

Abstract
Murine models indicate that interleukin-10 (IL-10) can suppress viral clearance, and interventional blockade of IL-10 activity has been proposed to enhance immunity in chronic viral infections. Increased IL-10 levels have been observed during HIV infection and IL-10 blockade has been shown to enhance T-cell function in some HIV-infected subjects. However, the categories of individuals in whom the IL-10 pathway is up-regulated are poorly defined, and the cellular sources of IL-10 in these subjects remain to be determined. Here we report that blockade of the IL-10 pathway augmented in vitro proliferative capacity of HIV-specific CD4 and CD8 T cells in individuals with ongoing viral replication. IL-10 blockade also increased cytokine secretion by HIV-specific CD4 T cells. Spontaneous IL-10 expression, measured as either plasma IL-10 protein or IL-10 mRNA in peripheral blood mononuclear cells (PBMCs), correlated positively with viral load and diminished after successful antiretroviral therapy. IL-10 mRNA levels were up-regulated in multiple PBMC subsets in HIV-infected subjects compared with HIV-negative controls, particularly in T, B, and natural killer (NK) cells, whereas monocytes were a major source of IL-10 mRNA in HIV-infected and -uninfected individuals. These data indicate that multiple cell types contribute to IL-10–mediated immune suppression in the presence of uncontrolled HIV viremia.
Introduction
Persistent viral infection often results in T-cell impairment due to combined effects of changes in antigen-specific lymphocytes and antigen-presenting cell (APC) populations.1–4 Recent studies have highlighted the role of inhibitory immunoregulatory pathways in T-cell exhaustion during chronic viral infection. For example, the cosignaling molecule PD-1 has been shown to mediate a reversible dysfunction of virus-specific cytotoxic T lymphocytes (CTLs) during chronic lymphocytic choriomeningitis virus (LCMV) infection in mice,5,6 as well as during HIV,7–9 hepatitis B virus (HBV),10 and hepatitis C virus (HCV)11,12 infection in humans. Besides PD-1, other surface molecules can also mediate reversible T-cell dysfunction in chronic HIV infection, as shown by recent reports of the involvement of cytotoxic T lymphocyte–associated antigen 4 (CTLA-4) in HIV-specific CD4 T-cell impairment13 and TIM-3 in HIV-specific CTL dysfunction.14 Studies in murine models suggest that other cosignaling molecules expressed on the cell surface are also involved.6
Soluble factors, such as chemokines and cytokines, also regulate T-cell function.15 Among these soluble factors, the cytokine interleukin 10 (IL-10) has been shown to play a critical role in the balance between immunopathology and protective responses in infectious diseases (reviewed in Couper et al16 and Moore et al17). IL-10 is a pleiotropic cytokine that can be produced by multiple cell populations, with diverse effects on many hematopoietic cell types. The ability of IL-10 to inhibit cytokine production by T cells and natural killer (NK) cells is thought to be largely indirect, by alteration of monocyte/macrophage function.18 IL-10 decreases major histocompatibility complex (MHC) class II and CD80/CD86 expression on monocytes and macrophages and limits production of proinflammatory cytokines. However, IL-10 can also stimulate the proliferation of B cells and enhance their maturation into plasma cells.19 Inflammatory bowel disease and other excessive inflammatory responses occurring in IL-10−/− mice indicate that IL-10 is critically involved in limiting inflammatory responses in vivo.20,21 The impact of IL-10 on host immunity in the setting of infectious diseases is complex. IL-10 knockout or blockade can enhance resistance to infection with pathogens such as Listeria monocytogenes,22 Mycobacterium avium,23 and Trypanozoma cruzi.24 IL-10 plays a key role in facilitating establishment of chronic Leishmania major infection.25 However, absence of IL-10 signaling results in severe and often fatal immunopathology in Toxoplasma gondii,26 malaria,27 and herpes simplex virus28 infections. These studies indicate that a delicate balance between proinflammatory mechanisms and dampening of excessive immune activation through IL-10 is critical for successful clearance of a pathogen without harm to the host.
Recent studies in the murine LCMV model29,30 have shed new light on the role of IL-10 in the establishment of chronic viral infection. Treating mice with an IL-10Rα blocking antibody shortly after acute infection, or infecting IL-10−/− mice, resulted in enhancement of T-cell responses and rapid viral clearance of an LCMV strain that is usually associated with viral persistence.29,30
These findings in murine models suggest that manipulation of the IL-10 pathway might be used to treat chronic infections in humans. Elevated IL-10 levels (IL-10 protein and/or mRNA) have been observed for several chronic infectious diseases in humans, including visceral leishmaniasis,31 leprosy,32 and tuberculosis33 (reviewed in Couper et al16 and Moore et al17). The observation that the herpesviruses Epstein-Barr virus (EBV)34 and cytomegalovirus (CMV)35 encode for IL-10 analogues suggest that IL-10 can indeed facilitate viral persistence in humans. In chronic HIV infection, the concentration of IL-10 protein in plasma increases over time.36 Research by Clerici et al37 first indicated that mitogen-stimulated PBMCs of HIV-infected individuals produced more IL-10 than did PBMCs of HIV-negative controls. Furthermore, blockade of the IL-10 pathway with an IL-10 blocking antibody enhanced some proliferative responses to HIV Env peptides. Further studies showed that IL-10 blockade could restore Env-specific T-cell proliferative responses in subjects with relatively preserved CD4 T-cell counts, but was lost in individuals with advanced HIV infection38 and that increased IL-10 levels correlated with a more advanced disease stage and the presence of synctium-inducing virus.39
Based upon the recent LCMV results,29,30 interventional studies to block IL-10/IL-10Rα have been proposed as a strategy to enhance underlying immunity in chronic viral infections, such as HIV, and as a potential adjuvant to vaccination regimens.40 However, prior to considering such trials in humans, a better understanding of the role of IL-10 in HIV infection is essential. We therefore undertook a detailed study of IL-10 activity and IL-10 expression in a cohort of 80 individuals. First, we assessed the ability of antibody-mediated blockade of IL-10Rα to restore virus-specific CD4 and CD8 T-cell proliferative capacity in HIV-infected persons and to augment cytokine secretion by HIV-specific CD4 T cells. To define the categories of subjects who respond best to IL-10 blockade, we associated differences with clinical markers of disease status. We also compared the impact of IL-10 blockade on T-cell responses specific for HIV and for CMV, a virus that causes lifelong infection but is usually controlled by the immune system. Second, we quantified IL-10 protein in plasma and spontaneous mRNA expression in PBMCs from HIV-infected individuals at various stages of disease in cross-sectional and longitudinal studies. Finally, we identified cell populations in PBMCs from uninfected and HIV-infected individuals that expressed IL-10 mRNA. Based on the results of these studies, we conclude that IL-10 production correlates with HIV disease progression, is up-regulated in multiple cell types, and acts as a reversible mechanism to suppress both HIV-specific CD4 and CD8 T-cell responses in individuals with uncontrolled viremia.
Methods
Study subjects
Chronically HIV-infected individuals were enrolled at the Massachusetts General Hospital (MGH) or Lemuel Shattuck Hospital, in Boston, MA. Subjects identified during acute HIV infection were enrolled at the Jessen Praxis Clinic, in Berlin, Germany. These studies were approved by the respective institutional review boards of each center. Untreated persons and individuals on antiretroviral (ARV) therapy were enrolled in the study (range of viral loads: < 50 to 750 000 HIV RNA copies/mL plasma). “Elite controllers” were defined as individuals with a plasma viremia below the limit of detection of standard HIV-RNA assays (< 50 or 75 copies/mL) in the absence of therapy. After written informed consent, in accordance with the Declaration of Helsinki, whole blood was collected from each subject, plasma was collected, and peripheral blood mononuclear cells (PBMCs) were isolated by density centrifugation (Ficoll-Histopaque; Sigma-Aldrich) according to standard protocols. PBMCs were used as fresh or frozen cells, as indicated in the results.
HLA tissue typing
DNA was extracted from peripheral blood cells using Puregene DNA Isolation Kit for blood (Gentra Systems). High- and intermediate-resolution HLA class I typing was performed by sequence-specific polymerase chain reaction (PCR) according to standard procedures.
Proliferation assays with IL-10Rα blockade
For measurement of CD4 T-cell responses, 5- (and 6-)carboxyfluorescein diacetate succinimidyl ester (CFSE)–based proliferation assays were performed as described previously.7,13 Freshly isolated PBMCs were depleted of CD8 T cells during Ficoll preparation using RosetteSep CD8 Depletion reagents (StemCell Technologies) to maximize sensitivity of the assays.13,41 Cells were labeled with 1.25 μM CFSE dye (Molecular Probes) for 7 minutes, washed, and incubated with antigen in the presence of IL-10 receptor alpha chain (IL-10Rα) blocking antibody (clone 37607; R&D Systems) or IgG1 isotype control antibody at 10 μg/mL. Conditions of stimulation tested were no antigen, phytohemagglutinin (PHA; 5 μg/mL; Sigma-Aldrich), recombinant HIV p24 protein (10 μg/mL; Protein Sciences), or CMV lysate (1 μg/mL; Advanced Biotechnologies). Cells were incubated for 7 days in RPMI 1640 medium (Invitrogen) supplemented with 10% human AB serum (Gemini Bioproducts) at 37°C, 5% CO2, stained with antibodies (CD3-phycoerythrin [PE]-Cy7, CD8–Alexa 700, and CD4-allophycocyanin [APC]; BD Biosciences), and analyzed by flow cytometry (LSRII flow cytometer; BD Biosciences).
CD8 T-cell proliferation was determined as described previously.7 PBMCs were left undepleted, and peptides corresponding to human leukocyte antigen (HLA)–matched optimal CD8 T-cell epitopes were used as HIV antigens (0.2 μg/mL; MGH Peptide Synthesis Core Facility). No antigen and PHA were included as controls. At the end of the 7-day incubation period, cells were stained with the relevant PE- or APC-labeled HLA class I tetramer (Beckman Coulter) or pentamer (ProImmune) refolded with the respective CD8 T-cell epitopes (listed in supplemental Table 1, available on the Blood website; see the Supplemental Materials link at the top of the online article). After 30 minutes of incubation at 37°C, cells were washed and stained with surface antibodies (CD3-PE-Cy7, CD8-APC-H7, and CD4–Alexa 700; BD Biosciences) before acquisition (LSRII; BD Biosciences).
Cytokine secretion assays with IL-10Rα blockade
For measurement of CD4 T-cell responses, freshly isolated PBMCs were depleted of CD8 T cells during Ficoll preparation as for the proliferation assays. Cells (106) were then incubated with antigen in the presence of IL-10Rα blocking antibody (clone 37607; R&D Systems) or IgG1 isotype control antibody at 10 μg/mL. After 48 hours of incubation at 37°C, 5% CO2, supernatants were collected and IFN-γ and IL-2 levels were detected using a multiplex immunoassay (Milliplex beads; Millipore) on a Bio-Plex 200 array system (Bio-Rad Laboratories).
Plasma IL-10 detection
Plasma IL-10 cytokine concentration was determined by luminex assay using the Bio-Plex Precision Pro Human Cytokine 10-Plex Panel (Bio-Rad Laboratories) according to the manufacturer's instructions on a Bio-Plex 200 array system (Bio-Rad Laboratories).
IL-10 mRNA detection
PBMCs were resuspended in RNA lysis solution (RLT buffer; QIAGEN) containing 1% β-mercaptoethanol (Sigma-Aldrich). Total RNA was extracted using the RNeasy Mini kit (QIAGEN). Complementary DNA (cDNA) was generated from 0.5 μg total RNA using 0.5 μM oligo dT primer and the MultiScribe reverse transcriptase enzyme and reagents (Applied Biosystems) for 60 minutes at 42°C. A 1:10 dilution of cDNA was analyzed by quantitative PCR using Brilliant II SYBR green enzyme mix (Stratagene) on an Mx3005P real-time PCR instrument (Stratagene; 40 cycles at 95°C for 15 seconds, 60°C for 15 seconds, and 72°C for 15 seconds). To minimize detection of genomic DNA, gene-specific primers were designed to recognize spliced transcripts for human IL-10 gene (IL10/NM_000572) (F, cgtggagcaggtgaagaatg; R, agagccccagatccgatttt; 195 nt) and the control gene human hypoxanthine guanine phosphoribosyltransferase 1 (HPRT/NM_000194) (F, gaccccacgaagtgttggat; R, ggcgatgtcaataggactcca; 209 nt). Cycle threshold (Ct) values for each reaction were compared with a standard curve containing a known copy number of plasmid DNA encoding each product. Results are shown as ratio of IL-10 copies per HPRT copies for each sample.
Isolation of PBMC subsets with cell sorter
Two panels of antibodies were used to isolate live cells subsets according to combinations of surface markers. In the first panel, PBMCs were stained with antibodies CD3-PE-Cy7, CD4-PerCp-Cy5.5, CD8–fluorescein isothiocyanate (FITC), CD14-PacBlue, CD19-APC-Cy7, and CD56-APC (BD Biosciences). A second panel was used to isolate dendritic cell (DC) subsets. PBMCs were stained with a lineage exclusion antibody cocktail: FITC, CD11c-PE, CD123-PE-Cy5, and HLA-DR-APC-Cy7 (BD Biosciences). T-cell subsets, B cells, NK cells, monocytes, and DC subsets were sorted as outlined in supplemental Figure 3 on a FACS Aria (BD Biosciences) equipped for biohazardous material. The FACS Aria was operated at 70 pounds per square inch with a 70-μm nozzle. For all populations, 20 000 cells were collected directly into RLT lysis buffer. IL-10 mRNA expression was assessed by reverse-transcription (RT)–PCR as described for IL-10 mRNA detection in unseparated PBMCs.
Statistical analysis
Flow cytometry data were analyzed with FlowJo software version 8.7 for Apple Macintosh (TreeStar). All statistical analyses were nonparametric and were performed using Prism 4.0 (GraphPad Software). Comparisons of cellular proliferation responses between groups were made using the Mann-Whitney U test. Pairwise comparisons were verified using the Wilcoxon matched pairs test. Correlation coefficients were calculated using the Spearman rank sum test. Analysis of IL-10 protein and mRNA expression among 3 or more groups was done using the Kruskall-Wallis test, followed by the Dunn posttest. IL-10 expression in cell subsets from HIV-negative and HIV+ patients was evaluated using the Mann-Whitney U test. All tests were 2-tailed and P values less than .05 were considered significant.
Results
Blockade of the IL-10/IL-10Rα pathway restores proliferative and effector CD4 T-cell functions in individuals with uncontrolled viremia
To define the role of IL-10 in virus-specific CD4 T-cell dysfunction, we first examined the impact of inhibition of the IL-10 pathway on HIV-specific T-cell proliferation in a cross-sectional study of 45 HIV-infected individuals. These included elite controllers who spontaneously suppressed HIV replication and chronically infected subjects—either viremic off therapy or aviremic on ARV therapy. PBMCs were labeled with CFSE and stimulated with either recombinant HIV p24 protein or CMV lysate for 7 days in the presence of IL-10Rα blocking antibody or an isotype control (Figure 1A). Blockade of IL-10Rα significantly enhanced CD4 T-cell proliferation to HIV p24 in subjects with ongoing viral replication (P < .001; Wilcoxon; Figure 1B), consistent with previous results obtained using HIV Env peptides in a smaller number of subjects.37 Enhancement of HIV-specific CD4 T-cell proliferative response by IL-10Rα blockade was restricted to viremic individuals and was not observed in aviremic subjects, including elite controllers and subjects on successful ARV therapy (P = .001, Kruskall-Wallis; Figure 1C). A significant positive correlation was observed between viral load at the time of the proliferation assay and fold increase of HIV p24–specific proliferation in the presence of blocking antibody (P < .001, Spearman; Figure 1D). No significant association was seen between absolute CD4 T-cell count and p24-specific proliferation after IL-10Rα blockade (P > .05, Spearman; Figure 1E). To determine whether the impact of IL-10Rα blockade was restricted to HIV-specific T-cell responses, we assessed CMV-specific CD4 T-cell responses in a subgroup of HIV+ subjects. IL-10Rα blockade enhanced CMV-specific CD4 T-cell proliferation in 17 of 30 individuals (supplemental Figure 1B); however, this increase was not statistically significant for the entire group. Furthermore, we observed no significant difference in enhancement of CMV-specific CD4 T-cell responses between HIV viremic and aviremic individuals (P > .05; Mann-Whitney) and no correlation between the impact of IL-10Rα blockade and HIV viral load (P > .05; Spearman). In addition to assessing the restoration of proliferative capacity, we also examined the effect of IL-10Rα blockade on cytokine secretion by HIV-specific CD4 T cells. PBMCs were depleted of CD8 T cells and then cultured in the absence of antigen or stimulated with an HIV Gag peptide pool in the presence of an isotype control antibody or IL-10Rα blocking antibody. After 48 hours, supernatants were collected and the secreted IFN-γ and IL-2 levels were measured (Figure 2A-C). Overall, there was a significant increase in the amounts of both IFN-γ (Figure 2D; P = .004, Wilcoxon) and IL-2 (Figure 2E; P = .026, Wilcoxon) that were produced after IL-10Rα blockade. These data demonstrated that blockade of the IL-10 pathway not only restored proliferative responses, but also augmented direct effector functions of HIV-specific CD4 T cells.
Figure 1.

IL-10Rα blockade restores HIV-specific CD4 T-cell function in individuals with uncontrolled viremia. (A) CD4 T-cell proliferation was measured by flow cytometry of CFSE-labeled CD8 T cell–depleted PBMCs incubated with no antigen or HIV p24, plus isotype control antibody or IL-10Rα antibody. Numbers in upper left quadrants indicate the percentage of CD3+CD4+CFSElow cells in each condition. Blockade of the IL-10 pathway significantly increased proliferation of p24 antigen–specific CD4 T cells, as shown in this representative sample. (B) Statistical analysis of CFSE results obtained in a cohort of 27 viremic subjects with chronic untreated infection (P < .001; Wilcoxon-matched pairs test). (C) Statistical comparison of impact of IL-10Rα blockade on HIV p24–specific CD4 T-cell proliferation in chronically HIV-infected subjects with viral loads suppressed to less than 50 RNA copies by antiretroviral therapy (ARV; n = 9), elite controllers (ELITE; n = 6), and chronically infected individuals with viral load of greater than 1000 RNA copies/mm3 (UNTREATED; n = 24) suggested a significant difference among groups (P = .001; Kruskall Wallis test, followed by Dunn posttest for paired comparisons). The vertical axis (IL-10 proliferation index) corresponds to the ratio of the fraction of proliferating (%CD3+CD4+CFSElow) cells in the presence of the IL-10Rα blocking antibody versus isotype control. Short horizontal bars indicate median proliferation index. (D,E) Statistical analysis of data on untreated subjects in panel C (elite controllers and chronic untreated subjects) indicated a significant correlation between viral load (R = 0.5636; P < .001) in panel D, but not CD4 count (P > .05; E) with the effect of IL-10Rα blockade on HIV-specific CD4 T-cell proliferation (Spearman rank sum test).
Figure 2.

IL-10-Rα blockade enhances cytokine secretion by HIV-specific CD4+ T cells. (A-C) Cytokine secretion by HIV-specific CD4 T-cell proliferation was measured by multiplex immunoassay in supernatants of CD8 T cell–depleted PBMCs incubated with no antigen or an HIV Gag peptide pool, plus isotype control antibody or IL-10Rα antibody. Blockade of the IL-10 pathway increased IFN-γ and IL-2 cytokine secretion by HIV-specific CD4 T cells, as shown in these 3 representative subjects. (D-E) Statistical analysis of data on 14 untreated subjects indicated a significant increase of both IFN-γ (P = .004; D) and IL-2 (P = .026; E) in the presence of IL-10Rα blockade (Wilcoxon matched pairs test).
Blockade of the IL-10/IL-10Rα pathway restores HIV-specific CTL function in individuals with uncontrolled viremia
Because of the link between CTL function and control of viremia in HIV infection (reviewed in Deeks and Walker42), we next investigated the effect of IL-10Rα blockade on HIV-specific CTL responses. We stimulated PBMCs with optimal HIV epitopes in the presence of anti–IL-10Rα antibody or isotype control. After a 7-day incubation, cells were stained with the relevant HIV class I tetramer or pentamer to identify the frequency of epitope-specific CTLs. Two representative examples are shown in Figure 3A. Similar to HIV-specific CD4 T-cell responses, the fold increase of HIV-specific CTLs in the presence of IL-10Rα blocking antibody versus isotype control was significantly higher in viremic individuals than in aviremic persons (P = .009; Mann-Whitney; Figure 3B). Furthermore, we observed a significant correlation between viral load and increase of CTL responses in untreated subjects after IL-10 blockade (P = .013, Spearman; Figure 3C). Because these assays were done after removal of patient serum that may contain high levels of IL-10 protein, our data show that in vitro expression of IL-10 by PBMCs directly impacts the proliferative capacity of T cells.
Figure 3.

IL-10Rα blockade restores HIV-specific CTL function in individuals with uncontrolled viremia. (A) Flow cytometry was used to assess proliferation of HIV-specific HLA class I tetramer–labeled CD8 T cells from 2 subjects incubated with the cognate HIV epitope, plus isotype control antibody or IL-10Rα blocking antibody. Representative data from 2 subjects is shown. Numbers in upper right quadrants indicate the percentage of CD3+CD8+Tetramerhigh cells for each condition. (B) Statistical comparison of impact of IL-10Rα blockade on HIV p24–specific Tet+ CD8 T-cell proliferation in chronically HIV-infected subjects with undetectable viral loads (AVIR; n = 7), and chronically infected individuals with viral loads of greater than 1000 RNA copies/mm3 (VIR; n = 13) demonstrated a significant difference between these groups (P = .009; Mann-Whitney U test). The vertical axis (IL-10 proliferation index) corresponds to the ratio of the fraction of Tet+ cells (%CD3+CD8+Tethigh) cells in the presence of the IL-10Rα blocking antibody versus isotype control. Short horizontal bars indicate median proliferation index. (C) Statistical analysis of the data on subjects in panel B indicated a significant correlation between untreated viral load and the effect of IL-10Rα blockade on HIV-specific CD8 T-cell proliferation (R = 0.5445, P = .013; Spearman rank sum test).
IL-10 protein and mRNA levels correlate with HIV viremia and response to IL-10Rα blockade
Since antibody blockade experiments demonstrated that the negative impact of IL-10 was most apparent in individuals displaying higher HIV viral load, we assessed cytokine expression in HIV-infected subjects presenting with different levels of plasma viremia. IL-10 protein was measured in plasma samples collected from 35 untreated HIV-infected individuals. We observed that IL-10 concentration correlated strongly with HIV viral load (R = 0.6017; P = .001, Spearman; Figure 4A).
Figure 4.

IL-10 expression correlates with HIV viremia and response to IL-10Rα blockade. The level of IL-10 protein in plasma and mRNA expression in bulk PBMCs were examined in 35 untreated HIV+ individuals. IL-10 protein concentration (A) and IL-10 mRNA expression values (B) correlated significantly with plasma viral load measurements (R = 0.6017 and R = 0.6754, respectively). Next, the impact of IL-10Rα blockade on HIV p24–specific CD4 T cells was correlated with IL-10 protein or mRNA results. The IL-10 proliferation index, defined as the ratio of the fraction of proliferating (%CD3+CD4+CFSElow) cells in the presence of the IL-10Rα blocking antibody versus isotype control, indicated a significant correlation between an individual's responsiveness to IL-10 pathway blockade and both plasma IL-10 protein (R = 0.5324, P = .034) (C) or PBMC IL-10 mRNA (R = 0.6951, P < .001). (D) Correlation coefficients suggest that IL-10 mRNA expression in PBMCs is a better predictor of response to IL-10 blockade than plasma IL-10 protein concentration. All statistical analyses used the Spearman rank sum test.
To assess cytokine gene expression, we measured IL-10 mRNA in PBMCs using quantitative RT-PCR. IL-10 mRNA expression in PBMCs correlated significantly with HIV plasma viral load (Figure 4B; P = .001, Spearman). In a subset of 20 untreated HIV-infected individuals, IL-10 mRNA expression in PBMCs correlated well with IL-10 protein levels in serum (P = .001, Spearman; data not shown). Our results for IL-10 protein and mRNA expression are consistent with previous studies.36,43–47 However, the correlation between IL-10 expression and response to IL-10 blockade has not been defined. We observed that plasma IL-10 protein levels (Figure 4C) and IL-10 mRNA expression in PBMCs (Figure 4D) both significantly correlated with the CD4 T-cell proliferative response after IL-10Rα blockade (R = 0.5234 and R = 0.6951, respectively; Spearman).
Relationships between IL-10 mRNA expression and disease stage
To further examine the impact of HIV infection and treatment status on IL-10 expression, we measured IL-10 mRNA in PBMCs collected from 10 healthy HIV-negative subjects and 4 cohorts of chronically HIV-infected individuals (Figure 5): 15 elite controllers; 20 viremic, untreated subjects; 12 persons with uncontrolled viremia on ARV therapy; and 11 individuals with successful ARV treatment. IL-10 mRNA levels differed among these groups (P = .002; Kruskal-Wallis), with a significant elevation of IL-10 mRNA in individuals with ongoing viremia, either untreated or on ARV, compared with elite controllers and uninfected controls (P < .01 for each, comparison Dunn posttest; Figure 5). In contrast, IL-10 mRNA expression was similar in HIV controllers and uninfected individuals. Intermediate levels of IL-10 mRNA were observed in subjects with viral loads lower than 50 copies/mL on ARV therapy, compared with the other 4 groups. Taken together, these results suggest that the reduced IL-10 expression observed upon successful ARV therapy is a consequence of decreased HIV viral load and does not result from a direct effect of ARV drugs on IL-10 expression.
Figure 5.

Relationships between IL-10 mRNA expression and disease stage. IL-10 mRNA levels in bulk PBMCs were measured in HIV-negative subjects (n = 10; HIV-neg), HIV-infected elite controllers (n = 15; ELITE), HIV+ untreated patients (n = 20; UNTREATED), and antiretroviral drug-treated individuals, which included some subjects with suppressed viremia less than 50 copies/mL on HAART (n = 11; AVIR ARV) and others who remained viremic despite therapy (n = 12; VIR ARV). A significant difference was seen among these groups (P < .001; Kruskal Wallis test), reflecting a significant increase in IL-10 expression in HIV+ untreated and viremic-treated individuals compared with HIV-negative and elite controller subjects (P < .01 for all, Dunn posttest).
Variations in IL-10 expression during periods of rapid viral load changes
Previous studies have indicated that plasma IL-10 protein levels are elevated during acute HIV infection.48 To better understand the link between HIV viremia and IL-10 expression during non–steady-state disease, we first examined IL-10 protein concentration in longitudinal samples collected from 3 individuals, beginning in acute infection (Figure 6A-C). Substantial levels of plasma IL-10 protein were detected at the earliest time point available; they declined rapidly during the first 6 months after infection in the 2 untreated subjects (Figure 6A-B) as well as in the treated individual (Figure 6C), suggesting that resolution of acute infection was associated with a decline in plasma IL-10, even in the absence of viral control. This analysis was extended to a total of 10 individuals for whom acute/early (within 30 days after infection) and chronic (6-12 months after infection) samples were available, and we saw a significant decline in IL-10 protein concentration at the later time point (P = .004, Wilcoxon; Figure 6D). Next, we analyzed IL-10 mRNA and protein levels in longitudinal samples collected from 2 individuals who discontinued ARV therapy (Figure 6E-F). In the absence of treatment, we observed a rapid rise in HIV viremia, but only a gradual increase of plasma IL-10 protein and mRNA expression in PBMCs for both individuals. Reinitiation of ARV treatment resulted in a marked reduction of both IL-10 protein and mRNA, suggesting that the pathogenic process responsible for IL-10 induction is at least partially reversible.
Figure 6.

Longitudinal analysis during acute infection and the impact of therapy on IL-10 suggest an indirect link with plasma viremia. Plasma IL-10 protein concentration was measured in 3 individuals identified during acute/early HIV infection. Longitudinal samples indicated that IL-10 protein levels (solid line) were initially high, and decreased over time in 2 untreated (A-B) as well as 1 treated subject (C), corresponding with a decline in HIV viral load (dashed line). A statistically significant decline in IL-10 protein (P = .004; Wilcoxon matched pairs test) in panel D was observed in a cross-sectional analysis of 10 HIV+ individuals captured at acute/early (within the first 30 days after infection) and chronic (6-12 months after infection) time points. IL-10 protein and mRNA expression were analyzed in 2 chronically infected individuals who interrupted antiretroviral therapy. Initially, both subjects displayed undetectable plasma viral loads while on ARV, which increased substantially in the absence of treatment (E-F). Plasma IL-10 protein concentration (●) as well as IL-10 mRNA expression (□) increased slowly over time in both individuals while off of therapy, but declined after reinitiation of suppressive antiretroviral treatment. Time points on ARV therapy are indicated by shaded regions in panels B-C and E-F.
IL-10 is up-regulated by multiple PBMC subsets in HIV-infected individuals
Monocytes have been reported to be major producers of IL-10 in HIV-infected individuals,37 but numerous cell types can express this cytokine (reviewed in Couper et al16 and Moore et al17). To better identify IL-10–producing cell populations, we first used antibody-conjugated magnetic beads to isolate cell subsets in PBMCs from HIV-infected subjects and healthy uninfected controls. We measured IL-10 mRNA expression using quantitative RT-PCR and observed IL-10 mRNA up-regulation in all cellular subsets isolated from HIV-infected subjects versus controls (supplemental Figure 2), which were statistically significant for total PBMCs and cellular populations isolated using antibodies recognizing CD3, CD11b, CD11c, CD14, CD16, CD19, and CD56 (all P < .05; Mann-Whitney). These results suggest that in addition to CD14+ monocytes, several other cell types contribute to the increased IL-10 expression observed during chronic HIV infection.
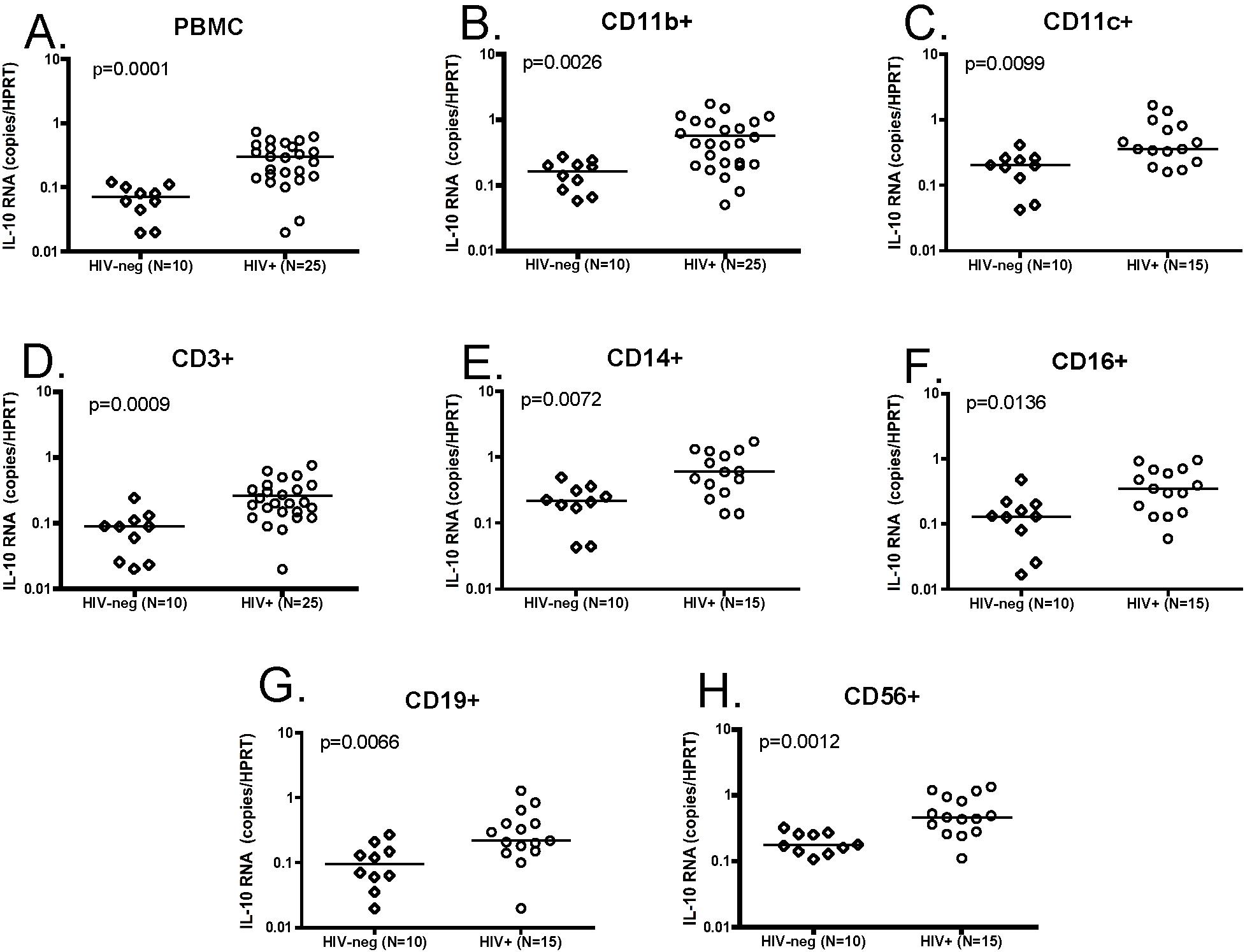
To confirm findings obtained using bead-sorted cell subsets and to define the IL-10–producing populations with great accuracy, we used a live-cell fluorescence-activated cell sorting (FACS) sorting approach to isolate CD123+ plasmacytoid dendritic cells (pDCs), CD11c+ myeloid dendritic cells (mDCs), CD14+ monocytes, CD4+ and CD8+ T cells, CD19+ B cells, and CD56+ NK cells from 10 HIV-infected individuals and 9 uninfected controls (for gating strategies, see supplemental Figure 3). Despite major differences in the frequencies of the cell populations sorted (range: < 0.1% for DC subsets to > 10% for T-cell subsets), purities of these sorted populations were comparable and consistently higher than 98%. IL-10 mRNA expression was analyzed in PBMCs and in each purified subset using quantitative RT-PCR (Figure 7). IL-10 mRNA levels in the total PBMCs from these HIV-infected individuals were significantly elevated relative to healthy controls (P = .028, Mann Whitney). In addition, we observed a significant elevation of IL-10 in CD14+ monocytes (P = .035), CD4+ T cells (P = .001), CD8+ T cells (P = .002), CD19+ B cells (P < .001), and CD56+ NK cells (P = .004) in HIV+ individuals (Figure 7D-H; all statistics: Mann Whitney). In contrast, we did not see a significant difference in IL-10 expression for the CD123+ pDC subset (P = .161; Figure 7B) in HIV-infected individuals compared with HIV-negative controls, and observed a decline in IL-10 expression in CD11c+ mDCs (P = .028). The difference between IL-10 mRNA levels observed in CD11c+ isolated by beads and CD11c+ DCs sorted by FACS is likely due to the mixed populations in bead-sorted cells that contain a majority of CD11c-expressing monocytes and a minority of CD11c+ DCs. These data demonstrate that IL-10 expression is induced in multiple PBMC subsets in HIV-infected individuals and identify potential candidate suppressor cell populations.
Figure 7.

Broad expression of IL-10 mRNA in FACS-sorted cell subsets from HIV-infected subjects. CD11c+ myeloid DCs, CD123+ plasmacytoid DCs, CD14+ monocytes, CD4+ T cells, CD8+ T cells, CD19+ B cells, and CD56+ NK cells were isolated from total PBMCs using live cell FACS sorting from 9 HIV-negative and 10 HIV-infected individuals, as described in supplemental Figure 3. IL-10 mRNA expression was determined in PBMCs and each subset by quantitative RT-PCR, and values are presented as IL-10 copies relative to the HPRT housekeeping gene (A-H). Significant up-regulation of IL-10 mRNA was observed in HIV+ individuals compared with HIV-negative controls for total PBMCs (P = .028) in panel A, CD14+ monocytes (P = .035) in panel D, CD4+ T cells (P = .001) in panel E, CD8+ T cells (P = .002) in panel F, CD19+ B cells, (P < .001) in panel G, and CD56+ NK cells (P = .004) in panel H. A significant reduction in IL-10 expression was observed in CD11c+ mDCs (P = .038) (C), whereas no change was seen in CD123+ pDCs (P = .161) in panel B. All statistics were calculated using the Mann-Whitney U test.
Discussion
In this study, we examined IL-10 expression and the ability of this cytokine to suppress HIV-specific T-cell function in HIV-infected individuals. Our results indicate that IL-10 mRNA expression is increased in the setting of chronic uncontrolled HIV infection and that IL-10 mRNA expression levels correlate with plasma viremia in infected persons. Increased IL-10 mRNA levels were identified in multiple different hematopoietic cells, and IL-10Rα blockade restored not only HIV-specific CD4 cell proliferation but also antigen-specific CD8 T-cell proliferation, although these effects were seen only in viremic subjects, as well as cytokine secretion by HIV-specific CD4 T cells. Both IL-10 mRNA expression and IL-10plasma levels were reduced through successful ARV treatment, indicating a direct effect of viral antigen load on IL-10 production. These data show that IL-10 contributes to a reversible T-cell dysfunction in HIV infected persons, and that viral antigen is a major driver of the increased levels of IL-10 observed.
Blockade of the IL-10 pathway significantly increased HIV-specific CD4 and CD8 T-cell proliferation, and also augmented effector T-cell functions, as shown by the enhanced secretion of the cytokines IFN-γ and IL-2 by antigen-specific CD4 T cells upon IL-10Rα blockade. Although less consistent, IL-10Rα blockade also increased CMV-specific CD4 T proliferation in HIV-infected individuals. These data show that IL-10 contributes to a broad reversible T-cell dysfunction. Significant positive correlations were observed between the fold increase in HIV p24–specific proliferation after blockade and plasma viral load or IL-10 mRNA. The variable enhancement of T-cell responses after IL-10Rα blockade observed in individuals with similar HIV viremia is consistent with a significant heterogeneity in the degree of T-cell exhaustion and activity of different inhibitory pathways among HIV-infected subjects. It is notable that we observed enhancement of HIV-specific T-cell proliferative responses upon IL-10Rα blockade only in individuals with uncontrolled viral replication. Elite controllers and individuals who suppressed viral replication while on successful ARV treatment failed to respond to IL-10Rα blockade. An association between IL-10 and viremia has been observed previously,39 and this may help to explain inconsistent results of studies where individuals were not stratified on the basis of viral load.49
Consistent with results of IL-10Rα blockade, we observed that both plasma IL-10 protein levels and IL-10 mRNA expression in PBMCs were elevated in chronically HIV-infected individuals. A significant positive correlation between IL-10 protein and IL-10 mRNA suggests that IL-10 expression by PBMCs is representative of IL-10 production in cellular compartments that contribute to plasma IL-10. IL-10 protein and mRNA levels were significantly higher in viremic individuals than in subjects with undetectable viral load, and directly correlated with plasma viremia. We performed longitudinal analyses to further address the relationship between viral load and IL-10 up-regulation. Our data from 10 individuals confirm previous work demonstrating elevated plasma IL-10 levels at the time of acute infection,48 concurrent with very high HIV viral load, which declined significantly after resolution of primary infection, regardless of continued viremia. Conversely, our results from 2 additional patients illustrated that interruption of ARV therapy resulted in elevation of HIV viral loads and subsequent increases in IL-10 protein and mRNA levels. Reinstitution of ARV treatment led to a substantial decline in both measures of IL-10. In each of these cases, changes in IL-10 were delayed relative to changes in plasma viremia. Additional studies will be necessary to define whether direct stimulation of the IL-10 pathway in immune cells by viral products, indirect feedback mechanisms due systemic immune hyperactivation, or both, lead to up-regulation of IL-10 in viremic individuals.
These data raise important questions about the cellular sources of IL-10 in viremic individuals. In the murine LCMV model, recent studies by Brooks et al29 and Ejrnaes et al30 reach different conclusions, either favoring the hypothesis that DCs from chronically infected mice induce IL-10 up-regulation by CD4 T cells, or concluding that regulatory T cells are the major source of IL-10 during chronic LCMV infection, respectively. A similar controversy exists for HIV infection in humans as well. Early studies assessing cytokines in culture supernatants of PBMCs stimulated with PHA suggested an up-regulation of IL-10 and a switch from a Th1 to a Th2 profile in T cells.50 This hypothesis was subsequently challenged by other reports.49 Trabattoni et al51 showed that monocytes, T cells, and B cells from HIV-infected individuals produce more IL-10 than HIV-uninfected controls, but IL-10 production by other subsets has been poorly characterized.
Our results provide a detailed assessment of IL-10 mRNA expression by several PBMC subsets. Consistent with previous studies,51 our assays suggest that CD14+ monocytes are a major source of IL-10 in both HIV-infected and HIV-uninfected individuals. However, analysis of highly purified cellular subsets obtained by FACS indicated that CD14+ monocytes showed a less dramatic up-regulation of IL-10 mRNA in HIV-infected subjects than some other subsets examined in our study, including CD19+ B cells and T cells. In addition, CD11c+ myeloid DCs, which are known to produce significant amounts of IL-10 under certain physiological conditions, appeared to produce less IL-10 in chronic HIV-infected individuals compared with uninfected controls. This result is in contrast with data obtained at the time of acute viral infection in the LCMV model29 and suggests that the IL-10–producing cell populations may differ among viral diseases, or perhaps vary in tissue location or change over time after acute infection. Such differences in IL-10–producing subsets may contribute to the broad spectrum of outcomes observed upon blockade of the IL-10 pathway in several murine models of infectious diseases. Previous reports have indicated that up-regulation of IL-10 by monocytes may be due to a direct effect of HIV Env52 and that the Env of different HIV strains could induce various degrees of IL-10 secretion by monocyte-derived dendritic cells (MDDCs).53 However, we found no significant increase of IL-10 mRNA in pDCs and mDCs. The HIV Tat protein has also been implicated in up-regulation of IL-10,54 and soluble Tat may correlate with HIV viral load in untreated infection.55 Additional studies will be necessary to determine whether IL-10–secreting cell populations differ in acute and chronic HIV infections and to define the cell types that are critical for IL-10–mediated T-cell inhibition. In particular, the role of regulatory T cells remains to be better defined. Altogether, our data demonstrate that IL-10 expression by several diverse cell populations, rather than induction of IL-10 by a single cell type, may be a key factor leading to the increase in IL-10 levels observed in HIV-infected subjects.
Interventional studies to block IL-10 activity have been proposed to enhance immunity in chronic human viral infections. Besides its impact on T-cell responses and viral load in established LCMV infection,29,30 blockade of the IL-10 pathway also allowed an otherwise ineffective therapeutic DNA vaccine to stimulate antiviral immunity and enhance viral clearance.40 IL-10 blockade may therefore be a promising adjuvant therapy to boost preventive or therapeutic vaccines. However, caution is needed with such approaches in HIV infection, since the relationship between IL-10 and HIV disease progression is complex. First, IL-10 blockade does not enhance HIV-specific T-cell function in our assays for subjects optimally treated with ARV therapy. If these findings in vitro correlate with activity of the IL-10 pathway in vivo, this may suggest that the potential impact of IL-10 blockade as a therapy would be limited in an era when optimal suppression of viral replication is the goal of patient care. Second, whereas our study, and others, show that IL-10 levels increase with disease progression and can mediate T-cell dysfunction, a genetic polymorphism in the IL-10 promoter that leads to a decrease in IL-10 expression (−592C > A) has been associated with more rapid disease progression in late stages of HIV.56 A second IL-10 promoter polymorphism (−1082A > G) that may increase IL-10 expression has been associated with slower CD4 decline and longer survival in HIV+ individuals.57 Given the crucial role of immune activation in HIV disease progression,58 it is possible that despite its inhibitory effect on T cells, IL-10 has a net beneficial impact during later stages of HIV infection by limiting systemic immune hyperactivation and CD4 T-cell loss. Third, the direct effects of IL-10 on virus production are complicated, as IL-10 has been reported to inhibit HIV replication in monocytes,52,59 but also to induce expression of CCR5 in CD4 T cells,60 which may hasten infection. A placebo-controlled trial investigating the tolerance and impact of recombinant IL-10 therapy on parameters of disease progression in HIV-infected individuals did not show significant changes in either viral load or CD4 T-cell counts during 4 weeks of treatment.61 Additional studies are required to better understand the overall impact of the IL-10 pathway in viremic individuals, which likely combines the detrimental effect of IL-10 on HIV-specific CD4 and CD8 T-cell immunity with a beneficial down-regulation of systemic immune activation. Our data showing IL-10 mRNA up-regulation in multiple cell types of HIV-infected subjects suggest that, beyond T-cell responses, IL-10 could broadly alter innate and adaptive immune functions in HIV infection. It will therefore be important to develop new technical approaches selectively targeting this pathway in specific cellular subsets. Such tools will provide additional insight into HIV pathogenesis and may pave the path toward novel therapeutic interventions to enhance virus-specific immunity.
Supplementary Material
Acknowledgments
We thank the clinical and laboratory staff at the Massachusetts General Hospital and that of The International HIV Controllers Study (http://www.hivcontrollers.org). We thank all study participants for their invaluable role in this project.
This study was supported by the National Heart, Lung and Blood Institute of the National Institutes of Health (Bethesda, MD; R01-HL092565 [D.E.K.]), the Concerned Parents for AIDS Research Foundation (D.E.K., B.D.W.), the Howard Hughes Medical Institute (B.D.W.), the Bill and Melinda Gates Foundation (Seattle, WA), and a gift from the Mark and Lisa Schwartz Foundation.
Footnotes
An Inside Blood analysis of this article appears at the front of this issue.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: D.E.K. was responsible for the overall design and conduct and provided supervision; M.A.B., D.S.K., D.G.K., B.D.W., and D.E.K provided intellectual input and contributed to the experimental design; M.A.B., D.S.K., D.P.T., D.F.P., P.C.R., J.S., F.P., S.L.G., and D.E.K. performed experiments; S.L.G., K.M., H.J., and F.P. provided clinical samples; M.T.W. provided technical assistance for flow cytometry and cell sorting; and M.A.B., D.S.K., and D.E.K wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Daniel E. Kaufmann, Massachusetts General Hospital East, Partners AIDS Research Center, Rm 6618B, 149 13th St, Charlestown, MA 02129; e-mail: dkaufmann@partners.org.
References
- 1.Munier ML, Kelleher AD. Acutely dysregulated, chronically disabled by the enemy within: T-cell responses to HIV-1 infection. Immunol Cell Biol. 2007;85:6–15. doi: 10.1038/sj.icb.7100015. [DOI] [PubMed] [Google Scholar]
- 2.Shin H, Wherry EJ. CD8 T cell dysfunction during chronic viral infection. Curr Opin Immunol. 2007;19:408–415. doi: 10.1016/j.coi.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 3.Chougnet C, Gessani S. Role of gp120 in dendritic cell dysfunction in HIV infection. J Leukoc Biol. 2006;80:994–1000. doi: 10.1189/jlb.0306135. [DOI] [PubMed] [Google Scholar]
- 4.Piguet V, Steinman RM. The interaction of HIV with dendritic cells: outcomes and pathways. Trends Immunol. 2007;28:503–510. doi: 10.1016/j.it.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barber DL, Wherry EJ, Masopust D, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682–687. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- 6.Wherry EJ, Ha SJ, Kaech SM, et al. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity. 2007;27:670–684. doi: 10.1016/j.immuni.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 7.Day CL, Kaufmann DE, Kiepiela P, et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature. 2006;443:350–354. doi: 10.1038/nature05115. [DOI] [PubMed] [Google Scholar]
- 8.Petrovas C, Casazza JP, Brenchley JM, et al. PD-1 is a regulator of virus-specific CD8+ T cell survival in HIV infection. J Exp Med. 2006;203:2281–2292. doi: 10.1084/jem.20061496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Trautmann L, Janbazian L, Chomont N, et al. Upregulation of PD-1 expression on HIV-specific CD8+ T cells leads to reversible immune dysfunction. Nat Med. 2006;12:1198–1202. doi: 10.1038/nm1482. [DOI] [PubMed] [Google Scholar]
- 10.Maier H, Isogawa M, Freeman GJ, Chisari FV. PD-1:PD-L1 interactions contribute to the functional suppression of virus-specific CD8+ T lymphocytes in the liver. J Immunol. 2007;178:2714–2720. doi: 10.4049/jimmunol.178.5.2714. [DOI] [PubMed] [Google Scholar]
- 11.Golden-Mason L, Palmer B, Klarquist J, Mengshol JA, Castelblanco N, Rosen HR. Upregulation of PD-1 expression on circulating and intrahepatic hepatitis C virus-specific CD8+ T cells associated with reversible immune dysfunction. J Virol. 2007;81:9249–9258. doi: 10.1128/JVI.00409-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Urbani S, Amadei B, Tola D, et al. Restoration of HCV-specific T cell functions by PD-1/PD-L1 blockade in HCV infection: effect of viremia levels and antiviral treatment. J Hepatol. 2008;48:548–558. doi: 10.1016/j.jhep.2007.12.014. [DOI] [PubMed] [Google Scholar]
- 13.Kaufmann DE, Kavanagh DG, Pereyra F, et al. Upregulation of CTLA-4 by HIV-specific CD4(+) T cells correlates with disease progression and defines a reversible immune dysfunction. Nat Immunol. 2007;8:1246–1254. doi: 10.1038/ni1515. [DOI] [PubMed] [Google Scholar]
- 14.Jones RB, Ndhlovu LC, Barbour JD, et al. Tim-3 expression defines a novel population of dysfunctional T cells with highly elevated frequencies in progressive HIV-1 infection. J Exp Med. 2008;205:2763–2779. doi: 10.1084/jem.20081398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Masopust D, Kaech SM, Wherry EJ, Ahmed R. The role of programming in memory T-cell development. Curr Opin Immunol. 2004;16:217–225. doi: 10.1016/j.coi.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 16.Couper KN, Blount DG, Riley EM. IL-10: the master regulator of immunity to infection. J Immunol. 2008;180:5771–5777. doi: 10.4049/jimmunol.180.9.5771. [DOI] [PubMed] [Google Scholar]
- 17.Moore KW, de Waal Malefyt R, Coffman RL, O'Garra A. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol. 2001;19:683–765. doi: 10.1146/annurev.immunol.19.1.683. [DOI] [PubMed] [Google Scholar]
- 18.de Waal Malefyt R, Haanen J, Spits H, et al. Interleukin 10 (IL-10) and viral IL-10 strongly reduce antigen-specific human T cell proliferation by diminishing the antigen-presenting capacity of monocytes via downregulation of class II major histocompatibility complex expression. J Exp Med. 1991;174:915–924. doi: 10.1084/jem.174.4.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rousset F, Garcia E, Defrance T, et al. Interleukin 10 is a potent growth and differentiation factor for activated human B lymphocytes. Proc Natl Acad Sci U S A. 1992;89:1890–1893. doi: 10.1073/pnas.89.5.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Berg DJ, Davidson N, Kuhn R, et al. Enterocolitis and colon cancer in interleukin-10-deficient mice are associated with aberrant cytokine production and CD4(+) TH1-like responses. J Clin Invest. 1996;98:1010–1020. doi: 10.1172/JCI118861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Davidson NJ, Leach MW, Fort MM, et al. T helper cell 1-type CD4+ T cells, but not B cells, mediate colitis in interleukin 10-deficient mice. J Exp Med. 1996;184:241–251. doi: 10.1084/jem.184.1.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dai WJ, Kohler G, Brombacher F. Both innate and acquired immunity to Listeria monocytogenes infection are increased in IL-10-deficient mice. J Immunol. 1997;158:2259–2267. [PubMed] [Google Scholar]
- 23.Denis M, Ghadirian E. IL-10 neutralization augments mouse resistance to systemic Mycobacterium avium infections. J Immunol. 1993;151:5425–5430. [PubMed] [Google Scholar]
- 24.Reed SG, Brownell CE, Russo DM, Silva JS, Grabstein KH, Morrissey PJ. IL-10 mediates susceptibility to Trypanosoma cruzi infection. J Immunol. 1994;153:3135–3140. [PubMed] [Google Scholar]
- 25.Belkaid Y, Hoffmann KF, Mendez S, et al. The role of interleukin (IL)-10 in the persistence of Leishmania major in the skin after healing and the therapeutic potential of anti-IL-10 receptor antibody for sterile cure. J Exp Med. 2001;194:1497–1506. doi: 10.1084/jem.194.10.1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gazzinelli RT, Wysocka M, Hieny S, et al. In the absence of endogenous IL-10, mice acutely infected with Toxoplasma gondii succumb to a lethal immune response dependent on CD4+ T cells and accompanied by overproduction of IL-12, IFN-gamma and TNF-alpha. J Immunol. 1996;157:798–805. [PubMed] [Google Scholar]
- 27.Couper KN, Blount DG, Wilson MS, et al. IL-10 from CD4CD25Foxp3CD127 adaptive regulatory T cells modulates parasite clearance and pathology during malaria infection. PLoS Pathog. 2008;4:e1000004. doi: 10.1371/journal.ppat.1000004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sarangi PP, Sehrawat S, Suvas S, Rouse BT. IL-10 and natural regulatory T cells: two independent anti-inflammatory mechanisms in herpes simplex virus-induced ocular immunopathology. J Immunol. 2008;180:6297–6306. doi: 10.4049/jimmunol.180.9.6297. [DOI] [PubMed] [Google Scholar]
- 29.Brooks DG, Trifilo MJ, Edelmann KH, Teyton L, McGavern DB, Oldstone MB. Interleukin-10 determines viral clearance or persistence in vivo. Nat Med. 2006;12:1301–1309. doi: 10.1038/nm1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ejrnaes M, Filippi CM, Martinic MM, et al. Resolution of a chronic viral infection after interleukin-10 receptor blockade. J Exp Med. 2006;203:2461–2472. doi: 10.1084/jem.20061462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ghalib HW, Piuvezam MR, Skeiky YA, et al. Interleukin 10 production correlates with pathology in human Leishmania donovani infections. J Clin Invest. 1993;92:324–329. doi: 10.1172/JCI116570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yamamura M, Uyemura K, Deans RJ, et al. Defining protective responses to pathogens: cytokine profiles in leprosy lesions. Science. 1991;254:277–279. doi: 10.1126/science.254.5029.277. [DOI] [PubMed] [Google Scholar]
- 33.Zhang M, Gong J, Iyer DV, Jones BE, Modlin RL, Barnes PF. T cell cytokine responses in persons with tuberculosis and human immunodeficiency virus infection. J Clin Invest. 1994;94:2435–2442. doi: 10.1172/JCI117611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu Y, de Waal Malefyt R, Briere F, et al. The EBV IL-10 homologue is a selective agonist with impaired binding to the IL-10 receptor. J Immunol. 1997;158:604–613. [PubMed] [Google Scholar]
- 35.Kotenko SV, Saccani S, Izotova LS, Mirochnitchenko OV, Pestka S. Human cytomegalovirus harbors its own unique IL-10 homolog (cmvIL-10). Proc Natl Acad Sci U S A. 2000;97:1695–1700. doi: 10.1073/pnas.97.4.1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stylianou E, Aukrust P, Kvale D, Muller F, Froland SS. IL-10 in HIV infection: increasing serum IL-10 levels with disease progression–down-regulatory effect of potent anti-retroviral therapy. Clin Exp Immunol. 1999;116:115–120. doi: 10.1046/j.1365-2249.1999.00865.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Clerici M, Wynn TA, Berzofsky JA, et al. Role of interleukin-10 in T helper cell dysfunction in asymptomatic individuals infected with the human immunodeficiency virus. J Clin Invest. 1994;93:768–775. doi: 10.1172/JCI117031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Landay AL, Clerici M, Hashemi F, Kessler H, Berzofsky JA, Shearer GM. In vitro restoration of T cell immune function in human immunodeficiency virus-positive persons: effects of interleukin (IL)-12 and anti-IL-10. J Infect Dis. 1996;173:1085–1091. doi: 10.1093/infdis/173.5.1085. [DOI] [PubMed] [Google Scholar]
- 39.Clerici M, Balotta C, Salvaggio A, et al. Human immunodeficiency virus (HIV) phenotype and interleukin-2/interleukin-10 ratio are associated markers of protection and progression in HIV infection. Blood. 1996;88:574–579. [PubMed] [Google Scholar]
- 40.Brooks DG, Lee AM, Elsaesser H, McGavern DB, Oldstone MB. IL-10 blockade facilitates DNA vaccine-induced T cell responses and enhances clearance of persistent virus infection. J Exp Med. 2008;205:533–541. doi: 10.1084/jem.20071948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boritz E, Palmer BE, Wilson CC. Human immunodeficiency virus type 1 (HIV-1)-specific CD4+ T cells that proliferate in vitro detected in samples from most viremic subjects and inversely associated with plasma HIV-1 levels. J Virol. 2004;78:12638–12646. doi: 10.1128/JVI.78.22.12638-12646.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Deeks SG, Walker BD. Human immunodeficiency virus controllers: mechanisms of durable virus control in the absence of antiretroviral therapy. Immunity. 2007;27:406–416. doi: 10.1016/j.immuni.2007.08.010. [DOI] [PubMed] [Google Scholar]
- 43.Orsilles MA, Pieri E, Cooke P, Caula C. IL-2 and IL-10 serum levels in HIV-1-infected patients with or without active antiretroviral therapy. APMIS. 2006;114:55–60. doi: 10.1111/j.1600-0463.2006.apm_108.x. [DOI] [PubMed] [Google Scholar]
- 44.Barcellini W, Rizzardi GP, Borghi MO, Fain C, Lazzarin A, Meroni PL. TH1 and TH2 cytokine production by peripheral blood mononuclear cells from HIV-infected patients. AIDS. 1994;8:757–762. doi: 10.1097/00002030-199406000-00006. [DOI] [PubMed] [Google Scholar]
- 45.Fakoya A, Matear PM, Filley E, et al. HIV infection alters the production of both type 1 and 2 cytokines but does not induce a polarized type 1 or 2 state. AIDS. 1997;11:1445–1452. doi: 10.1097/00002030-199712000-00008. [DOI] [PubMed] [Google Scholar]
- 46.Imami N, Antonopoulos C, Hardy GA, Gazzard B, Gotch FM. Assessment of type 1 and type 2 cytokines in HIV type 1-infected individuals: impact of highly active antiretroviral therapy. AIDS Res Hum Retroviruses. 1999;15:1499–1508. doi: 10.1089/088922299309784. [DOI] [PubMed] [Google Scholar]
- 47.Trabattoni D, Lo Caputo S, Biasin M, et al. Modulation of human immunodeficiency virus (HIV)-specific immune response by using efavirenz, nelfinavir, and stavudine in a rescue therapy regimen for HIV-infected, drug-experienced patients. Clin Diagn Lab Immunol. 2002;9:1114–1118. doi: 10.1128/CDLI.9.5.1114-1118.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Norris PJ, Pappalardo BL, Custer B, Spotts G, Hecht FM, Busch MP. Elevations in IL-10, TNF-alpha, and IFN-gamma from the earliest point of HIV Type 1 infection. AIDS Res Hum Retroviruses. 2006;22:757–762. doi: 10.1089/aid.2006.22.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Graziosi C, Pantaleo G, Gantt KR, et al. Lack of evidence for the dichotomy of TH1 and TH2 predominance in HIV-infected individuals. Science. 1994;265:248–252. doi: 10.1126/science.8023143. [DOI] [PubMed] [Google Scholar]
- 50.Clerici M, Hakim FT, Venzon DJ, et al. Changes in interleukin-2 and interleukin-4 production in asymptomatic, human immunodeficiency virus-seropositive individuals. J Clin Invest. 1993;91:759–765. doi: 10.1172/JCI116294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Trabattoni D, Saresella M, Biasin M, et al. B7-H1 is up-regulated in HIV infection and is a novel surrogate marker of disease progression. Blood. 2003;101:2514–2520. doi: 10.1182/blood-2002-10-3065. [DOI] [PubMed] [Google Scholar]
- 52.Schols D, De Clercq E. Human immunodeficiency virus type 1 gp120 induces anergy in human peripheral blood lymphocytes by inducing interleukin-10 production. J Virol. 1996;70:4953–4960. doi: 10.1128/jvi.70.8.4953-4960.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shan M, Klasse PJ, Banerjee K, et al. HIV-1 gp120 mannoses induce immunosuppressive responses from dendritic cells. PLoS Pathog. 2007;3:e169. doi: 10.1371/journal.ppat.0030169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Badou A, Bennasser Y, Moreau M, Leclerc C, Benkirane M, Bahraoui E. Tat protein of human immunodeficiency virus type 1 induces interleukin-10 in human peripheral blood monocytes: implication of protein kinase C-dependent pathway. J Virol. 2000;74:10551–10562. doi: 10.1128/jvi.74.22.10551-10562.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Huigen MC, Kamp W, Nottet HS. Multiple effects of HIV-1 trans-activator protein on the pathogenesis of HIV-1 infection. Eur J Clin Invest. 2004;34:57–66. doi: 10.1111/j.1365-2362.2004.01282.x. [DOI] [PubMed] [Google Scholar]
- 56.Shin HD, Winkler C, Stephens JC, et al. Genetic restriction of HIV-1 pathogenesis to AIDS by promoter alleles of IL10. Proc Natl Acad Sci U S A. 2000;97:14467–14472. doi: 10.1073/pnas.97.26.14467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Erikstrup C, Kallestrup P, Zinyama-Gutsire RB, et al. Reduced mortality and CD4 cell loss among carriers of the interleukin-10-1082G allele in a Zimbabwean cohort of HIV-1-infected adults. AIDS. 2007;21:2283–2291. doi: 10.1097/QAD.0b013e3282f153ed. [DOI] [PubMed] [Google Scholar]
- 58.Silvestri G, Feinberg MB. Turnover of lymphocytes and conceptual paradigms in HIV infection. J Clin Invest. 2003;112:821–824. doi: 10.1172/JCI19799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kollmann TR, Pettoello-Mantovani M, Katopodis NF, et al. Inhibition of acute in vivo human immunodeficiency virus infection by human interleukin 10 treatment of SCID mice implanted with human fetal thymus and liver. Proc Natl Acad Sci U S A. 1996;93:3126–3131. doi: 10.1073/pnas.93.7.3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Juffermans NP, Paxton WA, Dekkers PE, et al. Up-regulation of HIV coreceptors CXCR4 and CCR5 on CD4(+) T cells during human endotoxemia and after stimulation with (myco)bacterial antigens: the role of cytokines. Blood. 2000;96:2649–2654. [PubMed] [Google Scholar]
- 61.Angel JB, Jacobson MA, Skolnik PR, et al. A multicenter, randomized, double-blind, placebo-controlled trial of recombinant human interleukin-10 in HIV-infected subjects. AIDS. 2000;14:2503–2508. doi: 10.1097/00002030-200011100-00012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}