

Abstract
In Escherichia coli, the largest class of small regulatory RNAs binds to the RNA chaperone Hfq and regulates the stability and/or translation of specific mRNAs. While recent studies have shown that some mRNAs could be subject to posttranscriptional regulation by sRNAs (e.g. mRNAs found by co-immunoprecipitation with Hfq), no method has yet been described to identify small RNAs that regulate them. We developed a method to easily make translational fusions of genes of interest to the lacZ reporter gene, under the control of a PBAD inducible promoter. A multicopy plasmid library of the E. coli genome can then be used to screen for small RNAs that affect the activity of the fusion. This screening method was first applied to the dpiB gene from the dpiBA operon, which encodes a two-component signal transduction system involved in the SOS response to β-lactams. One small RNA, RybC, was found to negatively regulate the expression of dpiB. Using mutants in the dpiB-lacZ fusion and compensatory mutations in the RybC sRNA, we demonstrate that RybC directly basepairs with the dpiBA mRNA.
Keywords: SroB, MicM, DpiA/B, sRNA, citB
Introduction
In recent years, numerous studies have shown that non-coding RNAs act as a major class of post-transcriptional regulators and play crucial roles in cell physiology, developmental control and adaptation in bacteria, archaea and eukaryotes. A number of genome-wide searches helped to uncover close to one hundred of these small regulatory RNAs (sRNAs) in E. coli (reviewed in (Vogel and Sharma, 2005)). However, only a small fraction of these sRNAs have been characterized thus far and shown to affect various physiological pathways (reviewed in (Gottesman et al., 2006)).
In E. coli, the largest class of sRNAs exert their post-transcriptional control by base-pairing to specific mRNA targets and controlling either their stability and/or translation (Aiba, 2007). For example, the sRNAs DsrA and RprA stimulate the translation of rpoS, encoding the stress sigma factor of E. coli, by antagonizing the formation of an inhibitory stem loop structure in the 5’ UTR of the rpoS mRNA (Majdalani et al., 1998; Majdalani et al., 2005). Other sRNAs, such as SgrS and RyhB, affect the stability of their mRNA targets. SgrS pairs with ptsG mRNA, which encodes a glucose transporter (Vanderpool and Gottesman, 2004). RyhB, which is induced in iron limiting conditions, acts on many mRNA targets encoding proteins involved in the use and storage of iron by bacteria (Massé et al., 2003; Massé et al., 2005; Massé et al., 2007). For both SgrS and RyhB, pairing of the sRNAs to their mRNA targets occurs near the ribosome binding site in the 5’ end of the mRNA transcripts and leads to their degradation by RNase E.
Most of the trans-acting sRNAs that function by base pairing to their mRNA targets have been shown to bind the Hfq protein. Hfq is an RNA chaperone belonging to the Sm-like family of proteins that, in eukaryotes and archaea, is involved in various aspects of RNA metabolism, including splicing and mRNA decay (Seraphin, 1995). In E. coli and related bacteria, Hfq is an abundant protein that forms hexamers and binds RNA at A/U rich regions preceded or followed by a stem loop structure (reviewed in (Brennan and Link, 2007)). The proposed role of Hfq in sRNA post-transcriptional regulation is to facilitate base pairing between sRNAs and their mRNA targets.
Mechanisms underlying the pairing of sRNAs to their mRNA targets are not fully understood yet, rendering prediction of mRNA targets difficult. Attempts at developing computational predictions of mRNA targets of sRNAs in different microorganisms have yielded mixed results (Mandin et al., 2007; Tjaden et al., 2006). While they can be useful, they often predict incorrect targets or fail to predict known ones, suggesting that further work will be necessary to improve these approaches. Reciprocally, even though some mRNAs are believed to be subject to post-transcriptional regulation by sRNAs (e.g. mRNAs found by co-immunoprecipitation with Hfq), no reliable method has yet been described to identify sRNAs that could regulate these mRNAs.
The DpiA/B two-component signal transduction system of E. coli is an ortholog of the CitA/B system that regulates citrate uptake and utilization in Klebsiella pneumoniae (Bott et al., 1995). In E, coli, this system has been shown to modulate the replication and destabilize the inheritance of pSC101 and certain other plasmids (Ingmer et al., 1998). This is due to the ability of DpiA to bind A/T-rich sequences at the origin, perturbing DNA replication and inducing the SOS response (Miller et al., 2003). It was subsequently shown that DpiA/B induces SOS in response to certain β-lactams, thereby providing a way for the bacteria to mitigate the lethality of these antibiotics (Miller et al., 2004).
We developed a new method that allows screening for post-transcriptional regulators, including sRNAs, regulating genes of interest. The protocol begins with easy construction of translational fusions of the 5’ ends of genes of interest to the lacZ reporter gene in the bacterial chromosome. A multicopy plasmid library of the E. coli genome can then be screened to isolate clones that can affect the activity of the fusion. Here we describe this screening method as first applied to the dpiB gene from the dpiBA operon. RybC, a small RNA of previously unknown function, was found to negatively regulate the levels of a dpiB-lacZ fusion. Using mutants in the dpiB-lacZ fusion and compensatory mutations in the RybC sRNA, we demonstrate that RybC acts post-transcriptionally by directly pairing with the dpiBA mRNA.
Results
Selection of candidates for sRNA regulation
Pairing between sRNAs and mRNAs is usually found to occur close to or overlapping the Ribosome Binding Site (RBS) in the 5’ end of mRNAs. In most cases, mRNAs subject to regulation by sRNAs display unusually long 5’ untranslated regions (UTR) for which sequence is conserved within closely related species. A previous study had shown that RNAs bound to Hfq can be co-immunoprecipitated with the protein (Zhang et al., 2003). RNAs were subsequently detected using Affymetrix microarrays, thereby identifying new sRNAs as well as more than 20 potential mRNA targets. We searched the Hfq immunoprecipitation microarray data for genes displaying long or conserved UTRs that might be subject to sRNA regulation.
To improve the likelihood of sRNA regulation and keep the number of genes to analyze manageable, we initially focused our analysis on operons for which all the genes were enriched after Hfq immunoprecipitation and microarray analysis. In a first non-exhaustive screen, a few candidates were selected for further analysis (data not shown), of which only the dpiBA genes are discussed here. dpiA and dpiB encode a response regulator and a sensory histidine kinase, respectively, of a two component system orthologous to the CitA/B system which modulates citrate metabolism in K. pneumoniae (Bott et al., 1995). These genes are adjacent on the chromosome, overlapping by 31 nt, and previous studies indicated that they are part of the same transcriptional unit (Miller et al., 2003). dpiA and dpiB were enriched by 21 and 23 fold respectively in samples treated for co-immunoprecipitation with Hfq-specific antiserum and detected on microarrays, as compared to total RNA samples (Zhang et al., 2003). Taken together, these data prompted us to analyze more closely the dpiBA system for possible regulation by sRNAs.
Screening of a dpiB-lacZ fusion using a multicopy plasmid library
To study the post-transcriptional regulation and to screen for sRNAs regulating the dpiBA genes, we constructed translational fusions with the lacZ reporter gene. We chose to place the desired gene fusions under the control of the arabinose inducible PBAD promoter, so that transcription would be independent of the natural promoters, enabling us to look only at posttranscriptional regulation such as that exerted by sRNAs.
When this project was initiated, the transcriptional start site of dpiBA had not been reported. As described below, 5’ RACE combined with direct cloning to create a translational fusion, as well as classical 5’ RACE, was used to determine the likely transcriptional start site. Most of the data presented here was carried out using these translational fusions. However, we began a screening approach to find sRNAs regulating dpiB before the 5’ ends were mapped. We therefore used a different strain in which the PBAD-dpiB-lacZ translational fusion transcriptional start site was arbitrarily chosen at - 89 nt upstream of the ATG of dpiB (called PBAD-(-89) dpiB-lacZ, beginning at bolded G in Figure 1C). As described below, the 5’end of this fusion turned out to be upstream of the natural start site. This construct was introduced in single copy at the lambda attachment site of a Δlac strain. The resulting strain was white (Lac-) on MacConkey lactose plates containing no arabinose and slightly red on plates containing 0.0005% arabinose, indicating that the expression of this fusion is arabinose dependent. Therefore, any possible internal promoter was silent under these conditions and changes in color in our screen should primarily reflect post-transcriptional regulatory events, although a regulator of a cryptic promoter could have been identified.
Figure 1. 5’RACE coupled to direct cloning of lacZ fusions identifies two 5’ ends for the dpiBA mRNA.
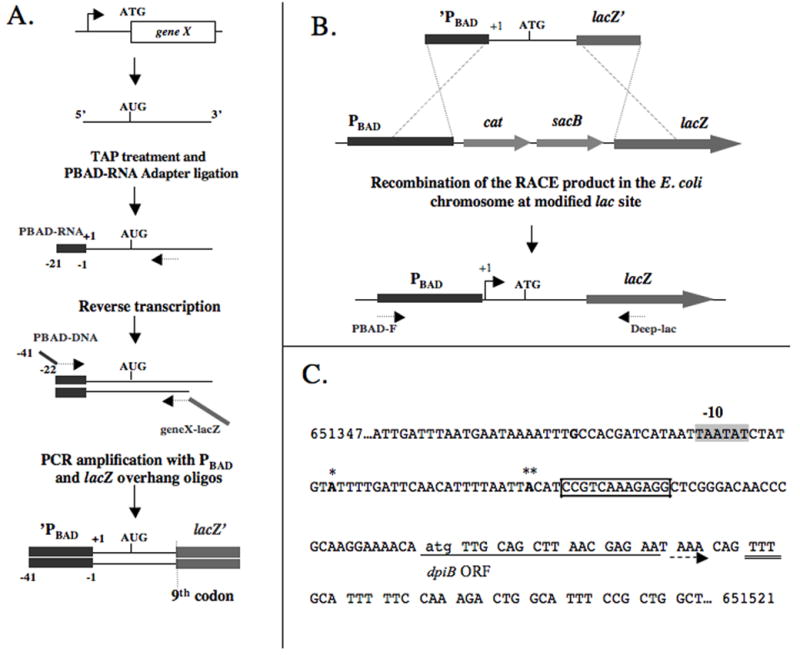
(A.) Schematic representation of 5’RACE with modified oligonucleotides. First, RNA 5′ tri-phosphates are converted to 5′ monophosphates using tobacco acid pyrophosphatase (TAP) and the PBAD-RNA adapter is ligated to mRNAs isolated from the cells. After a reverse transcription step, the cDNA is amplified by PCR using DNA primers PBAD-DNA and geneX-lacZ, generating a PCR product with 40 nt homologous sequences to PBAD on one side and lacZ on the other. (B.) The PCR product obtained in (A) is recombined into the chromosome of strain PM1205 by lambda Red-mediated recombination. Strain PM1205 harbors a PBAD-cat-sacB cassette in front of lacZ, enabling one to select for loss of the sacB gene on sucrose plates containing X-Gal and resulting in the desired lacZ fusion. Sequencing of the resulting strain with oligos PBAD-F and Deep-lac allows the determination of the 5’extremities. (C.) Results obtained by applying the technique to the dpiBA mRNA. Two different 5’ extremities were obtained: (*) 5’extremity located 63 upstream of the initiation codon for the translation of dpiB (indicated by lower case letter); (**) 5’ extremity located 41 nt upstream of the atg of dpiB. The resulting LacZ translational fusions were called dpiB-L (strain PM1211) and dpiB-S (strain PM1213), respectively. The shaded sequence indicates the sequence of a putative -10 box for transcription starting at -63 nt. The bolded G located at -89 nt indicates the transcriptional start site chosen in the initial dpiB-lacZ fusion used for the plasmid library screening. Arrow indicates the beginning and the direction of translation of the dpiB open reading frame (ORF); the double underline indicates the last codon of dpiB used in all the dpiB-lacZ fusions of this study. Boxed nucleotides: sequence included in the predicted pairing with RybC (see Figure 5).
The PBAD-(-89) dpiB-lacZ fusion gave increased activity in an hfq mutant (data not shown), suggesting that post-transcriptional regulation was likely, and that such regulation might be negative. Thus, this strain was transformed with a pBR322-based E. coli genomic library (Ulbrandt et al., 1997) with the rationale that overproduction of a putative sRNA from the plasmid should give white (Lac-) colonies on MacConkey Lactose ampicillin plates containing arabinose. Approximately 6000 colonies were analyzed. White or lighter colonies were isolated and the activity of the plasmids purified from these colonies was confirmed by retransformation. No plasmids stimulating the activity of the fusion were found. The plasmid inserts were subsequently determined by sequencing and are listed in Table 1. Three classes of plasmids were found.
Table 1.
| Class | Number of isolates | Sequence of inserts | Phenotype |
|---|---|---|---|
| 1 | 2 | ‘ybaK, rybC, ybaP, (ybaQ) | - |
| 2 | 2 | ‘mdtF, gadW, gadY, gadX, gadA’ | - |
| 3 | 1 | ‘yhcB, degQ, degS | (-) |
The number of isolates refers to overlapping (non-identical) plasmid isolates. Genes in brackets are present in some of the isolates only and a prime on one side of a gene indicates that it is interrupted on this side.
- : refers to a decreased β-galactosidase activity of the dpiB-lacZ (PM1011) fusion on MacConkey lactose agar plates for colonies carrying the plasmid as compared to the pHDB3 vector.
Two identical plasmids were isolated for the first class; these gave a white, Lac−, phenotype on MacConkey lactose plates. Both plasmids contained a 5’ truncated ybaK gene, a truncated version of ybaP, encoding a protein of unknown function conserved among various bacterial species, and rybC (also named sroB), an sRNA of unknown function, previously identified in genome-wide searches and whose levels were shown to be Hfq-dependent in two different studies (Vogel et al., 2003; Zhang et al., 2003). Two plasmids isolated in the second class also gave a white (Lac−) phenotype on MacConkey plates. Both contained a truncated mdtF gene, which is unlikely to be functional, gadW and gadX, two AraC-like regulators of the glutamate based acid resistance in E. coli (Castanie-Cornet et al., 1999; Ma et al., 2002; Tramonti et al., 2002), gadY, a sRNA previously shown to regulate gadX (Opdyke et al., 2004), and a short fragment of the 5’end of gadA, unlikely to be functional in this plasmid.
Finally, the third class contained one isolate that had a milder Lac−phenotype as compared to the other classes. This plasmid contained degQ and degS, encoding the two periplasmic proteases (Waller and Sauer, 1996). Colonies of this isolate were mucoid so that the Lac phenotype was difficult to interpret. This isolate was therefore not characterized in this study. The activities of plasmids in class one and two were further analyzed, using the translational fusions described below.
Construction of dpiB-lacZ translational fusions coupled to 5’ RACE identification of 5’ ends
As the transcriptional start site of the dpiBA operon had not been previously reported, we developed a new method allowing the simultaneous identification of 5’ ends and direct cloning of desired genes under the control of a PBAD promoter in the chromosome, in translational fusion with lacZ (Figure 1 A and B).
The first step of the process consists of a modified 5’RACE, derived from (Urban and Vogel, 2007), where the RNA adapter and the RT-PCR primers have been modified so that the final PCR product harbors 40 nt homologous to the PBAD promoter at the 5’end of the gene (from nt -1 to -41 relative to the transcription start site of the PBAD promoter), and 40 nt of the lacZ gene on its 3’end (from nt 25 to 65 relative to the ATG of lacZ) (see Figure 1A and Material and Methods). The reverse primer is designed so that the coding sequence of the gene amplified is in frame with the lacZ ORF at the 9th codon. Using mini-lambda mediated recombineering (Court et al., 2003), the PCR product is then directly recombined with the chromosome of a modified E. coli wild type strain (PM1205) which carries a PBAD-cat-sacB cassette inserted in front of lacZ at the 9th codon (Figure 1 B). Recombinants are selected for loss of the cat-sacB genes, resulting in the translational fusion of the desired gene to lacZ under the transcriptional control of the PBAD promoter. Additional modifications introduced in PM1205 include the deletion of the araBAD genes (but retaining araC) so that arabinose is used only as an inducer of the PBAD promoter and not as a metabolite, and the introduction of a mal∷lacIq allele to allow better control of expression of plasmid-borne genes from PLAC promoters (see below).
We used this method to find the 5’end(s) of the dpiBA mRNA and make translational fusions of dpiB to lacZ. Three types of clones were obtained after recombination of the final 5’RACE PCR products and selection on sucrose plates containing X-Gal to report on β-galactosidase activity. Some 25% of the colonies were white (Lac-), as is the parental strain, suggesting that the expected recombination had not occurred. Consistent with this, PCR and sequencing using primers inside the PBAD promoter and lacZ revealed no insertion in these isolates. The other clones showed a clear Lac+ phenotype, ~75% of which gave a strong dark blue color and ~25% a lighter blue color. PCR was performed on 8 clones of each type, four of which were also sequenced; PCR profiles were distinct for the two categories of clones (data not shown). The 5’ end of the darker clones was subsequently mapped at -41nt from the ATG of dpiB (Figure 1C). The lighter blue clones had a 5’ end located at -63nt from the ATG of dpiB (Figure 1C). The resulting translational fusions were thus called dpiB-lacZ-Short (dpiB-S, strain PM1213) and dpiB-lacZ-Long (dpiB-L, strain PM1211) respectively. Both 5’end were also confirmed by classical 5’RACE followed by PCR-TOPO cloning; thus the direct cloning method gave the same start sites as classical 5’ RACE.
A sequence (5’-TAATAT-3’) resembling a consensus -10 box (5’-TATAAT-3’) was found at an appropriate distance upstream of the longer form of the UTR but not in front of the shorter one (Figure 1C), suggesting that the longer form may be the transcriptional start site of the dpiBA mRNA and that the shorter form may arise from a post-transcriptional processing event. However, we could not identify any sequence that would correspond to a -35 consensus sequence for either form of the mRNA. Note that the PBAD-(-89) dpiB-lacZ fusion used in the first part of this work would not be expected to contain the full promoter for a -63 start, but might for a -41 start. However, no promoter activity was detected in the PBAD-(-89) dpiB-lacZ fusion in the absence of arabinose, again suggesting that the -41 nt 5’ end may not be a transcriptional start site but may arise from post-transcriptional cleavage.
Expression of the dpiB-lacZ fusions vary in an hfq mutant
We first tested the expression of the dpiB-L and dpiB-S fusions in LB liquid medium containing 0.0005% arabinose in early stationary phase (Figure 2). Consistent with the pattern we had seen on plates containing X-Gal, β-galactosidase activity of the dpiB-S fusion was ~2-fold higher than that of dpiB-L (columns 1 and 3).
Figure 2. β-galactosidase assay on the dpiB-lacZ fusions in the presence or absence of hfq.
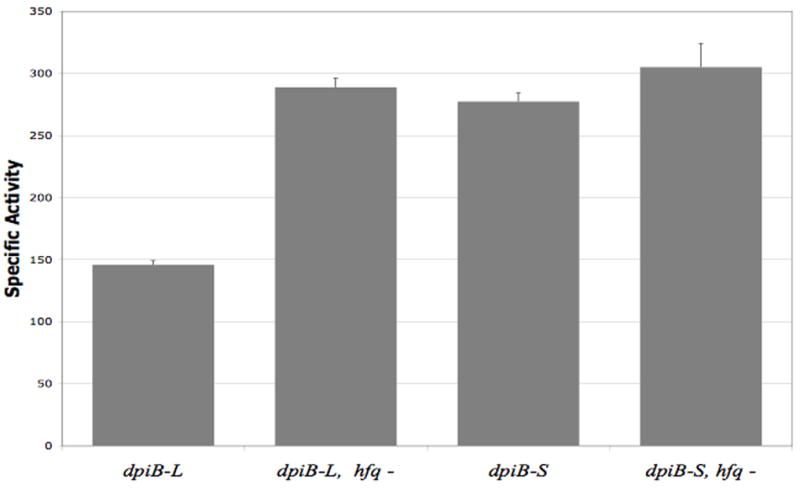
Overnight cultures of strain PM1211 (dpiB-L), PM1213 (dpiB-S) and the respective hfq∷cat derivatives PM1224 (dpiB-L hfq-) and PM1244 (dpiB-S hfq-) were diluted 500-fold in fresh LB medium containing 0.0005% arabinose and grown at 37°C to stationary phase (OD=3). Samples were then taken and β-galactosidase assays were performed as previously described (Majdalani et al., 1998).
Trans-acting sRNAs have been shown to require the protein Hfq for their function. As noted above, the PBAD-(-89) dpiB-lacZ fusion showed higher expression in an hfq deletion mutant. An hfq mutation was introduced into the dpiB-L and dpiB-S fusion strains and the resulting strains were tested for their β-galactosidase activity in the same conditions as used above (Figure 2). As seen on Figure 2, the activity of the dpiB-L fusion increased by 2-fold in an hfq mutant background, to a level almost identical to the dpiB-S fusion in the wild-type strain. In contrast, there was almost no change in the β-galactosidase activity of the dpiB-S fusion after introduction of the hfq mutation. Taken together, these results indicate that one or several sRNAs could down-regulate the longer form of the dpiB mRNA. The shorter form is less sensitive to an hfq mutation, indicating that it may lack sequence and/or structure requirements needed for this regulation.
Analysis of potential small RNA regulators
At this step it was not clear whether one of the sRNAs, RybC or GadY, carried by the two first classes of plasmids isolated with the original fusion or one of the other genes on these plasmids was responsible for down-regulation of the dpiB-lacZ fusion when over-expressed. To directly address this question, we constructed plasmids carrying either sRNA under the transcriptional control of a PLAC promoter. These plasmids were introduced into the dpiB-L and dpiB-S translational fusion strains, which also contain a mal∷lacIq allele, allowing a tighter control of the expression of the sRNAs from the PLAC promoter. As described below, the over-expression of RybC, but not of GadY, had a clear effect on the fusions and was therefore further investigated.
RybC, an hfq dependent sRNA
To further characterize the RybC sRNA, we examined its expression profile from log phase, late log phase and in stationary phase in the wild-type E. coli MG1655 strain grown in LB. As seen in Figure 3, RybC was easily detected at all phases of growth and its expression peaked in stationary phase, in line with previous studies (Vogel et al., 2003). We could not detect RybC in an hfq mutant, suggesting that RybC becomes unstable in the absence of Hfq as with many other Hfq-dependent sRNAs. This was consistent with previous experiments that found RybC bound to Hfq by co-immunoprecipitation experiments (Zhang et al., 2003).
Figure 3. Northern blot analysis of RybC expression.
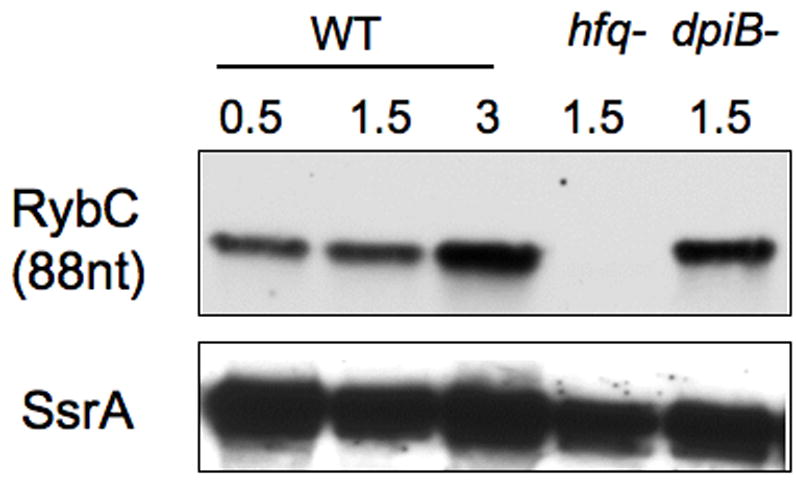
Wild type (WT) strain MG1655, or its hfq∷cat (PM1203) and dpiBA (PM1251) deletion derivatives were grown in LB medium at 37°C and samples were removed at appropriate OD600, as indicated. RNA was extracted by the hot phenol method, fractionated on a 10% TBE-urea gel, and transferred to a nylon membrane. A 5’-biotinylated probe was used to detect RybC; the membrane was then stripped and probed against SsrA RNA as a loading control.
RybC is an 84 nt RNA, as determined by 5’RACE and Northern blots (Figure 3 and (Vogel et al., 2003; Zhang et al., 2003)), and is well conserved among several γ-proteobacteria (Figure 4). Two stem loop structures are predicted to form in the RybC RNA using the Mfold RNA secondary structure prediction program (Zuker, 2003). The first stem loop is located in its 5’ end (nt +4 to +19) and is followed by an A/T rich region, a motif resembling predicted Hfq binding regions (Brennan and Link, 2007). Interestingly, the sequence of this predicted stem loop structure is not very well conserved in related bacterial species other than Shigella flexneri, but structural conservation is kept, as the same region of the RNA is predicted to fold into a stem loop in Citrobacter koseri, Klebisiella pneumoniae, Salmonella typhimurium and Enterobacter sakazakii.
Figure 4. Alignment of rybC and its promoter region from several γ-proteobacteria.
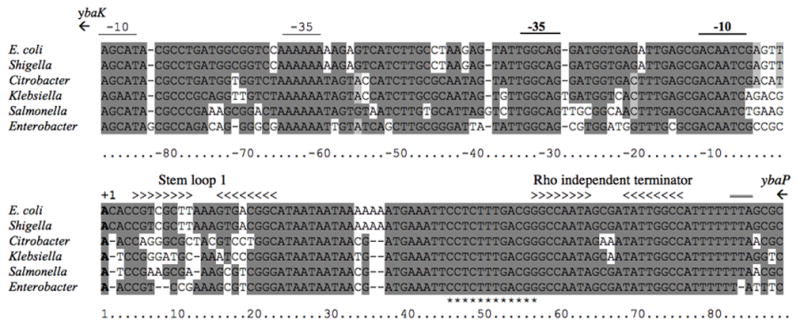
DNA sequence identities are shaded in grey. The transcription start site mapped by 5’ RACE (Vogel et al., 2003) and the deduced -10 and -35 regions of the rybC promoter are denoted by the text above the sequence. Mfold (Zuker, 2003) predicted stem loop structures and Rho independent terminator are indicated by >. (*) indicates residues that are predicted to be involved in the pairing with the dpiBA mRNA. The TAA stop codon for ybaP is, double underlined above the sequence (TTA at end of the rybC terminator). The start codon for ybaK begins 35 nt upstream of the sequence shown.
The second predicted stem loop structure, located at the 3’ end (nt +58 to +75) of the RNA, is followed by a stretch of 7 Us and is therefore most likely a Rho-independent transcription terminator. This predicted terminator and a ~15 nt region upstream are particularly well conserved among closely related species and even in the more remotely related E. sakazakii.
RybC is located in a 203 bp intergenic region between and on the opposite strand from the ybaK and ybaP genes. rybC is encoded divergently from ybaK, encoding a cys tRNA deacylase (Ruan and Soll, 2005), with a distance of 122 nt between the start codon of ybaK and the start of RybC. In the promoter region, the -10 (5’-GCGACAATCG-3’) and -35 (5’-TATTGGCAGG-3’) sequences of rybC are similar to consensus -10 (5’-TATAAT-3’) and -35 (5’-TTGACA-3’) boxes and are highly conserved among other bacterial species (Figure 4). It was previously suggested that the pattern of expression of RybC was reminiscent of regulation by the stress sigma factor RpoS (Vogel et al., 2003). However, no effect of an rpoS deletion mutant was seen on a transcriptional reporter fusion of RybC (data not shown). We also used this transcriptional fusion construct and Northern blots to explore the possibility of a feedback loop regulation of RybC by the DpiB/A two-component system but no transcriptional regulation was found (Figure 3, lane 5 and data not shown). The region upstream of the -35 is also well conserved; however, transcriptional fusions carrying only 45 nt upstream of the +1 gave significant levels of expression, suggesting that these upstream regions are not essential for rybC promoter expression. One reason for the conservation may be the presence of the -10 and -35 sequences for ybaK within this region. Candidate sequences for these are marked in Fig. 4. It is possible that ybaK and rybC share a binding site for a common (non-essential) regulator or that the promoters otherwise interact, but this has not been explored. The stop codon of ybaP is at the end of the rybC terminator (see Fig. 4); possibly the stem-loop for rybC acts as a terminator for ybaP. The genes neighboring RybC are conserved in the organisms in which RybC is found (Salmonella, Klebsiella, Citrobacter, and Enterobacter).
RybC directly regulates dpiB
To better characterize the regulation of dpiBA by RybC, we constructed a plasmid, pRybC, carrying RybC under the control of a PLAC promoter. We then checked the effect of over-expressing the sRNA from this plasmid on the β-galactosidase activity of the dpiB-L and dpiB-S translational fusions. Both fusion strains, carrying the pBR-pLAC vector or pRybC, were grown in LB liquid medium containing arabinose, ampicillin, and IPTG at 37°C. β-galactosidase activity was measured from samples taken in stationary phase (OD600of 3). As seen in Figure 5, expression of RybC from the pRybC plasmid reduced the expression of dpiB-L and dpiB-S by 2 and 1.5 fold respectively in a rybC+ host (WT, left side of Fig.5), as compared to the vector alone, confirming the results we saw with the plasmid from the library.
Figure 5. RybC regulates the dpiB-L and dpiB-S fusions.
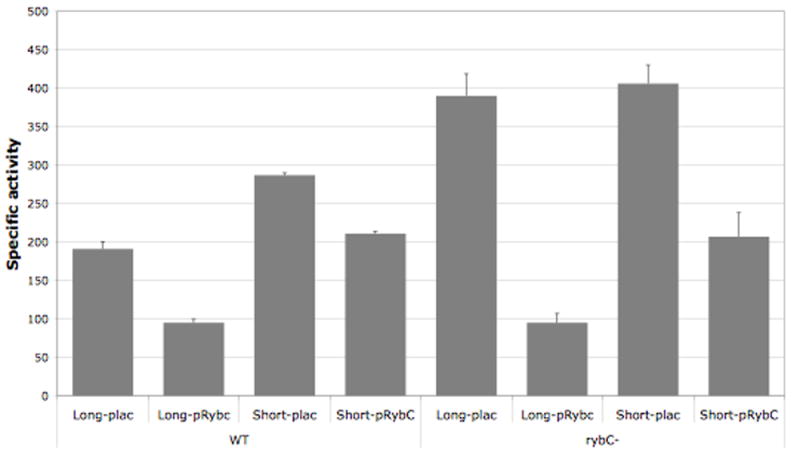
Overnight cultures of strains PM1211 (dpiB-L fusion), PM1213 (dpiB-S fusion) (four first bars) and their respective rybC deletion derivatives PM1222 and PM1242 (four last bars), containing either the pBRplac (plac) control vector or the plasmid expressing RybC (pRybC), were diluted 500-fold in fresh LB medium containing ampicillin and 0.0005% arabinose. Cultures were grown at 37°C to exponential phase at which point IPTG was added to the medium at a final concentration of 100 μM. Samples were then taken at early stationary phase (OD600 of 3) and β-galactosidase assays were performed as previously described (Majdalani et al., 1998).
Northern Blots had shown that RybC is well expressed in stationary phase (Figure 3). We conducted the same tests in derivatives of the dpiB-L and dpiB-S strains deleted for the chromosomal copy of rybC (right side of Fig. 5). The basal level of β-galactosidase activity in the ΔrybC- dpiB-L and dpiB-S derivatives with the vector control increased by 2 and 1.5 fold respectively as compared to the wild type fusion strains, bringing them to a very comparable level. These results suggest that the difference seen in the wild type strain between the two different forms of the fusion reflects regulation by the chromosomally-expressed RybC, and that the long fusion is more sensitive to this than the short fusion. As in the rybC+ host, expression of RybC from the plasmid reduced the expression of dpiB-L and dpiB-S by 4 and 2 fold respectively (Figure 5). Note that the level of RybC expressed from the plasmid was 5-10 fold higher than that from the chromosome (data not shown). In each case, expression of RybC either from the chromosome or from the plasmid had a stronger effect on the dpiB-lacZ fusion having a longer UTR (Figure 5).
We used the Mfold program (Zuker, 2003) with minor modifications to predict base pairing between RybC and the dpiBA mRNA. The best predicted base pairing is a 12 nucleotide stretch, without any mismatch or bulge between the two RNAs (Figure 6A). Base pairing is predicted to occur at the 5’ end of the dpiBA mRNA, from nts -37 to -26 upstream of the ATG of dpiB. This region is therefore present in both the long and short forms of the dpiB-lacZ fusions (see Fig. 1C). On RybC, the predicted base pairing involves a region located just upstream (and overlapping by 1nt) the Rho independent terminator (from nt +45 to +57). Interestingly, this is one of the most highly conserved regions of RybC (Figure 4), suggesting that this region plays an important role in its function.
Figure 6. RybC exerts its regulation on the dpiB mRNA in a direct manner and through base pairing.
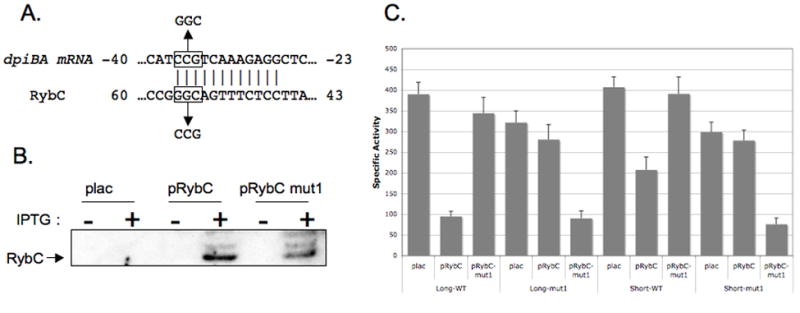
A. Schematic representation of the predicted base pairing in between the dpiBA mRNA and RybC. Numbers indicate the position of the nucleotides from the initiation of translation of the dpiBA mRNA or from the initiation of transcription of RybC. Nucleotides predicted to be involved in the base pairing are shaded in grey. Mutations in both RNAs used in the following β-galactosidase assays are depicted in the boxes above and below the sequences of the RNAs. B. The rybC deletion strain PM1252 harboring an empty vector (pBR-plac) or a plasmid containing either rybC (pRybC) or the mutant form of rybC C55G G56C G57C (pRybC-mut1) under the PLAC promoter were grown at 37°C in LB liquid medium containing ampicillin to an OD600 of 0.5 and a sample of each culture was taken. IPTG was then added to each culture and additional samples were removed 15 mins after the addition of IPTG. RNA was extracted using the hot phenol method, fractionated on a 10% TBE-urea gel, and transferred to a nylon membrane. In a side-by-side comparison, expression of RybC from the IPTG-inducible plasmid in this experiment is 5-10 fold in excess of that seen from the single chromosomal copy (data not shown). C. Overnight cultures of strains PM1222 (dpiB-L rybC∷kan, bars 1-3) and PM1242 (dpiB-S rybC∷kan, bars 7-9) and their respective derivatives where compensatory mutations were introduced, PM1232 (dpiB-L-mut1 rybC∷kan, bars 4-6) and PM1246 (dpiB-S-mut1 rybC∷kan, bars 10-12) containing either the pBRplac (plac) control vector or the plasmid expressing RybC (pRybC) or the mutant version of RybC (pRybC-mut1), were diluted 500-fold in fresh LB medium containing ampicillin and 0.0005% arabinose. Cultures were grown at 37°C to exponential phase at which point 100 μM IPTG was added to the medium. Samples were then taken at early stationary phase (OD=3) and β-galactosidase assays were performed as previously described (Majdalani et al., 1998).
Based on these predictions, we used site directed mutagenesis on the original pRybC plasmid to create a plasmid carrying a mutant form of the sRNA, giving rise to plasmid pRybC-mut1. This mutant contains a 3 nt substitution, C55G G56C G57C, as indicated in Figure 6A. Steady state levels of the mutant RybC-mut1 sRNA were compared to wild-type RybC after a 15 min induction by IPTG. While RybC-mut1 levels were somewhat lower than wild-type RybC levels, significant amounts of the sRNA accumulated (Figure 6B). The lower levels of the sRNA may reflect a less stable terminator; we note higher molecular weight forms, present for both wild type and mut1, possibly arising from inefficient termination by the RybC terminator. However, since significant amounts of the expected size RNA were produced, we were able to use these constructs to test their ability to negatively regulate the translational fusions as described above. The mut1 substitutions in RybC abolished its ability to negatively regulate both the wild type dpiB-L and dpiB-S translational fusions (Figure 6C; lanes 3 and 9). To test whether this was a result of disrupting the base pairing between RybC and the dpiBA mRNA, compensatory mutations were introduced in both dpiB-lacZ translational fusions (dpiB-L-mut1 and dpiB-S-mut1) that would restore pairing with RybC-mut1 if the predicted pairing was correct (Figure 6A). Consistent with the predicted pairing, mutations in either dpiB-lacZ fusion drastically reduced the ability of wild-type RybC to down regulate them (Figure 6C, lanes 5 and 11). In contrast, expression of RybC-mut1 was able to reduce the activity of both mutant fusions in a manner very comparable to that observed with wild type forms of the sRNA on the wild-type fusions (Figure 6C, lanes 6 and 12, compared to lanes 2 and 8). Taken together, these data confirm our model of a direct regulation of the dpiBA mRNA by RybC via base-pairing.
The GadY sRNA does not regulate the dpiB-lacZ fusion
In our original screen, one plasmid was isolated containing the gadW and gadX genes, encoding transcriptional regulators, and gadY, encoding an sRNA. Using β-galactosidase assays, we confirmed that this plasmid had an effect on the dpiB-L fusion and that this effect was RybC-independent (supplemental Fig. S1). To test if this effect was due to the GadY sRNA, we constructed a plasmid overexpressing GadY from a PLAC promoter. We subsequently confirmed expression of GadY from the plasmid by Northern blot, and validated its activity on the gadX transcript, a known target for GadY (data not shown) (Opdyke et al., 2004). Overexpression of GadY from this plasmid had no effect on the expression of the dpiB-L fusion (Figure 7). Similar results were obtained with the dpiB-S fusion (data not shown), ruling out a potential regulation of the fusions by this sRNA. To test if the effect seen with the initial plasmid could be due to transcriptional regulation by GadX and/or GadW, we introduced gadX, gadW and a triple gadXYW deletion mutant in both dpiB-lacZ fusions. As seen in Figure 7, either the gadX or gadW mutation modestly increased the activity of the dpiB-L fusion by 1.5 fold. Interestingly this effect was no longer seen when all three gadXYW genes were deleted. Taken together, these data indicate that GadX and GadW affect the dpiB fusion by a mechanism that is still unclear. In any case, this regulation is independent of RybC since none of the gad mutations had an effect on the expression of RybC as seen by Northern Blot (data not shown). Furthermore, overexpression of RybC from the pRybC plasmid efficiently decreased the activity of the dpiB-L fusion whether gadX and/or gadW were present on the chromosome or not (Figure 7), clearly indicating that RybC regulation is independent of the gad genes.
Figure 7. GadY does not regulate the dpiB-lacZ fusion.
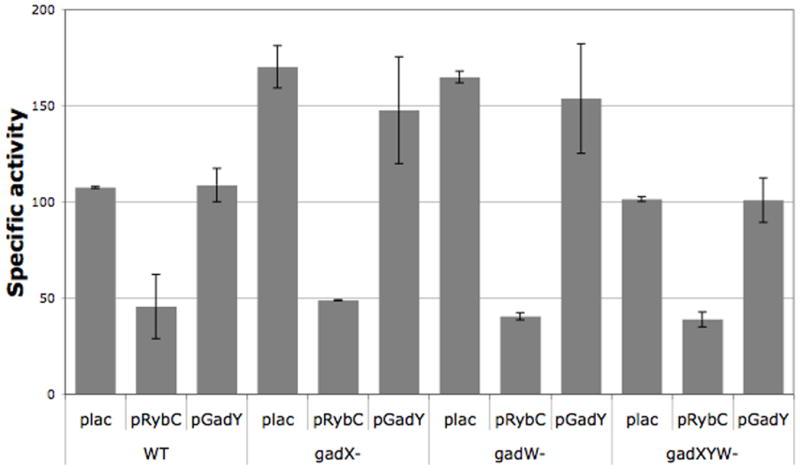
Overnight cultures of strains PM1211 (dpiB-L, WT), and their respective gadX (PM1291), gadW (PM1292) and gadXYW (PM1293) deletion derivatives, containing either the pBRplac (plac) control vector or the plasmid expressing RybC (pRybC) or GadY (pGadY), were diluted 500-fold in fresh LB medium containing ampicillin and 0.0005% arabinose. Cultures were grown at 37°C to exponential phase, at which point 100 μM IPTG was added to the medium. Samples were then taken at early stationary phase (OD600 of 3) and β-galactosidase assays were performed as previously described (Majdalani et al., 1998).
Discussion
While the identification of mRNA targets of sRNAs has been the subject of numerous investigations in the last few years, a smaller number of studies have focused on designing approaches to finding sRNAs regulators for specific genes of interest. We describe here a new genetic screen allowing such identification and validate its utility by finding an sRNA, RybC, regulating the DpiA/B two-component system.
A useful system for identifying post-transcriptional regulators
The system developed in this study allows rapid determination of the 5’ end of a gene of interest, via 5’ RACE, and the simultaneous creation of a translational fusion of the 5’ UTR and first part of the gene to lacZ. This fusion is created in single copy in the chromosome, and designed to be expressed from the PBAD promoter, rather than the native promoter. As a result, transcription of the fusion can be easily modulated via changes in arabinose concentration and, in most cases, only post transcriptional regulation should change expression of the fusion. Changes in the read-out from these reporters, expression of β-galactosidase, should reflect changes in mRNA stability, changes in translation, or both. It is possible, however, that repressor sites, fully contained within the transcribed region, might contribute to regulation in some cases. Note also that the constructed strain is free of antibiotic markers, allowing introduction of marked mutations as needed.
The advantages of this approach are many. While a previously identified sRNA of unknown function was found in this set of experiments, it should be possible to identify novel sRNA regulators of particular genes as well. It is therefore a particularly useful approach for organisms not as thoroughly mined for regulatory RNAs as E. coli. Furthermore, indirect regulators and/or proteins that carry out post-transcriptional regulation should also be detected. In this work, we identified gad genes as possible indirect regulators of dpiBA. While we used a multicopy library in this work, it should also be possible to identify regulators via mutagenesis of a host carrying a fusion of interest.
A major advantage provided by the use of an inducible promoter is the ability to study genes for which expression is low or of unknown regulation. A widely used approach to identify targets for a specific sRNA has been to express the sRNA from a regulated promoter and examine changes in microarrays (e.g. (De Lay and Gottesman, 2009; Massé et al., 2005)). Because the mRNA levels of the dpiBA operon are quite low even in the absence of RybC expression (data not shown), dpiBA might not have been identified as negatively regulated by RybC using the typical microarray approaches. Finally, the use of translational fusions allows the isolation of RNAs that affect translation without affecting mRNA stability, which would not be revealed by the microarray approach.
If specific target mRNAs are to be studied, the next question becomes which mRNAs are appropriate candidates for this approach. We chose to investigate the dpiBA encoding a two-component system, since its mRNA was found to bind the RNA chaperone Hfq in a previous set of experiments (Zhang et al., 2003). Further obvious candidates for sRNA regulation could be other mRNAs that have been shown to bind Hfq. Immunoprecipitation of Hfq and isolation of the bound RNAs, followed by detection on microarrays or by deep sequencing (Sittka et al., 2008; Zhang et al., 2003) can provide a list of candidate genes. However, signals for messages in such experiments should not be considered an essential pre-condition for choosing a given gene. While these techniques have allowed the identification of sRNAs with some success, expression conditions as well as technical issues (e.g. stability of the mRNAs during the course of the experiment) may render a global analysis of the mRNA targets more difficult, explaining why many known targets of sRNAs were not found in those studies.
Even though sRNAs represent a fraction of the genome of E. coli (~2%), they may control a much larger proportion of genes inside the cell. One of the best examples of the pleiotropic regulation that can be exerted by an sRNA comes from RyhB, which controls the expression of at least 56 genes organized in 18 transcriptional units, involved in iron metabolism (Massé et al., 2005). It is reasonable to assume that numerous genes will be found to be under the control of sRNAs even though they have not yet been shown to be associated with Hfq. Genes with conserved 5’ UTRs or for which unexplained post-transcriptional regulation has been observed, whether or not they have been shown to bind Hfq, are additional classes of genes likely to be regulated by sRNAs or other translational regulators. By providing a simple and effective way for making translational fusions of any gene to lacZ, our method could be an excellent way to evaluate such regulation relatively easily.
dpiBA as a target for small RNA regulation
Two-component regulators are among the most widespread and numerous of bacterial regulatory systems. In general, they consist of a sensor kinase capable of receiving an environmental signal that leads to changes in phosphorylation, and therefore activity, for a response regulator, generally a DNA binding protein. Frequently but not always, sensor kinase and response regulator are in the same operon, and in some cases, the promoter of this operon is regulated by the encoded regulators, providing a positive and/or negative feedback loop. Downstream gene expression is also tightly and reversibly regulated by the state of phosphorylation of the response regulator.
Recent studies suggest that post-transcriptional regulation by sRNAs provides an additional component to the regulatory architecture of these systems. The Hfq-binding binding sRNAs OmrA and B negatively regulate the two component system OmpR/EnvZ, which in turn are necessary for OmrA and OmrB synthesis, providing a negative feedback loop (Guillier and Gottesman, 2008). Another example of a negative feedback loop is provided by the Qrr sRNAs in Vibrio cholerae, which negatively regulate LuxO, the sigma54 dependent response regulator for Qrr synthesis, providing an important component of the mechanisms that keep the redundant Qrr sRNAs at appropriate levels (Svenningsen et al., 2008).
Regulation of dpiBA by RybC provides a different, and not totally understood paradigm for sRNA regulation of a two-component system. Unlike EnvZ/OmpR, there is no evidence that RybC, the regulator of dpiA/B, is itself regulated by DpiA/B, so that this is not a feedback system. In addition, because RybC is well expressed under most conditions and our attempts to find critical positive regulatory sites were unsuccessful, RybC may be relatively unique in not being an sRNA expressed under stress conditions but in being constitutively expressed. Attempts to find the signals or regulators controlling the expression of RybC using a transcriptional fusion of the promoter of the sRNA to lacZ (data not shown) were not successful, and deletion of all sequences upstream of the -35 region did not significantly decrease expression, suggesting the absence of upstream activators. If RybC is constitutively expressed, what is its role?
One hypothesis could be that RybC is used to tightly control the basal expression of DpiA/B. DpiA’s binding site on the DNA is similar to the A/T rich sites found in replication origins (Miller et al., 2003). As a result, in E. coli, over-expression of DpiA or induction of its expression by sub-lethal concentration of some β-lactams has been shown to trigger the SOS response, presumably by binding to inappropriate sites and interrupting DNA replication (Miller et al., 2004). While induction of the SOS response is a benefit to the cell in the latter case by providing a way to mitigate lethal effects from the antibiotics, inappropriate expression of the DpiA/B two-component system could lead to SOS-induced toxicity. It is thus possible that RybC may be produced to prevent expression of the two-component system in conditions when it could be toxic to the cell. While no obvious deficiency in cell growth or increase in expression of the dpiBA transcript from the chromosome could be observed in a rybC mutant strain when grown in rich medium (data not shown), it is possible that either a subpopulation of cells is expressing toxic levels of DpiA, or that expression is induced under specific conditions. In this model, redundant mechanisms may keep dpiBA expression low in normal growth conditions independently of RybC.
We carried out a variety of experiments to try to further test a role for RybC in the β-lactam-induced SOS response. RybC expression, measured either by Northern Blot or with the prybC-lacZ transcriptional fusion, was not changed by treatment with beta-lactams or affected by SOS induction (data not shown). We also saw no reproducible difference in viability of the rybC mutant, compared to the wild-type, in response to either beta-lactam treatment or SOS induction, but had difficulty replicating the basic Dpi-dependent SOS induction observations, making it impossible to evaluate the role of RybC in this process (data not shown). The low level of transcription, combined with the finding that RybC negatively regulates gene expression, suggests that long-term expression of these genes needs to be stringently regulated.
Processing of the dpiBA message may play a role in regulation. Using our method combining 5’RACE and recombineering, we were able to define two 5’ extremities for the dpiBA mRNA, located 22 nt apart. Strikingly, we found that the shorter form of the transcript, which we believe is due to processing rather than a second start site, is less susceptible to the negative regulation exerted by RybC than the longer form (Figure 5), even though both forms contain the sequence involved in the pairing with RybC (Figure 1C). These data indicate that the sequence present only in the longer form may be required for the action of RybC. One hypothesis that could explain these results is that the longer form is able to bind Hfq as opposed to the shorter form. Stem loops preceded or followed by A/U rich sequences have been described to be binding sites for Hfq (Brennan and Link, 2007). In line with the hypothesis that only the longer form binds Hfq, both UTRs are predicted to form a relatively loose stem loop structure when using the Mfold program (Zuker, 2003), but the longer form displays an A rich sequence in the 5’ end of this stem loop that would be absent in the shorter form (supplemental Figure 2). A further extension of this idea would reflect the recognition of similar sites to those bound by Hfq for RNase E cleavage (Morita et al., 2005). If the AT rich sequence just upstream of the second start site is in fact cut by RNase E, this might explain the origin of this 5’ end; Hfq binding might protect this site from RNase E. Thus any process that led to further RNase E cutting would lead to loss of stringent RybC regulation. Whether or not this cleavage event is regulated, and how it is regulated, remains to be determined. We note that the putative Hfq-binding sequence is well conserved in related organisms, although the RybC pairing to dpiB is not as well conserved. This suggests that dpi may be regulated by sRNAs other than RybC in other organisms.
Other roles for RybC
As with many other sRNAs acting in trans, it is likely that RybC has more than a single mRNA target. Identification of these other targets, as well as the signals (if any) that control the expression of RybC, may help to further elucidate its function and whether it may be related to the control of the SOS response through DpiA/B and possibly other unknown genes. As mentioned above, while RybC is found in Salmonella, Klebsiella, and Citrobacter, the pairing found between E. coli RybC and dpiB is not conserved in the leaders of the dpiB homologs in these organisms. Thus, it is likely that other targets have driven the conservation of this sRNA.
We used the TargetRNA (Tjaden et al., 2006) program to look in silico for other putative targets of RybC. By focusing the search on mRNAs that would bind to the same portion of RybC involved in the pairing with the dpiBA mRNA, one other gene, ybfM, a gene encoding a putative outer membrane porin of unknown function, is predicted to be a target of RybC. The predicted pairing involves the same sequence as the one involved in the pairing with DpiB; in this case, pairing is conserved in other organisms (Klebsiella, Citrobacter, Salmonella, Enterobacter). Recently, the regulation of ybfM by RybC was confirmed by P.Valentin-Hansen; they have renamed RybC MicM (Rasmussen, Johansen, Nielsen, Overgaard, Kallipolitis and Valentin-Hansen, submitted).
The approach and the regulatory interaction revealed by the work presented in this study provide assurance of even wider roles for regulatory RNAs than those described thus far. How these interactions have evolved and what roles they play in cell physiology continues to be a challenge, but will be important to address if an intensively studied organism like E. coli is ever to be fully understood.
Experimental procedures
Bacterial Strains and Plasmids
All E. coli strains used in this study are derivatives of E. coli K-12 MG1655 and are listed in Table 2. Plasmids were generally introduced into strains by TSS transformation (Chung and Miller, 1988). The ΔrybC∷kan, ΔgadX∷kan, ΔgadW∷kan and ΔgadXYW∷kan strains were generated by PCR amplification of the kan cassette of strain CRB316 (Ranquet and Gottesman, 2007) with the kan-for and kan-rev oligos listed in Table 3 followed by lambda Red recombinase-mediated gene replacement. Marked mutations were obtained and moved into the desired strain background using bacteriophage P1 transduction (Silhavy et al., 1984). Oligonucleotides used as probes and for polymerase chain reaction are described in Table 3. For cloning procedures, PCR amplification was carried out using the Expand High Fidelity PCR system (Roche) and DH5a was used as the recipient strain.
Table 2.
Strains and plasmids used in this study
| Strain or plasmid | Relevant features | Source or Reference |
|---|---|---|
| Strains | ||
| MG1655 | E. coli WT | Laboratory strain |
| CRB316 | MG1655 lacI’∷kan-PBAD-rpoS990-lacZ | (Ranquet and Gottesman, 2007) |
| NC397 | lacI’∷kan-Ter-cat sacB-lacZYA | (Svenningsen et al., 2005) |
| SG30115 | MG1655 mal∷lacIq | This study |
| PM1001 | MG1655Δlac ΔaraBAD araC+ | This study |
| PM1002 | PM1001 lacI’∷kan-PBAD-rpoS990-lacZ | This study |
| PM1004 | PM1002 lacI’∷kan-PBAD-cat-sacb:lacZ, miniλtetR | This study |
| PM1011 | PM1001 attλ∷PBAD-(-89)dpiB-lacZ | This study |
| PM1021 | PM1001 attλ∷PBAD-(-89)dpiB-lacZΔhfq∷cat | This study |
| PM1106 | PM1001 attλ∷rybC(-245)-lacZ | This study |
| PM1151 | PM1004 lacI’∷kan-rybC(-101)-lacZ | This study |
| PM1152 | PM1004 lacI’∷kan-rybC(-45)-lacZ | This study |
| PM1203 | SG30115 ΔaraBAD araC+ | This study |
| PM1205 | PM1203 lacI’∷ PBAD-cat-sacB:lacZ, miniλtetR | This study |
| PM1211 | PM1205 lacI’∷PBAD-dpibL-lacZ | This study |
| PM1212 | PM1205 lacI’∷ PBAD-dpibL-mut1-lacZ | This study |
| PM1213 | PM1205 lacI’∷ PBAD-dpibS-lacZ | This study |
| PM1214 | PM1205 lacI’∷ PBAD-dpibS-mut1-lacZ | This study |
| PM1222 | PM1211 ΔrybC∷kan | This study |
| PM1224 | PM1211 Δhfq∷cat | This study |
| PM1232 | PM1212 ΔrybC∷kan | This study |
| PM1242 | PM1213 ΔrybC∷kan | This study |
| PM1244 | PM1213 Δhfq∷cat | This study |
| PM1246 | PM1214 ΔrybC∷kan | This study |
| PM1251 | MG1655 ΔdpiBA∷kan | This study |
| PM1252 | MG1655 ΔrybC∷kan | This study |
| PM1253 | MG1655 Δhfq∷cat | This study |
| PM1291 | PM1211 ΔgadX∷kan | This study |
| PM1292 | PM1211 ΔgadW∷kan | This study |
| PM1293 | PM1211 ΔgadXYW∷kan | This study |
| Plasmids | ||
| pBR-plac | Ampr, plac promoter based expression vector | (Guillier et al., 2006) |
| pRybC | AatII-EcoRI rybC containing fragment cloned into pBR-plac | This study |
| pRybC-mut1 | C55G G56C G57C site directed mutation in pRybC | This study |
| pGadY | AatII-EcoRI gadY containing fragment cloned in pBR-plac | This study |
Table 3.
Primers and probes used in this study
| Primer or probe | sequence (5’ to 3’) | |
|---|---|---|
| Primers | ||
| PBAD-cat-for | ACCTGACGCTTTTTATCGCAACTCTCTACTGTTTCTCCATAATGAGACGTTGATCGGCACG | |
| lacZ-sacB-rev | TAACGCCAGGGTTTTCCCAGTCACGACGTTGTAAAACGACACTGTCCATATGCACAGATG | |
| lacI-PBAD-for | CGAAGCGGCATGCATTTACGTTGACACCATCGAATGGCGCGCGCTTCAGCCATACTTTTC | |
| Kan-rybC-for | ACGCCTGATGGCGGTCCAAAAAAAAGAGTCATCTTGCCTAAAAGCCACGTTGTGTCTCAA | |
| Kan-rybC-rev | AATGGCCAATATCGCTATTGGCCCGTCAAAGAGGAATTTCTTAGAAAAACTCATCGAGCA | |
| Kan-gadX-for | GCTACATTAATAAACAGTAATATGTTTATGTAATATTAAGAAAGCCACGTTGTGTCTCAA | |
| Kan-gadX-rev | TATGTCTGAGTAAAACTCTATAATCTTATTCCTTCCGCAGTTAGAAAAACTCATCGAGCA | |
| Kan-gadW-for | AACCAGTCACCCCCCATGCCGCTGGTGACGGCGATTTTTGAAAGCCACGTTGTGTCTCAA | |
| EcoRI-rybC(-245)-for | CGACGAATTCGTTTTGTAGACCTGATCCGG | |
| BamHI-rybC(-245)-rev | CGAGCGGATCCGCGACGGTGTAACTCGATTGTC | |
| PBAD-dpiB(-89)-rev | CATAGATATTAATTATGATCGTGGCAATGGAGAAACAGTAGAGAGTTGCG | |
| dpiB(-89)-for | TGCCACGATCATAATTAATATCTATG | |
| SmaI-dpiB(-89)rev | CGATCCCGGGCTGTTTATTCTCGTTAAGCTGCAAC | |
| rybC(-45)-for | GAGGGTGGCGGGCAGGACGCCCGCCATAAACTGCCAGGAACAAAAAAACGAAGCAAGCAT | |
| rybC(-101)-for | GAGGGTGGCGGGCAGGACGCCCGCCATAAACTGCCAGGAACCTAAGAGTATTGGCAGGAT | |
| deeplac | CGGGCCTCTTCGCTA | |
| AatII-rybC-for | TAAGACGTCACACCGTCGCTTAAAGTGAC | |
| EcoRI-rybC-rev | TGGGAATTCAAAAAAATGGCCAATATCGCTATTG | |
| QC-rybc-mut1-for | AAATGAAATTCCTCTTTGAGCCGCCAATAGCGATATTGGCC | |
| QC-rybc-mut1-rev | GGCCAATATCGCTATTGGCGGCTCAAAGAGGAATTTCATTT | |
| AatII-gadY-for | TAAGACGTCACTGAGAGCACAAAGTTTCCCG | |
| EcoRI-gadY-rev | TGGGAATTCAAAAAAACCCGGCATAGGGG | |
| PBAD-RNA | ACUCUCUACUGUUUCUCCAU | |
| dpiB-RT1 | GGAAAAATGCAAACTGTTTATTCTCG | |
| PBAD-DNA | ACCTGACGCTTTTTATCGCAACTCTCTACTGTTTCTCCAT | |
| dpiB-RT2 | TAACGCCAGGGTTTTCCCAGTCACGACGTTGTAAAACGACCTGTTTATTCTCGTTAAGCT | |
| dpiB-L-mut1-for | ACCTGACGCTTTTTATCGCAACTCTCTACTGTTTCTCCATATTTTGATTCAACATTTTAATTACATGGCTCAAAGAGGCTCGGGA | |
| dpiB-S-mut1-for | ACCTGACGCTTTTTATCGCAACTCTCTACTGTTTCTCCATACATGGCTCAAAGAGGCTCG | |
| 5’-PBAD | CGACGAATTCGCGCTTCAGCCATACTTTTCATAC | |
| Probes | ||
| RybC | CATTTTTTTATTATTATGCCGTCACTTTAAGCGACGGTG |
Strain PM1004 was created by first amplifying the cat-sacB cassette from strain NC397 (Svenningsen et al., 2005) using primers PBAD-cat-For and lacZ-sacB-rev. The PCR product was then recombined in strain PM1002 using mini λ mediated recombination as previously described (Court et al., 2003). The resulting strain, PM1004, was lacI’∷kan-PBAD-cat-sacB-lacZ. Strain PM1205 was constructed by amplifying the PBAD-cat-sacB-lacZ cassette using oligonucleotides lacI-PBAD-for and lacZ-sacB-rev and strain PM1004 genomic DNA as a template. The resulting PCR DNA fragment was recombined as previously described (Court et al., 2003) in the chromosome of strain PM1203, giving rise to strain PM1205.
The rybC (-245)-lacZ and PBAD-(-89) dpiB-lacZ translational fusion were constructed as follows. A DNA fragment corresponding to nts -245 to +10 respective to the transcription start site of rybC was amplified using primers EcoRI-rybC(-245)-for and BamHI-rybC(-245)-rev. The PCR product was subsequently cloned in between the EcoRI and BamHI sites of the pRS415 plasmid (Simons et al., 1987), giving rise to pRSRybC. The PBAD-(-89) dpiB-lacZ translational fusion (strain PM1011) was obtained by first amplifying the PBAD promoter and a sequence corresponding to nts −89 to +10 respective to the ATG translation start codon of dpiB, using primers 5’PBAD and PBAD-dpiB(-89)-rev, and dpiB(-89)-for and SmaI-dpiB(-89)-rev, respectively. The two PCR fragments were then joined by an overlap extension PCR and the resulting PCR product was subsequently cloned in frame with lacZ between the EcoRI and SmaI sites of the pRS414 plasmid (Simons et al., 1987), resulting in pRSdpiB. The pRSRybC and pRSdpiB plasmids were then crossed with λRS468 bacteriophage and monolysogens were constructed in strain PM1001 as previously described (Simons et al., 1987), giving rise to strains PM1106 and PM1011, respectively.
The rybC (-101)-lacZ (PM1151) and rybC (-45)-lacZ (PM1152) lacZ fusions were constructed as follows. DNA fragments corresponding to nts -101 to +10 and to nts -45 to +10 respective to the transcription start site of rybC were amplified by PCR using oligonucleotides rybC(-101)-for or rybC(-45)-for and deeplac, and strain PM1106 as template. The PCR products were subsequently recombined in strain PM1004 as previously described (Court et al., 2003), resulting in strain PM1151 and PM1152, respectively.
The pRybC and pGadY plasmids were constructed by first PCR amplifying rybC and gadY from strain MG1655 using AatII-rybC-for and EcoRI-rybC-rev primers and AatII-gadY-for and EcoRI-gadY-rev primers, respectively. The PCR product was subsequently cloned into the pBR-plac vector digested with AatII and EcoRI. The C55G G56C G57C site directed mutants of pRybC was constructed using the Quickchange II site directed mutagenesis kit (Stratagene) following the manufacturers instructions, with pRybC as a template and primers QC-rybc-mut1-for and QC-rybc-mut1-rev.
5’Rapid Amplification of cDNA Ends (RACE) coupled to construction of lacZ fusions
5′ RACE was carried out as previously described in (Argaman et al., 2001) with some modifications. Major modifications include the use of a new RNA adapter and of new DNA primers for RT-PCR that were designed to insert a sequence at each end of the final PCR products allowing λ red mediated recombination in strain PM1205 (see figure 1A and B).
For the first 5’RACE step, RNA was extracted from strain PM1102, which carries ΔrybC∷kan mutation, grown to an OD600 of 1.5. RNA 5′ triphosphates were converted to 5′ monophosphates by treatment with tobacco acid pyrophosphatase (TAP, Epicentre Technologies) at 37°C for 30 min. For control reactions, RNA was incubated in the absence of TAP. After phenol chloroform extraction and ethanol/sodium acetate RNA precipitation, pellets were dissolved in water and mixed with 300 pmol of the 5′ RNA adapter PBAD-RNA, and heat-denatured at 95°C for 5 min. The adapter was ligated for 16 h at 17°C with 40 U of T4-RNA ligase (New England Biolabs). Ligated RNA was then reverse transcribed using the Superscript III (Invitrogen) reverse transcriptase according to the manufacturer’s instructions, using the dpiB-RT1 oligo, followed by RNase H (New England Biolabs, 1 U) treatment for 20 min at 37°C. The cDNA was then amplified by PCR using DNA primers PBAD-DNA and dpiB-RT2. A sample of the PCR products was then TOPO cloned into the pCR4-TOPO vector (Invitrogen), and transformants were analyzed and sequenced as previously described (Argaman et al., 2001). The remaining PCR product was treated as follows for direct recombination into strain PM1205.
After the 5’RACE, PCR products harbor a 40 nt sequence homologous to the PBAD promoter (from nt -1 to -41 relative to the transcription start site of the PBAD promoter) at the 5’end of the desired gene (here dpiB) and a 40 nt sequence homologous to the lacZ ORF on its 3’end (from nt 25 to 65 relative to the ATG of lacZ) (Figure 1B). The PCR product was therefore inserted into the chromosome of strain PM1205 by lambda red mediated recombination as described previously (Court et al., 2003). Recombinants were selected for loss of the cat-sacB cassette on sucrose minimal plates (M63 salts, 0.2% glycerol, 5% sucrose) containing 40μg/ml of 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside (X-Gal). Three types of colonies appeared reflecting different β-galactosidase activity (see results). Eight colonies of each were purified and checked for sensitivity on LB+ 10 μg/ml chloramphenicol plates. Four clones of each type were then purified on LB plates and the resulting lacZ fusion were subsequently sequenced using oligo primer 5’PBAD and deeplac.
The dpiBL-mut1-lacZ and dpiBS-mut1-lacZ translational fusions that harbor a 3 nucleotide substitution in the predicted pairing sequence with RybC were constructed by amplifying the respective sequences with forward primers carrying the desired mutations, dpiB-L-mut1-for or dpiB-S-mut1-for, respectively, and deeplac as the reverse primer, using strain PM1211 or PM1213 genomic DNA as templates. The resulting PCR fragments were then recombined in the chromosome of strain PM1205 to generate strain PM1212 (PBAD-dpibL-mut1-lacZ) and PM1214 (PBAD-dpibS-mut1-lacZ).
RNA extraction and Northern blotting experiments
Overnight cultures of the strains to be analyzed were grown in LB liquid medium, containing ampicillin when necessary, then diluted 500-fold in fresh medium and incubated at 37°C. At appropriate OD600, 800 μL samples were removed from each culture and RNA was extracted from the samples using the hot phenol method described in (Massé et al., 2003). Northern blots were performed with 3 to 5 μg total RNA separated on a BioRad Criterion 10% TBE-Urea polyacrylamide gel that was pre-run for 30 min at 55V and subsequently run at 55V for 2 h in 1xTBE. RNA was then transferred onto a positively charged nylon membrane (Zeta Probe, BioRad) at 200 mA for 1 h, in 0.5X TBE. After UV crosslinking, the membrane was hybridized with the 5’-biotinylated specific probes in ULTRAhyb solution (Ambion) at 42°C and sRNAs were detected using the Brightstar Biotect Kit (Ambion) according to the manufacturer’s instructions.
β-galactosidase assays
β-galactosidase assays were performed as previously described (Majdalani et al., 1998). Briefly, overnight cultures were diluted 500-fold in fresh LB medium containing 0.0005% arabinose with appropriate antibiotics and grown at 37°C. 100μl samples were taken during growth at regular time intervals and arrested by lysis in permeabilization buffer. The calculated specific activities correspond to kinetic measurements of Vmax/OD600 as read on a SpectraMax 250 microtiter plate reader (Molecular Devices).
Acknowledgments
We thank members of the laboratory and Gisela Storz for comments on the manuscript and advice and discussion throughout the work. We thank P. Valentin-Hansen for sharing unpublished information, and thank S. N. Cohen and J. Opdyke for strains and advice. This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
References
- Aiba H. Mechanism of RNA silencing by Hfq-binding small RNAs. Curr Opin Microbiol. 2007;10:134–139. doi: 10.1016/j.mib.2007.03.010. [DOI] [PubMed] [Google Scholar]
- Argaman L, Hershberg R, Vogel J, Bejerano G, Wagner EG, Margalit H, Altuvia S. Novel small RNA-encoding genes in the intergenic regions of Escherichia coli. Curr Biol. 2001;11:941–950. doi: 10.1016/s0960-9822(01)00270-6. [DOI] [PubMed] [Google Scholar]
- Bott M, Meyer M, Dimroth P. Regulation of anaerobic citrate metabolism in Klebsiella pneumoniae. Mol Microbiol. 1995;18:533–546. doi: 10.1111/j.1365-2958.1995.mmi_18030533.x. [DOI] [PubMed] [Google Scholar]
- Brennan RG, Link TM. Hfq structure, function and ligand binding. Curr Opin Microbiol. 2007;10:125–133. doi: 10.1016/j.mib.2007.03.015. [DOI] [PubMed] [Google Scholar]
- Castanie-Cornet MP, Penfound TA, Smith D, Elliott JF, Foster JW. Control of acid resistance in Escherichia coli. J Bacteriol. 1999;181:3525–3535. doi: 10.1128/jb.181.11.3525-3535.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung CT, Miller RH. A rapid and convenient method for the preparation and storage of competent bacterial cells. Nucleic Acids Res. 1988;16:3580. doi: 10.1093/nar/16.8.3580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Court DL, Swaminathan S, Yu D, Wilson H, Baker T, Bubunenko M, Sawitzke J, Sharan SK. Mini-lambda: a tractable system for chromosome and BAC engineering. Gene. 2003;315:63–69. doi: 10.1016/s0378-1119(03)00728-5. [DOI] [PubMed] [Google Scholar]
- De Lay N, Gottesman S. The Crp-activated small noncoding regulatory RNA CyaR (RyeE) links nutritional status to group behavior. J Bacteriol. 2009;191:461–476. doi: 10.1128/JB.01157-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottesman S, McCullen CA, Guillier M, Vanderpool CK, Majdalani N, Benhammou J, Thompson KM, FitzGerald PC, Sowa NA, FitzGerald DJ. Small RNA regulators and the bacterial response to stress. Cold Spring Harb Symp Quant Biol. 2006;71:1–11. doi: 10.1101/sqb.2006.71.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillier M, Gottesman S, Storz G. Modulating the outer membrane with small RNAs. Genes Dev. 2006;20:2338–2348. doi: 10.1101/gad.1457506. [DOI] [PubMed] [Google Scholar]
- Guillier M, Gottesman S. The 5’ end of two redundant sRNAs is involved in the regulation of multiple targets, including their own regulator. Nucleic Acids Res. 2008;36:6781–6794. doi: 10.1093/nar/gkn742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingmer H, Miller CA, Cohen SN. Destabilized inheritance of pSC101 and other Escherichia coli plasmids by DpiA, a novel two-component system regulator. Mol Microbiol. 1998;29:49–59. doi: 10.1046/j.1365-2958.1998.00895.x. [DOI] [PubMed] [Google Scholar]
- Ma Z, Richard H, Tucker DL, Conway T, Foster JW. Collaborative regulation of Escherichia coli glutamate-dependent acid resistance by two AraC-like regulators, GadX and GadW (YhiW) J Bacteriol. 2002;184:7001–7012. doi: 10.1128/JB.184.24.7001-7012.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majdalani N, Cunning C, Sledjeski D, Elliott T, Gottesman S. DsrA RNA regulates translation of RpoS message by an anti-antisense mechanism, independent of its action as an antisilencer of transcription. Proc Natl Acad Sci U S A. 1998;95:12462–12467. doi: 10.1073/pnas.95.21.12462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majdalani N, Vanderpool CK, Gottesman S. Bacterial small RNA regulators. Crit Rev Biochem Mol Biol. 2005;40:93–113. doi: 10.1080/10409230590918702. [DOI] [PubMed] [Google Scholar]
- Mandin P, Repoila F, Vergassola M, Geissmann T, Cossart P. Identification of new noncoding RNAs in Listeria monocytogenes and prediction of mRNA targets. Nucleic Acids Res. 2007;35:962–974. doi: 10.1093/nar/gkl1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massé E, Escorcia FE, Gottesman S. Coupled degradation of a small regulatory RNA and its mRNA targets in Escherichia coli. Genes Dev. 2003;17:2374–2383. doi: 10.1101/gad.1127103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massé E, Vanderpool CK, Gottesman S. Effect of RyhB small RNA on global iron use in Escherichia coli. J Bacteriol. 2005;187:6962–6971. doi: 10.1128/JB.187.20.6962-6971.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massé E, Salvail H, Desnoyers G, Arguin M. Small RNAs controlling iron metabolism. Curr Opin Microbiol. 2007;10:140–145. doi: 10.1016/j.mib.2007.03.013. [DOI] [PubMed] [Google Scholar]
- Miller C, Ingmer H, Thomsen LE, Skarstad K, Cohen SN. DpiA binding to the replication origin of Escherichia coli plasmids and chromosomes destabilizes plasmid inheritance and induces the bacterial SOS response. J Bacteriol. 2003;185:6025–6031. doi: 10.1128/JB.185.20.6025-6031.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller C, Thomsen LE, Gaggero C, Mosseri R, Ingmer H, Cohen SN. SOS response induction by beta-lactams and bacterial defense against antibiotic lethality. Science. 2004;305:1629–1631. doi: 10.1126/science.1101630. [DOI] [PubMed] [Google Scholar]
- Morita T, Maki K, Aiba H. RNase E-based ribonucleoprotein complexes: mechanical basis of mRNA destabilization mediated by bacterial noncoding RNAs. Genes Dev. 2005;19:2176–2186. doi: 10.1101/gad.1330405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opdyke JA, Kang JG, Storz G. GadY, a small-RNA regulator of acid response genes in Escherichia coli. J Bacteriol. 2004;186:6698–6705. doi: 10.1128/JB.186.20.6698-6705.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranquet C, Gottesman S. Translational regulation of the Escherichia coli stress factor RpoS: a role for SsrA and Lon. J Bacteriol. 2007;189:4872–4879. doi: 10.1128/JB.01838-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan B, Soll D. The bacterial YbaK protein is a Cys-tRNAPro and Cys-tRNA Cys deacylase. J Biol Chem. 2005;280:25887–25891. doi: 10.1074/jbc.M502174200. [DOI] [PubMed] [Google Scholar]
- Seraphin B. Sm and Sm-like proteins belong to a large family: identification of proteins of the U6 as well as the U1, U2, U4 and U5 snRNPs. Embo J. 1995;14:2089–2098. doi: 10.1002/j.1460-2075.1995.tb07200.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silhavy T, Berman M, Enquist L. Experiments with gene fusions. Cold Spring Harbor Laboratory; 1984. [Google Scholar]
- Simons RW, Houman F, Kleckner N. Improved single and multicopy lac-based cloning vectors for protein and operon fusions. Gene. 1987;53:85–96. doi: 10.1016/0378-1119(87)90095-3. [DOI] [PubMed] [Google Scholar]
- Sittka A, Lucchini S, Papenfort K, Sharma CM, Rolle K, Binnewies TT, Hinton JC, Vogel J. Deep sequencing analysis of small noncoding RNA and mRNA targets of the global post-transcriptional regulator, Hfq. PLoS Genet. 2008;4:e1000163. doi: 10.1371/journal.pgen.1000163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svenningsen SL, Costantino N, Court DL, Adhya S. On the role of Cro in lambda prophage induction. Proc Natl Acad Sci U S A. 2005;102:4465–4469. doi: 10.1073/pnas.0409839102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svenningsen SL, Waters CM, Bassler BL. A negative feedback loop involving small RNAs accelerates Vibrio cholerae’s transition out of quorum-sensing mode. Genes Dev. 2008;22:226–238. doi: 10.1101/gad.1629908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tjaden B, Goodwin SS, Opdyke JA, Guillier M, Fu DX, Gottesman S, Storz G. Target prediction for small, noncoding RNAs in bacteria. Nucleic Acids Res. 2006;34:2791–2802. doi: 10.1093/nar/gkl356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tramonti A, Visca P, De Canio M, Falconi M, De Biase D. Functional characterization and regulation of gadX, a gene encoding an AraC/XylS-like transcriptional activator of the Escherichia coli glutamic acid decarboxylase system. J Bacteriol. 2002;184:2603–2613. doi: 10.1128/JB.184.10.2603-2613.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulbrandt ND, Newitt JA, Bernstein HD. The E. coli signal recognition particle is required for the insertion of a subset of inner membrane proteins. Cell. 1997;88:187–196. doi: 10.1016/s0092-8674(00)81839-5. [DOI] [PubMed] [Google Scholar]
- Urban JH, Vogel J. Translational control and target recognition by Escherichia coli small RNAs in vivo. Nucleic Acids Res. 2007;35:1018–1037. doi: 10.1093/nar/gkl1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderpool CK, Gottesman S. Involvement of a novel transcriptional activator and small RNA in post-transcriptional regulation of the glucose phosphoenolpyruvate phosphotransferase system. Mol Microbiol. 2004;54:1076–1089. doi: 10.1111/j.1365-2958.2004.04348.x. [DOI] [PubMed] [Google Scholar]
- Vogel J, Bartels V, Tang TH, Churakov G, Slagter-Jager JG, Huttenhofer A, Wagner EG. RNomics in Escherichia coli detects new sRNA species and indicates parallel transcriptional output in bacteria. Nucleic Acids Res. 2003;31:6435–6443. doi: 10.1093/nar/gkg867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel J, Sharma CM. How to find small non-coding RNAs in bacteria. Biol Chem. 2005;386:1219–1238. doi: 10.1515/BC.2005.140. [DOI] [PubMed] [Google Scholar]
- Waller PR, Sauer RT. Characterization of degQ and degS, Escherichia coli genes encoding homologs of the DegP protease. J Bacteriol. 1996;178:1146–1153. doi: 10.1128/jb.178.4.1146-1153.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang A, Wassarman KM, Rosenow C, Tjaden BC, Storz G, Gottesman S. Global analysis of small RNA and mRNA targets of Hfq. Mol Microbiol. 2003;50:1111–1124. doi: 10.1046/j.1365-2958.2003.03734.x. [DOI] [PubMed] [Google Scholar]
- Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]