

Abstract
Tetrathiatriarylmethyl radicals are ideal spin probes for biological electron paramagnetic resonance (EPR) spectroscopy and imaging. The wide application of trityl radicals as biosensors of oxygen or other biological radicals was hampered by the lack of affordable large-scale syntheses. We report the large-scale synthesis of the Finland trityl radical using an improved addition protocol of the aryl lithium monomer to methylchloroformate. A new reaction for the formal one-electron reduction of trityl alcohols to trityl radicals using neat trifluroacetic acid is reported as well. Initial applications show that the compound is very sensitive to molecular oxygen. It has already provided high-resolution EPR images on large aqueous samples and should be suitable for a broad range of in vivo applications.
Reactive oxygen species (ROS) in low concentration play a vital role in the regulation of physiological processes, and are central in the initiation of pathological events in unregulated concentrations.1,2,3 It is well established that the generation of ROS in cellular systems is a result of chemical or enzymatic reduction of molecular oxygen, and is modulated by tissue oxygen levels.4,5,6,7 Therefore, it is critical to monitor the degree of intracellular and tissue oxygenation in vitro and in vivo. Conventional methods, such as optical and electrochemical techniques, have been employed in the monitoring of oxygen partial pressure (pO2) but are limited by their invasiveness and the coverage of area being measured. Electron paramagnetic resonance (EPR) spectroscopy and imaging using water-soluble paramagnetic probes, such as nitroxides and trityl radicals, as well as particulate-based probes, such as chars, india ink and lithium phthalocyanine, have been employed for the measurement of pO2 or dissolved O2 in the mouse tumor, brain, and heart.8,9,10 EPR imaging has also been employed in the investigation of drug delivery, toxicity of xenobiotics, and tissue redox status.11,12,13 In contrast to conventional methods, EPR spectroscopy and imaging are non-invasive techniques. Furthermore, these techniques can be applied to map oxygenation over a predefined area in various tissues. In particular, trityl radicals have been demonstrated to be very effective spin probes in measuring the oxygen concentration in vivo, and in vitro through line-width broadening analysis.14,15,16,17
Nycommed Innovations (now a subsidiary of GE Healthcare) initially designed the sterically crowded trityl radicals, otherwise known by the acronym TAM (tetrathiatriarylmethyl), for use as contrast agents in Overhauser Magnetic Resonance Imaging (Figure 1).18,19,20,21,22,23,24,25,26 Gomberg’s work over 100 years ago proved that trityl radicals are persistent at room temperature, hence the interest in these compounds.27 An ideal spin probe would exhibit a single, sharp EPR signal in order to obtain maximum sensitivity under in vivo conditions. To ensure practical use in biological systems, it is convenient to have trityl radicals that are soluble in water. By removing all sources of inhomogeneous hyperfine couplings, the radicals exhibit extremely narrow signals, allowing for high sensitivity (i.e. detection of nanomolar concentration of radicals by EPR spectroscopy in biological media). The trityl radicals designed by Nycommed Innovations are extremely efficient spin probes; however they are complex molecules, and their synthesis in large scale has proven challenging.28 This limits their utility, as biological, and especially in vivo animal studies and clinical applications require large quantities of these spin probes. Herein, we report a procedure for the large-scale synthesis of the Finland trityl radical (radical 7 in this paper).
Figure 1.

Representative trityl radicals used as spin probes
Using the patent literature, and the procedure published by Rawal and co-workers, milligram amounts of radical 7 were produced.29 The scale-up of this literature procedure did not give satisfactory results. The synthesis of compound 2 proceeded as described in the literature with some minor changes (Scheme 1). It was necessary to reflux for at least one day, rather than two hours in order to synthesize 70 g of compound 2. Compound 3 was obtained in large amount as reported in the literature, with minor modifications.28–29 Ethanol was used to precipitate pure compound 3 from the crude reaction mixture instead of acetonitrile and tetrahydrofurane. However, our attempts to produce the trityl alcohol 5 in large scale were unsuccessful, resulting in very low yield.
Scheme 1.

(a) Na, DMF, 2-methyl-2-propanethiol 61 % (b) HBF4 54% in Et2O, acetone, toluene 51 %; (c) n-BuLi 2.5 M in hexane 1 equiv. in Et2O; than 0.3 equiv. of methyl chloroformate (d) n-BuLi 2.5M in hexanes 3 equiv., 3 equiv. of 3 in Et2O; than add the ketone 4; 70 % in two steps; (e) n-BuLi 2.5 M in hexanes and TMEDA 10 equiv. than excess Diboc 44 %; (f) CF3COOH 99%
The direct synthesis of trityl alcohol 5 was extremely slow because of the steric bulk of the aryl monomers. The ketone 4 was isolated first, as an intermediate toward the trityl alcohol 5.30 Subsequently it was reacted with the 3 equivalents of the aryl lithium monomer to afford trityl alcohol 5 in very good yield (method A). This procedure involved lengthy chromatographic purifications; however it gave us easy access to gram amounts of the target compound 5.31 Next, tert-butyl ester 6, rather than the ethyl ester described in the literature procedure, was synthesized in good yield.32
The rationale was that tert-butyl esters are not susceptible to multiple addition, and polymerization. Compound 6 was treated with neat trifluoroacetic acid over 20 h at rt in order to cleave the tert-butyl ester, and fortuitously during the same step the radical 7 was generated in quantitative yield. Evaporating the trifluoroacetic acid afforded the pure radical 7 in quantitative yield. This formal one-electron reduction of the central carbon was quite surprising. Other trityl alcohols that do not posses the tert-butyl ester functionality undergo formal one-electron reduction of the central carbon using neat trifluoroacetic acid just as easily and efficiently.33 We are looking into possible explanations for this particular reaction. In order to scale-up to any desired amount of product, further modifications were introduced. The extremely slow addition of methyl chloroformate afforded the trityl 5 from starting material 3 in one step, as described in method B.31 The procedures have been optimized, and all products are purified by precipitation, making possible the scale-up to any desired amount.
The radical 7 is a bulky molecule, surrounded by hydrophobic groups on most of its surfaces. However, the three carboxylic groups render radical 7 to be soluble in water. Due to aggregation, a lower concentration of up to 2 mM was found suitable for practical applications. Three equivalent of hydrochloric acid were required to neutralize one equivalent of radical 7. This number was calculated by potentiometric titration using the amount of HCl used to neutralize the trisodium tricarboxylate form of the radical 7 (ν(HCl)/7ml/[radical 7] = 34.3 μmole/7 ml/1.6 mM ~ 3). The obtained number supports the independent protonation of all three carboxyl groups of the radical 7 with similar pKa values in agreement with the large distance between these groups. Titration of the radical 7 was performed using X-band EPR spectroscopy as well (Figure 2).
Figure 2.

EPR spectra of 0.05 mM solution of the radical 7 in the presence of 1.5 mM citrate buffer at different pH values. The spectrometer settings were: sweep width, 30 G; time constant, 5.12 ms; conversion time, 40.96 ms; scan time, 83.89 s; 2048 points; modulation amplitude, 0.2 G; and microwave power, 5.0 mW. The pH of the samples was adjusted to the required value by the addition of 1M aqueous HCl.
The determination of the exact pKa value by potentiometric titration is complicated due to precipitate formation in acidic medium. Reversible EPR spectral changes were observed with the appearance of a broad signal with a line-width of 2.5 G in the acidic medium, apparently due to reversible aggregation of the radical 7 (Figure 2). To investigate the influence of pH of the solution on the EPR intensity as a result of aggregation the experiment in figure 3 was performed. The solid lines in figure 3 represent the best fits for the function [R−]pH = [R]0 – [R]0/(1+10(pH−pKagg)/q), where [R]0 is total radical 7 concentration, [R−]pH is the concentration of the anionic form of the radical 7 dissolved at given pH value, pKagg is the pH value when half of the radical 7 molecules are aggregated, and q is a numerical coefficient. The fittings yield the pKagg and q values to be equal to 3.7 and 0.37, respectively, for titration from basic to acidic medium. The corresponding values for titration from acidic to basic medium are 4.0 and 0.54, respectively. The data support the conclusion that [R−]pH = [R]0 if pH > 4.5, while in acidic conditions [R−]pH decreases exponentially, becoming close to zero at pH=3. The calculated partition coefficient of the radical 7 (defined as Kp=[Octanol]/[water]) at pH values equal to 1, 7.4, and 13 was found to be ≥ 4×104, 0.27±0.15, and 0.3±0.1 respectively. The Kp values provide further evidence for the observed low solubility of the radical 7 in acidic solutions.
Figure 3.
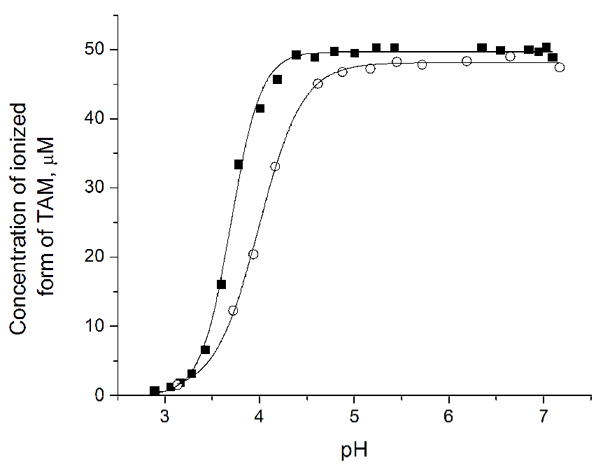
The dependence of the EPR peak signal intensity of radical 7 on the pH of the sample (50 μM radical 7 in the presence of 1.5 mM citrate buffer). The symbols (■) and (○) denote the data obtained upon titration from alkaline pH into the direction of acidic pH and in reverse direction, respectively.
The oxygen partial pressure is one of the most important parameters in the metabolic processes of the cell, thus it plays a key role in physiological processes. Molecular oxygen has two unpaired electrons, and the bimolecular collision with a persistent radical (spin probe) of known concentration causes the EPR line broadening of the persistent radical. This line broadening is measured; thus indirectly the concentration of oxygen in living tissue can be ascertained. At biological pH (pH=7.4) we estimate from the approximate pKa=3.7 that the radical 7 exists in ionized form (7−). The EPR line-width of the radical 7− was measured at different concentrations of oxygen, diluted with argon. The EPR spectrum in phosphate buffer (0.1 M, pH 7.4) consists of a single sharp peak with a peak-to-peak line-width of 210 mG in room air. The radical 7− exhibited excellent EPR characteristics with an anaerobic line-width of 90 mG, and 0.84 mG/mmHg linear sensitivity to oxygen. The line broadening effect is reversible upon exclusion of oxygen by argon, and the line-width is restored to its original value of 90 mG (Figure 4).
Figure 4.
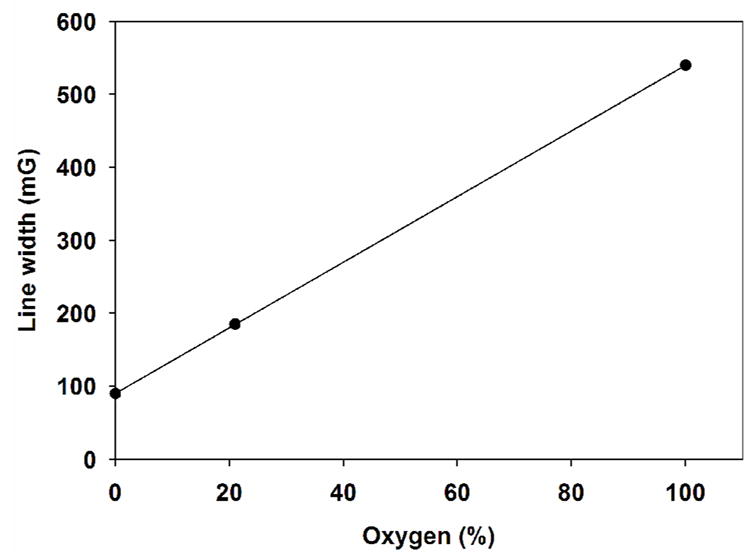
The dependency of the EPR line-width for radical 7− on the concentrations of oxygen
Radical 7− has already been used as a probe for EPR imaging experiments in large aqueous samples.34 These imaging experiments were performed in a home-built 1.2 GHz L-band EPR spectrometer. Several phantoms of defined geometry were used to develop new imaging software algorithms, and excellent image quality and fidelity were observed. These experiments indicate that this compound is suitable for high-quality, EPR imaging applications.
In conclusion, an efficient synthetic protocol for the large-scale synthesis of radical 7 is described. Our synthetic protocol involves no column chromatography, and is very reproducible. Within its solubility range, the ionized form of radical 7 (7−) exhibited excellent EPR characteristics with an anaerobic line-width of 90 mG, and 0.84 mG/mmHg linear sensitivity toward oxygen. Radical 7 was shown to be highly effective for EPR spectroscopy and imaging and should be well suited for ex vivo and in vivo biological applications.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health grants EB0890, EB4900, EB 03519, and HL81248.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Roberfroid M, Calderon PB. Free Radicals and Oxidation Phenomena in Biological Systems. Mercel Dekker; New York: 1995. [Google Scholar]
- 2.Halliwell B, Gutteridge JMC. Free Radicals in Biology and Medicine. Oxford University Press; Oxford: 1999. [Google Scholar]
- 3.Zweier JL, Talukder H. Cardiovascular Research. 2006;70(2):181. doi: 10.1016/j.cardiores.2006.02.025. [DOI] [PubMed] [Google Scholar]
- 4.Zweier JL, Kuppusamy P, Williams R, Rayburn BK, Smith D, Weisfeldt ML, Flaherty JT. J Biol Chem. 1989;264(32):18890. [PubMed] [Google Scholar]
- 5.Zweier JL, Broderick R, Kuppusamy P, Thompson-Gorman S, Lutty G. J Biol Chem. 1994;269:24156. [PubMed] [Google Scholar]
- 6.Zweier JL, Duke SS, Kuppusamy P, Sylvester JT, Gabrielson EW. FEBS Letters. 1989;252:12. doi: 10.1016/0014-5793(89)80881-6. [DOI] [PubMed] [Google Scholar]
- 7.Sanders SP, Zweier JL, Kuppusamy P, Harrison SJ, Bassett DJ, Gabrielson EW, Sylvester JT. J Clin Invest. 1993;91:46. doi: 10.1172/JCI116198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Williams BB, Parasca A, Mailer C, Pelizzari CA, Lewis MA, River JN, Karczmar GS, Barth ED, Halpern HJ. Magn Reson in Med. 2003;49:682. doi: 10.1002/mrm.10408. [DOI] [PubMed] [Google Scholar]
- 9.Liu S, Timmins GS, Shi H, Gasparovic CM, Liu KJ. NMR in Biomedicine. 2004;17(5):327. doi: 10.1002/nbm.899. [DOI] [PubMed] [Google Scholar]
- 10.Kuppusamy P, Zweier JL. NMR in Biomedicine. 2004;17(5):226. doi: 10.1002/nbm.912. [DOI] [PubMed] [Google Scholar]
- 11.Lurie D, Maeder K. Adv Drug Delivery Reviews. 2005;57:1171. doi: 10.1016/j.addr.2005.01.023. [DOI] [PubMed] [Google Scholar]
- 12.Hirata H, Fujii H. Current Organic Chemistry. 2006;10(5):521. [Google Scholar]
- 13.He G, Kutala VK, Kuppusamy P, Zweier JL. Free Radic Biol Med. 2004;36(5):665. doi: 10.1016/j.freeradbiomed.2003.11.024. [DOI] [PubMed] [Google Scholar]
- 14.Ardenkaer-Larsen JH, Laursen I, Leunbach I, Ehnholm G, Wistrand LG, Petersson JS, Golman K. J Magn Res. 1998;133:1. doi: 10.1006/jmre.1998.1438. [DOI] [PubMed] [Google Scholar]
- 15.Elas M, Ahn KH, Parasca A, Barth ED, Lee D, Haney C, Halpern HJ. Clin Cancer Res. 2006;12(14):4209. doi: 10.1158/1078-0432.CCR-05-0446. [DOI] [PubMed] [Google Scholar]
- 16.Kuppusamy P, Wang P, Chzhan M, Zweier JL. Magn Reson in Med. 1997;37:479. doi: 10.1002/mrm.1910370402. [DOI] [PubMed] [Google Scholar]
- 17.Williams BB, al Hallaq H, Chandramouli GVR, Barth ED, Rivers JN, Lewis M, Galtsev VE, Karczmar GS, Halpern HJ. Magn Reson in Med. 2002;47:634. doi: 10.1002/mrm.10089. [DOI] [PubMed] [Google Scholar]
- 18.Lurie D, Li H, Petryakov S, Zweier JL. Magn Reson in Med. 2002;47:181. doi: 10.1002/mrm.10029. [DOI] [PubMed] [Google Scholar]
- 19.Li H, Deng Y, He G, Kuppusamy P, Lurie D, Zweier JL. Magn Reson in Med. 2002;48(3):530. doi: 10.1002/mrm.10222. [DOI] [PubMed] [Google Scholar]
- 20.He G, Deng Y, Li H, Kuppusamy P, Zweier JL. Magn Reson in Med. 2002;47:571. doi: 10.1002/mrm.10077. [DOI] [PubMed] [Google Scholar]
- 21.He G, Evalappan S, Hirata H, Deng Y, Petryakov S, Kuppusamy P, Zweier JL. Magn Reson in Med. 2002;48:1057. doi: 10.1002/mrm.10302. [DOI] [PubMed] [Google Scholar]
- 22.Krishna MC, English S, Yamada K, Yoo J, Murugesan R, Devasahayam N, Cook JA, Golman K, Ardenkaer-Larsen JH, Subramanian S, Mitchel JB. Proc Natl Acad Sci USA. 2002;99(4):2216. doi: 10.1073/pnas.042671399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Andersson, S.; Radner, F.; Rydbeek, A.; Servin, R.; Wistrand, L. G. US Patent 5530140, 1996.
- 24.Ardenkjaer-Larsen JH, Leunbach I. PCT Int Appl Wo/9709633. 1997 [Google Scholar]
- 25.Andersson, S.; Radner, F.; Rydbeek, A.; Servin, R.; Wistrand, L. G. US Patent 5728370, 1998.
- 26.Thaning M. PCT Int Applwo/9839277. 1998 [Google Scholar]
- 27.Gomberg M. J Am Chem Soc. 1900;22(11):757. [Google Scholar]
- 28.Reddy TJ, Iwama T, Halpern HJ, Rawal VH. J Org Chem. 2002;67(14):4635. doi: 10.1021/jo011068f. [DOI] [PubMed] [Google Scholar]
- 29.Xia S, Villamena FA, Hadad CM, Kuppusamy P, Li Y, Zhu H, Zweier JL. J Org Chem. 2006;71(19):7268. doi: 10.1021/jo0610560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bis-(2,2,6,6-tetramethyl-benzo[1,2-d;4,5-d′]bis[1,3]dithiol-4-yl)-methanone (4). To a solution of 3 (9 g, 31.4 mmol) in 350 ml of diethyl ether were added 12.6 ml of n-butyl lithium (2.5 M in hexanes). The solution was stirred for at least two hours. Methyl chloroformate (0.97 ml, 12.5 mmol), dissolved in 60 ml of diethyl ether, was added drop wise over 120 min, and the solution was stirred over 24 hours. Saturated aqueous sodium bicarbonate was added, and the organic phase was separated. The aqueous phase was extracted again with 2×100 ml of diethyl ether. The combined organic phases were dried over sodium sulfate, and evaporated to afford the ketone 4 mixed with starting material 3. The title compound 4 was isolated in 70% yield (6.60 g) after flash chromatography on silica gel eluting first with hexanes to recover the starting material 3, than with 11:1 hexanes-ethyl acetate. mp =120–127 °C; 1H NMR (500 MHz, CDCl3) δ=7.19 (s, 2H), 1.78 (s, 12H); 13C (125 MHz, CDCl3) δ=193.6, 137.7, 137.6, 127.3, 119.4, 65.4, 31.2; MS (ESI, [M+Na]+ ): 620.9648 (observed), 620.9647 (calcd)..
- 31.Tris-(2,2,6,6-tetramethyl-benzo[1,2-d;4,5-d′]bis[1,3]dithiol-4-yl)-methanol (5). Method A. To a solution of 3 (9 g, 31.4 mmol) in 350 ml of diethyl ether were added 12.6 ml of n-BuLi (2.5 M in hexanes). The solution was stirred for at least two hours. The ketone 4 (5 g, 8.34 mmol) was added all at once, and the resulting mixture was stirred over 24 hours. Saturated aqueous sodium bicarbonate was added, and the organic phase was separated. The aqueous phase was extracted again with 2x100 ml of diethyl ether. The combined organic phases were dried over sodium sulfate, and evaporated. The mixture was purified by flash chromatography on silica gel eluting first with hexanes to remove the starting material 3, then with 20:1 to 10:1 hexanes-ethyl acetate. The title product 5 was isolated in 96 % yield (7.1 g). Method B. To a solution of 3 (12 g, 41.9 mmol) in 450 ml of diethyl ether were added 16.8 ml of n-BuLi (2.5 M in hexanes). The solution was stirred for at least two hours. Methyl chloroformate (1.29 ml, 16.7 mmol) was dissolved in 60 ml of diethyl ether, and added very slowly via a syringe pump over two days. Saturated aqueous sodium bicarbonate was added, and the organic phase was separated. The aqueous phase was extracted again with 2×100 ml of diethyl ether. The combined organic phases were dried over sodium sulfate, and evaporated. The thick residue was treated with 200 ml of pentane, and stirred vigorously until a yellow precipitate is obtained. The precipitate was filtered, and dried to afford the title product 5 in 69 % yield (8.5 g). mp = 145–154 °C (dec); 1H NMR (500 MHz, CDCl3) δ=7.19 (s, 3H), 6.22 (s, 1H), 1.82 (s, 9H), 1.80 (s, 9H), 1.77 (s, 9H), 1.70 (s, 9H); 13C (125 MHz, CDCl3) δ=139.2, 138.3, 137.8, 137.3, 131.9, 118.2, 83.7, 64.1, 63.4, 34.8, 32.3, 29.2, 27.6
- 32.Tris-(8-tert-butoxycarbonyl-2,2,6,6-tetramethyl-benzo[1,2-d;4,5-d′]bis[1,3]dithiol-4-yl)-methanol (6). To a solution of 5 (8.5 g, 9.6 mmol) in 40 ml of benzene and TMEDA (14.5 ml, 96 mmol) was added slowly n-butyl lithium (38.4 ml 2.5 M in hexanes, 96 mmol). The mixture was stirred for 2 hours at room temperature. The resulting mixture was added slowly via syringe to solid di-tert-butyl-dicarbonate (DiBoc, 136 g, 0.62 mol), which was previously charged in a 1L dry flask, and placed at 0 °C. The reaction mixture was allowed to reach room temperature, and stirred for 2 days. A reflux condenser was placed on the reaction flask, and methanol was added in small portions on top, via the condenser, until no more reaction was observed. The mixture was evaporated under high vacuum, treated with 150 ml of methanol, and the yellow precipitate was filtered, and washed with small portions of methanol. Compound 6 was obtained in 44% yield (5 g). mp = 200–210 °C (dec); 1H NMR (500 MHz, CDCl3) δ=6.72 (s, 1H), 1.78 (s, 9H), 1.75 (s, 9H), 1.68 (s, 45H); 13C (125 MHz, CDCl3) δ=165.4, 141.2, 140.9, 140.3, 139.1, 133.8, 129.6, 122.9, 115.4, 84.2, 60.8, 34.0, 31.1, 29.3, 28.6, 28.4, 28.3; MS (ESI, [M+Na]+): 1207.1218 (observed), 1207.1198 (calcd).
- 33.Bobko AA, Dhimitruka I, Zweier JL, Khramtsov VV. J Am Chem Soc. 2007;129:7240. doi: 10.1021/ja071515u. [DOI] [PubMed] [Google Scholar]
- 34.Deng Y, Petryakov S, He G, Kesselring E, Kuppussamy P, Zweier JL. J Mag Res. 2007;185:283. doi: 10.1016/j.jmr.2007.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.