separate Western blot analyses. B, bar graph showing mean changes in transient and sustained increase in [Ca2+]i caused by 10 μm CPA after re-addition of 2 mm Ca2+ in the presence of 10 μm nifedipine, in non-infected cells (filled bars, n= 61) and Ad-GFP cells (open bars, n= 62).
Figure 7. Overexpression of STIM1 enhances CCE in mouse PASMCs.

A, overexpression of STIM1 enhanced the increase in CPA-induced transient and sustained rise in fura-2 fluorescence ratio in the presence of 10 μm nifedipine. B, bar graph showing mean changes in transient and sustained increase in [Ca2+]i caused by 10 μm CPA after re-addition of 2 mm Ca2+ in the presence of 10 μm nifedipine, in Ad-GFP cells (filled bars, n= 56) and in Ad-GFP-STIM1 cells (open bars, n= 75). **P < 0.01, *P < 0.05 (unpaired t test). C, overexpression of STIM1 enhanced the increase in Mn2+ quench of fura-2 fluorescence caused by 10 μm CPA in the presence of 10 μm nifedipine. D, bar graph showing percentage change in fura-2 quench rate after store depletion in the presence of 10 μm nifedipine, in Ad-GFP cells (n= 88) and in Ad-GFP-STIM1 cells (n= 103). **P < 0.01 (unpaired t test).
To determine if STIM1 is functionally associated with TRPC1 in mediating CCE, the effects of TRPC1 antibody were investigated in the STIM1-overexpresssed cells subjected to store depletion in the presence of 10 μm nifedipine. In control experiments, TRPC1 antibody was pre-adsorbed with TRPC1 antigen peptide and incubated with Ad-GFP-STIM1 cells for 24 h before recording. Figure 8A shows that 10 μm CPA caused an increase in nifedipine-insensitive transient and sustained rise in [Ca2+]i in the Ad-GFP-STIM1 cells under control condition. The transient and sustained increase in [Ca2+]i were significantly reduced in cells treated with TRPC1 antibody, from 235 ± 35 nm (ΔR= 0.35 ± 0.06, n= 48) to 105 ± 23 nm (ΔR= 0.17 ± 0.04, n= 63, P < 0.01) and 113 ± 30 nm (ΔR= 0.18 ± 0.04, n= 48) to 20 ± 18 nm (ΔR= 0.05 ± 0.03, n= 63, P < 0.01), respectively (Fig. 8A and B). To further confirm that STIM1 is associated with TRPC1 in mediating CCE, we compared the effects of 10 μm CPA on Mn2+ quench of fura-2 fluorescence in Ad-GFP-STIM1 cells under control condition to cells treated with TRPC1 antibody (Fig. 8C and D). Figure 8C shows that 10 μm CPA caused a 160 ± 27% (n= 44) increase in Mn2+ quench of fura-2 in the presence of 10 μm nifedipine in Ad-GFP-STIM1 cells under control condition. This increase in Mn2+ quench rate was significantly reduced to 44 ± 8% (n= 31, P < 0.01) in cells treated with TRPC1 antibody (Fig. 8C and D).
Figure 8. STIM1 associates with TRPC1 to mediate CCE in mouse PASMCs.

A, in cultured mouse PASMCs overexpressing STIM1, TRPC1 antibody (1 : 100) reduced the CPA-induced transient and sustained increase in fura-2 fluorescence ratio in the presence of 10 μm nifedipine. B, bar graph showing mean changes in transient and sustained increase in [Ca2+]i caused by 10 μm CPA after re-addition of 2 mm Ca2+ in the presence of 10 μm nifedipine, in Ad-GFP-STIM1 cells under control condition (filled bars, TRPC1 Ab+peptide, n= 48) and in cells treated with TRPC1 antibody (open bars, n= 63). **P < 0.01 (unpaired t test). C, in cultured mouse PASMCs overexpressing STIM1, TRPC1 antibody (1 : 100) inhibited the increase in Mn2+ quench of fura-2 fluorescence caused by 10 μm CPA in the presence of 10 μm nifedipine. D, bar graph showing percentage change in fura-2 quench rate after store depletion in the presence of 10 μm nifedipine, in Ad-GFP-STIM1 cells under control condition (filled bar, TRPC1 Ab+peptide, n= 44) and in cells treated with TRPC1 antibody (open bar, n= 31). **P < 0.01 (unpaired t test).
TRPC1 and STIM1 form a molecular complex in mouse PASMCs
To investigate if store depletion affects the expression levels of TRPC1 and STIM1, we compared the expression levels of TRPC1 and STIM1 between control cells and cells subjected to store depletion. In cells subjected to store depletion, the cells were incubated with Ca2+-free PSS containing 10 μm CPA followed by re-admission of 2 mm Ca2+ in the presence of CPA. We found that store depletion did not affect the expression levels of TRPC1 (Fig. 9A) or STIM1 (Fig. 9B) as compared to the control cells. To determine if Stim1 is associated with TRPC1 channel in mouse PASMCs, a co-immunoprecipitation study was performed. Figure 9C shows that STIM1 co-immunoprecipitates TRPC1, indicating a molecular complex formed between STIM1 proteins and TRPC1 channels in mouse PASMCs. Interestingly, more TRPC1 was co-immunoprecipitated with STIM1 in cells subjected to store depletion as compare to the control cells (Fig. 9C). This data suggests that during store depletion, the association of STIM1 with TRPC1 is enhanced in mouse PASMCs.
Figure 9. TRPC1 interacts with STIM1 to form SOCs in mouse PASMCs.

A, left panel, TRPC1 was detected in cultured mouse PASMCs in the absence and presence of store depletion. Right panel, bar graph showing expression levels of TRPC1 measured relative to GAPDH in control cells (denoted as 1, filled bar), and in cells subjected to store depletion (open bar). Data are means ±s.e.m. of 3 separate Western blot analyses. B, left panel, STIM1 was detected in cultured mouse PASMCs in the absence and presence of store depletion. Right panel, bar graph showing expression levels of STIM1 measured relative to GAPDH in control cells (denoted as 1, filled bar), and in cells subjected to store depletion (open bar). Data are means ±s.e.m. of 3 separate Western blot analyses. C, STIM1 co-immunoprecipitated TRPC1 in cultured mouse PASMCs in the absence and presence of store depletion. STIM1 was first immunoprecipitated (IP) with EXBIO STIM1 antibody (10 μg) and the blot was subsequently probed with BD Biosciences STIM1 antibody (WB, 1 : 100). The blot was then probed for co-IP of TRPC1 expression using TRPC1 antibody (WB, 1 : 100, Alomone). Experiments were performed in 3 separate co-IP procedures and Western blot analyses.
Discussion
The present study provides the first direct evidence that TRPC1 mediates CCE through activation of STIM1 in mouse PASMCs. This was indicated by the inhibitory effects of TRPC1 antibody and STIM1 siRNA, and the enhanced effects of STIM1 overexpression on the dihydropyridine-insensitive sustained rise in [Ca2+]i and the increase in Mn2+ quench of fura-2 fluorescence caused by CPA. This rise in [Ca2+]i and the increase in Mn2+ quench rate were due to CCE because they were activated by store depletion and blocked by SKF 96365, Ni2+, La3+ and Gd3+, a characteristic property of SOCs in many tissues, including pulmonary arteries (Wang et al. 2003; Snetkov et al. 2003; Ng et al. 2008). These pharmacological properties are similar to TRPC1 channels, which are believed to play a prominent role in store-operated Ca2+ entry (see Beech, 2005; Rychkov & Barritt, 2007 for reviews). There is increasing evidence that TRPC1 functions as a SOC in pulmonary arteries. In human PASMCs, TRPC1 gene expression and CCE were significantly reduced in cells treated with TRPC1 antisense (Sweeney et al. 2002). In addition, overexpression of human TRPC1 in rat pulmonary arteries enhanced the contractile responses to CPA (Kunichika et al. 2004). Knockdown of TRPC1 protein with siRNA inhibited cation influx caused by thapsigargin in rat PASMCs (Lin et al. 2004), further supporting that TRPC1 is an important molecular candidate to form SOCs in PASMCs.
Over the past decade, many studies have been performed to examine the molecular signal(s) that activate SOCs (Parekh & Putney, 2005). These include (1) the generation of a ‘Ca2+ influx factor’ from the SR after store depletion, which induces activation of Ca2+-independent phospholipase A2 leading to the activation of SOCs; (2) conformational coupling of the SR IP3 receptors with SOCs on the cell membrane; and (3) fusion of SOC vesicles in the cell membrane leading to the increase in channel number on the membrane. However, the evidence for these hypotheses is controversial and remains to be elucidated (Parekh & Putney, 2005). Recently, the discovery of STIM1 has given a new direction in the search for a molecular intermediate involved in the activation of SOCs. STIM1 was found to act as a sensor of the SR Ca2+ stores when the stores are depleted of Ca2+ and it also activates SOCs in non-excitable cells (Roos et al. 2005; Zhang et al. 2005; Smyth et al. 2006; Spassova et al. 2006; Lewis, 2007). To date, several studies in smooth muscle cells have shown expression of STIM1 and its role in mediating CCE. These include human airway smooth muscle cells (Peel et al. 2006), human coronary arterial smooth muscle cells (Takahashi et al. 2007b), mouse aorta smooth muscle cells (Dietrich et al. 2007) and human saphenous vein cells (Li et al. 2008). Although STIM1 was recently found in rat PASMCs (Lu et al. 2008), our present findings that the rise in [Ca2+]i and cation influx activated by store depletion were reduced by STIM1 siRNA, and these responses to store depletion were enhanced in cells overexpressing STIM1 provide the first functional evidence that endogenous STIM1 contributes to CCE in PASMCs. Taken together, TRPC1 mediates CCE and requires activation of STIM1 in PASMCs. This is consistent with other studies in HEK293 cells (Huang et al. 2006), human platelets (López et al. 2006), human coronary arterial smooth muscle cells (Takahashi et al. 2007a) and human saphenous vein cells (Li et al. 2008), in which STIM1 and TRPC1 mediate depletion-activated Ca2+ entry.
Perhaps the most important finding in the present study is that STIM1 co-immunoprecipitates TRPC1 and the precipitation level of TRPC1 was increased during store depletion (Fig. 9C). Furthermore, overexpression of STIM1 increased CCE (Fig. 7) and this increase in CCE was significantly reduced by TRPC1 antibody (Fig. 8). These findings suggest a functional association of STIM1 and TRPC1 to mediate CCE in mouse PASMCs. Therefore, SOCs may consist of a molecular complex composed of TRPC1 and STIM1 in mouse PASMCs, and when the intracellular Ca2+ stores are depleted, STIM1 that resides in the cytosol may be recruited to the cell membrane and interact with more TRPC1 to enhance CCE. This molecular complex has not been described in pulmonary vascular smooth muscle cells but it is supported by a recent finding in human saphenous vein cells that STIM1 and TRPC1 interact and both contribute to CCE (Li et al. 2008).
In HEK293 cells, STIM1 was found to bind to TRPC1, TRPC4 and TRPC5 and directly regulate these channels, whereas the regulation of TRPC3 and TRPC6 by STIM1 was mediated by STIM1-dependent heteromultimerization of TRPC3 with TRPC1 and TRPC6 with TRPC4 (Yuan et al. 2007). Another interesting finding of the present study is that the dihydropyridine-insensitive transient rise in [Ca2+]i caused by CPA was not affected by TRPC1 antibody but was significantly reduced in PASMCs transfected with STIM1 siRNA (see Figs 4A and 5B). Therefore, it is likely that other TRPC channels may heteromultimerize with TRPC1 and STIM1 to function as SOCs in mouse PASMCs. It is also possible that the dihydropyridine-insensitive transient rise in [Ca2+]i may be mediated by Orai1, which has been shown to be a pore subunit of the calcium release activated calcium (CRAC) channel in non-excitable cells (Feske et al. 2006; Prakriya et al. 2006). Co-expression of Orai1 and STIM1 was found to cause a significant gain in CRAC channel function, suggesting that STIM1 interacts with Orai1 to cause CCE (Soboloff et al. 2006; Mercer et al. 2006). On the other hand, over-expression of Orai1 in HEK cells was found to interact with the store depletion insensitive channels TRPC3 and TRPC6 and confer store depletion sensitivity to these channels (Liao et al. 2007). Most recently, TRPC1 was shown to form a complex with STIM1 and Orai1 to activate SOCs in human salivary gland cells (Ong et al. 2007; Cheng et al. 2008). Thus, future studies on whether other TRPC channels or Orai1 interact with STIM1 and mediate the dihydropyridine-insensitive transient rise in [Ca2+]i in mouse PASMCs are warranted.
On the other hand, we have identified another Ca2+ entry pathway activated by store depletion in addition to CCE in cultured mouse PASMCs. Following store depletion in Ca2+-free conditions, a transient rise in [Ca2+]i was activated after readmission of 2 mm Ca2+, which was partially inhibited by 10 μM nifedipine (Fig. 1B and D), suggesting that the Ca2+ entry process was mediated at least in part through VOCCs. This is also known to occur in cultured canine and rat PASMCs (Ng et al. 2008; McDaniel et al. 2001). It is possible that the release of Ca2+ from intracellular stores during store depletion may inhibit Kv channels, leading to membrane depolarization and subsequent activation of VOCCs (Post et al. 1995). It is also possible that Ca2+ release from stores may activate Ca2+-dependent Cl− channels, leading to membrane depolarization and hence activation of VOCCs (Ng & Gurney, 2001).
In conclusion, store depletion causes activation of VOCCs and CCE in mouse PASMCs. These data provide the first direct evidence that CCE is mediated by the TRPC1 channel through activation of STIM1 in PASMCs. The evidence that TRPC1 and STIM1 form a molecular complex may be an important model for future identification of SOCs in PASMCs and they may be useful targets for the development of new drugs to treat pulmonary hypertension.
Acknowledgments
This work was supported by NIH grants HL 49254 (J.R.H.), P20RR15581 from NCRR (J.R.H.) and an American Heart Association Scientist Development Grant (L.C.N.).
Glossary
Abbreviations
- CCE
capacitative Ca2+ entry
- CPA
cyclopiazonic acid
- PASMC
pulmonary artery smooth muscle cell
- ROC
receptor-operated channel
- SERCA
SR Ca2+-ATPase
- SOC
store-operated channel
- STIM1
stromal-interacting molecule 1
- TRPC
transient receptor potential non-selective cation channel
- VOCC
voltage-operated Ca2+ channel
Author contributions
L.C.N. designed and coordinated the experiments, performed Ca2+ imaging study, Mn2+ quench study, RT-PCR, overexpression, siRNA transfection, data analysis and wrote the paper. M.D.M. contributed to data collection for Ca2+ imaging and Mn2+ quench study. J.A.A. and P.S.K. performed Western Blot analysis and co-IP study. C.A.S. generated recombinant STIM1 and GFP adenoviruses. X.M.S. performed PASMCs isolation and cell culture. J.R.H. supervised the experiments, discussed the results and commented on the manuscript. All authors read and approved the manuscript for publication.
References
- Ahmmed GU, Mehta D, Stephen V, Holinstat M, Paria BC, Tiruppathi C, Malik AB. Protein kinase Cα phosphorylates the TRPC1 channel and regulates store-operated Ca2+ entry in endothelial cells. J Biol Chem. 2004;279:20941–20949. doi: 10.1074/jbc.M313975200. [DOI] [PubMed] [Google Scholar]
- Albert AP, Large WA. Activation of store-operated channels by noradrenaline via protein kinace C in rabbit portal vein myocytes. J Physiol. 2002;544:113–125. doi: 10.1113/jphysiol.2002.022574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert AP, Large WA. Store-operated Ca2+-permeable non-selective cation channels in smooth muscle cells. Cell Calcium. 2003;33:345–356. doi: 10.1016/s0143-4160(03)00048-4. [DOI] [PubMed] [Google Scholar]
- Albert AP, Saleh SN, Peppiatt-Wildman CM, Large WA. Multiple activation mechanisms of store-operated TRPC channels in smooth muscle cells. J Physiol. 2007;583:25–36. doi: 10.1113/jphysiol.2007.137802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barritt GJ. Receptor activated Ca2+ inflow in animal cells: a variety of pathways tailored to meet different intracellular Ca2+ signaling requirements. Biochem J. 1999;337:153–169. [PMC free article] [PubMed] [Google Scholar]
- Beech DJ. TRPC1: store-operated channel and more. Pflugers Arch. 2005;451:53–60. doi: 10.1007/s00424-005-1441-3. [DOI] [PubMed] [Google Scholar]
- Bergdahl A, Gomez MF, Wihlborg AK, Erlinge D, Eyjolfson A, Xu SZ, Beech DJ, Dreja K, Hellstrand P. Plasticity of TRPC expression in arterial smooth muscle: correlation with store-operated Ca2+ entry. Am J Physiol Cell Physiol. 2005;288:C872–C880. doi: 10.1152/ajpcell.00334.2004. [DOI] [PubMed] [Google Scholar]
- Brueggemann LI, Markun DR, Henderson KK, Cribbs LL, Byron KL. Pharmacological and electrophysiological characterization of store-operated currents and capacitative Ca2+ entry in vascular smooth muscle cells. J Pharmacol Exp Ther. 2006;317:488–499. doi: 10.1124/jpet.105.095067. [DOI] [PubMed] [Google Scholar]
- Cheng KT, Liu X, Ong HL, Ambudkar IS. Functional Requirement for Orai1 in store-operated TRPC1-STIM1 channels. J Biol Chem. 2008;283:12935–12940. doi: 10.1074/jbc.C800008200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai YP, Bungalon S, Hatton WJ, Hume JR, Yamboliev IA. ClC-3 chloride channel is upregulated by hypertrophy and inflammation in rat and canine pulmonary artery. Br J Pharmacol. 2005;145:5–14. doi: 10.1038/sj.bjp.0706135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich A, Kalwa H, Storch U, Mederos y Schnitzler M, Salanova B, Pinkenburg O, Dubrovska G, Essin K, Gollasch M, Birnbaumer L, Gudermann T. Pressure-induced and store-operated cation influx in vascular smooth muscle cells is independent of TRPC1. Pflugers Arch. 2007;455:465–477. doi: 10.1007/s00424-007-0314-3. [DOI] [PubMed] [Google Scholar]
- Du J, Sours-Brothers S, Coleman R, Ding M, Graham S, Kong DH, Ma R. Canonical transient receptor potential 1 channel is involved in contractile function of glomerular mesangial cells. J Am Soc Nephrol. 2007;18:1437–1445. doi: 10.1681/ASN.2006091067. [DOI] [PubMed] [Google Scholar]
- Feske S, Gwack Y, Prakriya M, Srikanth S, Puppel SH, Tanasa B, Hogan PG, Lewis RS, Daly M, Rao A. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature. 2006;441:179–185. doi: 10.1038/nature04702. [DOI] [PubMed] [Google Scholar]
- Flemming R, Cheong A, Dedman AM, Beech DJ. Discrete store-operated calcium influx into an intracellular compartment in rabbit arteriolar smooth muscle. J Physiol. 2002;543:455–464. doi: 10.1113/jphysiol.2002.023366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham FL, Prevec L. Methods for construction of adenovirus vectors. Mol Biotechnol. 1995;3:207–220. doi: 10.1007/BF02789331. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Huang GN, Zeng W, Kim JY, Yuan JP, Han L, Muallem S, Worley PF. STIM1 carboxyl-terminus activates native SOC, Icrac and TRPC1 channels. Nat Cell Biol. 2006;8:1003–1010. doi: 10.1038/ncb1454. [DOI] [PubMed] [Google Scholar]
- Jho D, Mehta D, Ahmmed G, Gao XP, Tiruppathi C, Broman M, Malik AB. Angiopoietin-1 opposes VEGF-induced increase in endothelial permeability by inhibiting TRPC1-dependent Ca2+ influx. Circ Res. 2005;96:1282–1290. doi: 10.1161/01.RES.0000171894.03801.03. [DOI] [PubMed] [Google Scholar]
- Kunichika N, Yu Y, Remillard CV, Platoshyn O, Zhang S, Yuan JX. Overexpression of TRPCs enhances pulmonary vasoconstriction induced by capacitative Ca2+ entry. Am J Physiol Lung Cell Mol Physiol. 2004;287:L962–L969. doi: 10.1152/ajplung.00452.2003. [DOI] [PubMed] [Google Scholar]
- Leung FP, Yung LM, Yao X, Laher I, Huang Y. Store-operated calcium entry in vascular smooth muscle. Br J Pharmacol. 2007;153:846–857. doi: 10.1038/sj.bjp.0707455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis RS. The molecular choreography of a storeoperated calcium channel. Nature. 2007;446:284–287. doi: 10.1038/nature05637. [DOI] [PubMed] [Google Scholar]
- Li J, Sukumar P, Milligan CJ, Kumar B, Ma ZY, Munsch CM, Jiang LH, Porter KE, Beech DJ. Interactions, functions, and independence of plasma membrane STIM1 and TRPC1 in vascular smooth muscle cells. Circ Res. 2008;103:e97–e104. doi: 10.1161/CIRCRESAHA.108.182931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Erxleben C, Yildirim E, Abramowitz J, Armstrong DL, Birnbaumer L. Orai proteins interact with TRPC channels and confer responsiveness to store depletion. Proc Natl Acad Sci U S A. 2007;104:4682–4687. doi: 10.1073/pnas.0611692104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MJ, Leung GP, Zhang WM, Yang XR, Yip KP, Tse CM, Sham JS. Chronic hypoxia-induced upregulation of store-operated and receptor-operated Ca2+ channels in pulmonary arterial smooth muscle cells: a novel mechanism of hypoxic pulmonary hypertension. Circ Res. 2004;95:496–505. doi: 10.1161/01.RES.0000138952.16382.ad. [DOI] [PubMed] [Google Scholar]
- López JJ, Salido GM, Pariente JA, Rosado JA. Interaction of STIM1 with endogenously expressed human canonical TRP1 upon depletion of intracellular Ca2+ stores. J Biol Chem. 2006;281:28254–28264. doi: 10.1074/jbc.M604272200. [DOI] [PubMed] [Google Scholar]
- Lu W, Wang J, Shimoda LA, Sylvester JT. Differences in STIM1 and TRPC expression in proximal and distal pulmonary arterial smooth muscle are associated with differences in Ca2+ responses to hypoxia. Am J Physiol Lung Cell Mol Physiol. 2008;295:L104–L113. doi: 10.1152/ajplung.00058.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDaniel SS, Platoshyn O, Wang J, Yu Y, Sweeney M, Krick S, Rubin LJ, Yuan JX. Capacitative Ca2+ entry in agonist-induced pulmonary vasoconstriction. Am J Physiol Lung Cell Mol Physiol. 2001;280:L870–L880. doi: 10.1152/ajplung.2001.280.5.L870. [DOI] [PubMed] [Google Scholar]
- McElroy SP, Gurney AM, Drummond RM. Pharmacological profile of store-operated Ca2+ entry in intrapulmonary artery smooth muscle cells. Eur J Pharmacol. 2008;584:10–20. doi: 10.1016/j.ejphar.2008.01.018. [DOI] [PubMed] [Google Scholar]
- Mercer JC, Dehaven WI, Smyth JT, Wedel B, Boyles RR, Bird GS, Putney JW., Jr Large store-operated calcium selective currents due to co-expression of Orai1 or Orai2 with the intracellular calcium sensor, Stim1. J Biol Chem. 2006;281:24979–24990. doi: 10.1074/jbc.M604589200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng LC, Gurney AM. Store-operated channels mediate Ca2+ influx and contraction in rat pulmonary artery. Circ Res. 2001;89:923–929. doi: 10.1161/hh2201.100315. [DOI] [PubMed] [Google Scholar]
- Ng LC, Kyle BD, Lennox AR, Shen XM, Hatton WJ, Hume JR. Cell culture alters Ca2+ entry pathways activated by store-depletion or hypoxia in canine pulmonary arterial smooth muscle cells. Am J Physiol Cell Physiol. 2008;294:C313–C323. doi: 10.1152/ajpcell.00258.2007. [DOI] [PubMed] [Google Scholar]
- Ong HL, Cheng KT, Liu X, Bandyopadhyay BC, Paria BC, Soboloff J, Pani B, Gwack Y, Srikanth S, Singh BB, Gill DL, Ambudkar IS. Dynamic assembly of TRPC1STIM1-Orai1 ternary complex is involved in store-operated calcium influx. Evidence for similarities in store-operated and calcium release-activated calcium channel components. J Biol Chem. 2007;282:9105–9116. doi: 10.1074/jbc.M608942200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parekh AB, Putney JW. Store-operated calcium channels. Physiol Rev. 2005;85:757–810. doi: 10.1152/physrev.00057.2003. [DOI] [PubMed] [Google Scholar]
- Pedersen SF, Owsianik G, Nilius B. TRP channels: an overview. Cell Calcium. 2005;38:233–252. doi: 10.1016/j.ceca.2005.06.028. [DOI] [PubMed] [Google Scholar]
- Peel SE, Liu B, Hall IP. A key role for STIM1 in store operated calcium channel activation in airway smooth muscle. Respir Res. 2006;7:119–126. doi: 10.1186/1465-9921-7-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Post JM, Gelband CH, Hume JR. [Ca2+]i inhibition of K+ channels in canine pulmonary artery. Novel mechanism for hypoxia-induced membrane depolarization. Circ Res. 1995;77:131–139. doi: 10.1161/01.res.77.1.131. [DOI] [PubMed] [Google Scholar]
- Prakriya M, Feske S, Gwack Y, Srikanth S, Rao A, Hogan PG. Orai1 is an essential pore subunit of the CRAC channel. Nature. 2006;443:230–233. doi: 10.1038/nature05122. [DOI] [PubMed] [Google Scholar]
- Roos J, DiGregorio PJ, Yeromin AV, Ohlsen K, Lioudyno M, Zhang S, Safrina O, Kozak JA, Wagner SL, Cahalan MD, Veliçelebi G, Stauderman KA. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J Cell Biol. 2005;169:435–445. doi: 10.1083/jcb.200502019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rychkov G, Barritt GJ. TRPC1: Ca2+-permeable channels in animal cells. Handb Exp Pharmacol. 2007;179:23–52. doi: 10.1007/978-3-540-34891-7_2. [DOI] [PubMed] [Google Scholar]
- Sage SO, Brownlow SL, Rosado JA. TRP channels and calcium entry in human platelets. Blood. 2002;100:4245–4246. doi: 10.1182/blood-2002-08-2417. [DOI] [PubMed] [Google Scholar]
- Saleh SN, Albert AP, Peppiatt CM, Large WA. Angiotensin II activates two cation conductances with distinct TRPC1 and TRPC6 channel properties in rabbit mesenteric artery myocytes. J Physiol. 2006;577:479–495. doi: 10.1113/jphysiol.2006.119305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleh SN, Albert AP, Peppiatt-Wildman CM, Large WA. Diverse properties of store-operated TRPC channels activated by protein kinase C in vascular myocytes. J Physiol. 2008;586:2463–2476. doi: 10.1113/jphysiol.2008.152157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth JT, Dehaven WI, Jones BF, Mercer JC, Trebak M, Vazquez G, Putney JW., Jr Emerging perspectives in store-operated Ca2+ entry: roles of Orai, Stim and TRP. Biochim Biophys Acta. 2006;1763:1147–1160. doi: 10.1016/j.bbamcr.2006.08.050. [DOI] [PubMed] [Google Scholar]
- Snetkov VA, Aaronson PI, Ward JP, Knock GA, Robertson TP. Capacitative calcium entry as pulmonary specific vasoconstrictor mechanism in small arteries of the rat. Br J Pharmacol. 2003;140:97–106. doi: 10.1038/sj.bjp.0705408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soboloff J, Spassova MA, Tang XD, Hewavitharana T, Xu W, Gill DL. Orai1 and STIM reconstitute store-operated calcium channel function. J Biol Chem. 2006;281:20661–20665. doi: 10.1074/jbc.C600126200. [DOI] [PubMed] [Google Scholar]
- Spassova MA, Soboloff J, He LP, Xu W, Dziadek MA, Gill DL. STIM1 has a plasma membrane role in the activation of store-operated Ca2+ channels. Proc Natl Acad Sci U S A. 2006;103:4040–4045. doi: 10.1073/pnas.0510050103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeney M, Yu Y, Platoshyn O, Zhang S, McDaniel SS, Yuan JX. Inhibition of endogenous TRP1 decreases capacitative Ca2+ entry and attenuates pulmonary artery smooth muscle cell proliferation. Am J Physiol Lung Cell Mol Physiol. 2002;283:L144–L155. doi: 10.1152/ajplung.00412.2001. [DOI] [PubMed] [Google Scholar]
- Takahashi Y, Watanabe H, Murakami M, Ohba T, Radovanovic M, Ono K, Iijima T, Ito H. Involvement of transient receptor potential canonical 1 (TRPC1) in angiotensin II-induced vascular smooth muscle cell hypertrophy. Atherosclerosis. 2007a;195:287–296. doi: 10.1016/j.atherosclerosis.2006.12.033. [DOI] [PubMed] [Google Scholar]
- Takahashi Y, Watanabe H, Murakami M, Ono K, Munehisa Y, Koyama T, Nobori K, Iijima T, Ito H. Functional role of stromal interaction molecule 1 (STIM1) in vascular smooth muscle cells. Biochem Biophys Res Commun. 2007b;361:934–940. doi: 10.1016/j.bbrc.2007.07.096. [DOI] [PubMed] [Google Scholar]
- Trepakova ES, Gericke M, Hirakawa Y, Weisbrod RM, Cohen RA, Bolotina VM. The properties of a native cation channel activated by Ca2+ store depletion in vascular smooth muscle cells. J Biol Chem. 2001;276:7782–7790. doi: 10.1074/jbc.M010104200. [DOI] [PubMed] [Google Scholar]
- Walker RL, Hume JR, Horowitz B. Differential expression and alternative splicing of TRP channel genes in smooth muscles. Am J Physiol Cell Physiol. 2001;280:C1184–C1192. doi: 10.1152/ajpcell.2001.280.5.C1184. [DOI] [PubMed] [Google Scholar]
- Wang J, Shimoda LA, Sylvester JT. Capacitative calcium entry and TRPC channel proteins are expressed in rat distal pulmonary arterial smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2003;286:L848–L858. doi: 10.1152/ajplung.00319.2003. [DOI] [PubMed] [Google Scholar]
- Weirich J, Dumont L, Fleckenstein-Grun G. Contribution of capacitative and non-capacitative Ca2+-entry to M3-receptor-mediated contraction of porcine coronary smooth muscle. Cell Calcium. 2005;38:457–467. doi: 10.1016/j.ceca.2005.06.035. [DOI] [PubMed] [Google Scholar]
- Wilson SM, Mason HS, Smith GD, Nicholson N, Johnston L, Janiak R, Hume JR. Comparative capacitative calcium entry mechanisms in canine pulmonary and renal arterial smooth muscle cells. J Physiol. 2002;543:917–931. doi: 10.1113/jphysiol.2002.021998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu SZ, Beech DJ. TrpC1 is a membrane-spanning subunit of store-operated Ca2+ channels in native vascular smooth muscle cells. Circ Res. 2001;88:84–87. doi: 10.1161/01.res.88.1.84. [DOI] [PubMed] [Google Scholar]
- Xu SZ, Boulay G, Flemming R, Beech DJ. E3-targeted anti-TRPC5 antibody inhibits store-operated calcium entry in freshly isolated pial arterioles. Am J Physiol Heart Circ Physiol. 2006;291:H2653–H2659. doi: 10.1152/ajpheart.00495.2006. [DOI] [PubMed] [Google Scholar]
- Yuan JP, Zeng W, Huang GN, Worley PF, Muallem S. STIM1 heteromultimerizes TRPC channels to determine their function as store-operated channels. Nat Cell Biol. 2007;9:636–645. doi: 10.1038/ncb1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SL, Yu Y, Roos J, Kozak JA, Deerinck TJ, Ellisman MH, Stauderman KA, Cahalan MD. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature. 2005;437:902–905. doi: 10.1038/nature04147. [DOI] [PMC free article] [PubMed] [Google Scholar]