

Abstract
Diamine linkers for the synthesis of dendrimers based on melamine were identified using competition reactions. The relative reactivity of the cyclic monoamines surveyed vary in relative reactivity by 40 times, expanding the previously identified series to an overall relative reactivity range of 320 times. Azetidine is 40 times more reactive than the cyclic, nine-membered ring (C8H17N), and 320 times more reactive than benzylamine. Reactivity differences are attributed to pKa values and sterics. Incorporating these groups into diamines provides linkers that can be employed in dendrimer synthesis. Specifically, the nucleophilicity of the individual amine groups comprising 3-aminoazetidine, 3-aminopyrrolidine, and 4-aminopiperidine vary by 100 times, 70 times, and 20 times, respectively. These linkers are incorporated into a generation three dendrimer.
Our group has invested significant energies in the synthesis of dendrimers based on triazines, also referred to as dendrimers based on melamine deriving from our use of diamine linkers.1,2 Our early targets commonly incorporated p-aminobenzylamine because of the significant differences in the reactivity of the amines of this group. That is, during a convergent synthesis, protecting group manipulations and functional group interconversions could be avoided because the benzylic amine would react preferentially (essentially exclusively) with the monochlorotriazine dendron being elaborated.1,3–7 The low stability of these aniline derivatives required reasonable, but additional, care on handling. While the use of distilled solvents and inert atmospheres are not uncommon, nor is the requirement for refrigerated storage of intermediates, we recognized that these precautions could impact the broader acceptance of these materials.
The low cost and high reactivity of piperazine soon made it a linking diamine of choice for our investigations. However, when using piperazine, dimerization of monochlorotriazines was observed under non-ideal reaction conditions that were usually attributed to concentration, rate of addition, ineffective stirring or lack thereof, temperature of addition and the magnitude of stoichiometric excess.4,8–18 Reactions with either p-aminobenzylamine or piperazine could be readily followed by NMR. The shift of the benzylic protons on reaction with the monochlorotriazine dendron or the desymmetrization of methylene groups of the piperazine group were diagnostic. The use of piperazine led to the undesirable formation of dimers, unfortunately. Detecting these dimers by NMR proves difficult due to signal degeneracy between the desired asymmetric monoamine and the symmetric dimer.
The wealth of commercially available diamines led us to conduct a rational survey of reactivity. Our goal was to identify diamine linkers with the reactivity differences displayed in p-aminobenzylamine without the disadvantages previously described. Our original study examined a range of primary and secondary amines including the cyclic amines piperidine and two piperazine derivatives (A–F, Figure 1).1 From these studies, aminomethylpiperidine emerged as a diamine linker of choice and was used extensively.1,2,7,9,15–17,19,20 Using competition experiments, the relative reactivity difference measured for the cyclic secondary amine and primary amine of aminomethylpiperidine is ~ 20 times. Theoretically, 5% of the product formed might derive from reaction of the monochlorotriazine dendron with the primary amine instead of the desired secondary amine. This population difference approaches the limit of detection by NMR spectroscopy, and accordingly, the efforts described here were undertaken to identify other suitable diamine linkers. These amines are identified as G–K in Figure 1. The diamine linkers identified from the relative reactivity data were incorporated into a dendrimer as a proof-of-concept.
Figure 1.

Relative reactivity map for the substitution of monochlorotriazines. The reactivity difference between consecutive amines is shown on the scale.
To establish relative reactivity of different monoamines, reactions were conducted by treating 1 equiv. of a monochlorotriazine, DMTA, with 3 equiv. of two competing amines chosen from G–K at room temperature (Supporting Information). The reactivity map was obtained by determining the product ratios using 1H NMR after disappearance of DMTA. These values for individual competition studies are consistent multiplicatively across the range of amines within experimental error. Protons α to the amine generally appeared downfield from those of the free amine, and provided convenient signals for measuring product distributions. Including A and F allow us to compare this new series to the existing series.
The data show that as ring size decreases, the reactivity of the amine increases.21 The pKa of the conjugate acids of most reactive amines (G, H, and A) is 11.3, while the pKa of the conjugate acids of less reactive amines (I–K) is 10.8.22,23 Sterics can be used to rationalize the difference in reactivity within these subgroups.
From this data, we identified new diamine linkers based on three criteria. First, a minimum reactivity difference of ~ 20 times between the amines should reduce dimer formation. Second, one or more unique 1H NMR signals should facilitate characterization. Third, the diamine should be commercial availability or accessibility in a minimal number of steps. Diamine linkers L1–L3 (Scheme 1) were chosen because they meet the criteria and are commercially available. A single enantiomer of 3-aminopyrrolidine was used. These linkers were then evaluated by preparing dendrimer 1.
Scheme 1.
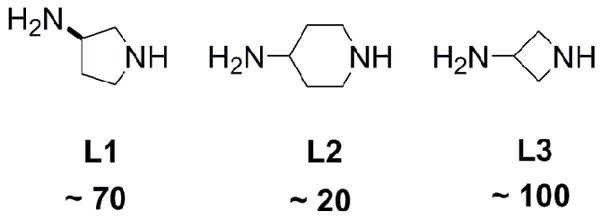
Three new diamine linkers with the relative reactivity difference of the diamines indicated.
Scheme 2 shows the convergent strategy used to synthesize dendrimer 1. Following selective protection of the primary amines of 3,3′–diaminodipropylamine with BOC–ON, treatment with cyanuric chloride affords monochlorotriazine 2. Intermediate 2 is treated with an excess of L1 to produce 3. In an iterative fashion, the synthesis continues with reaction of cyanuric chloride to form 4, then L2 to generate 5. Cyanuric chloride treated with a slight excess of 5 gives 6. While iteration with 3-aminoazetidine progresses the sequence, sterics precludes trimerization with a cyanuric chloride core.
Scheme 2.
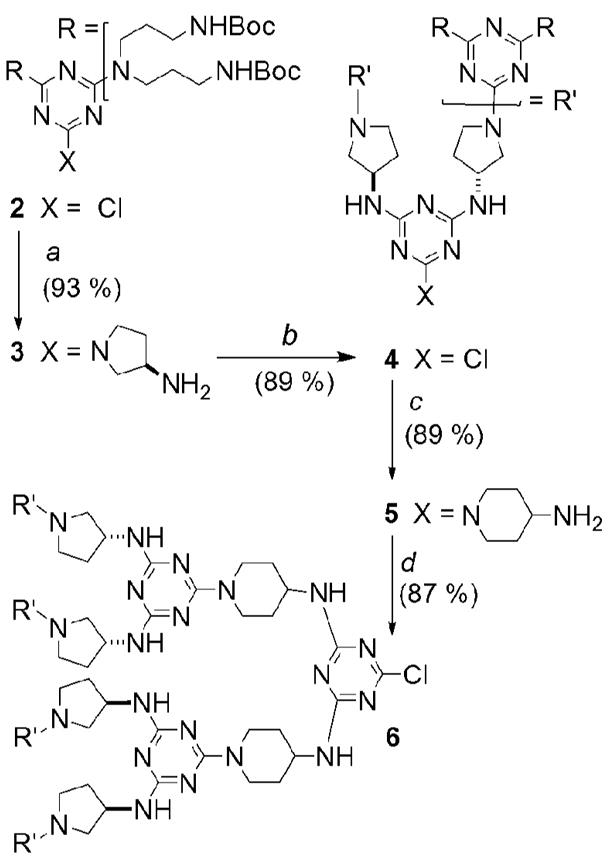
Synthesis of G3 dendron 6. a) THF, rt, overnight. b) THF, cyanuric chloride, Hunig’s Base (DIPEA), 40 °C, overnight. c) THF, rt, overnight. d) THF, cyanuric chloride, Hunig’s Base (DIPEA), 40 °C, 2d. e) THF, 8, BEMP resin, 70 °C, 7d.
Instead, a less sterically encumbered core, 8, is synthesized by treating cyanuric chloride with L3 followed by deprotection (Scheme 3).
Scheme 3.
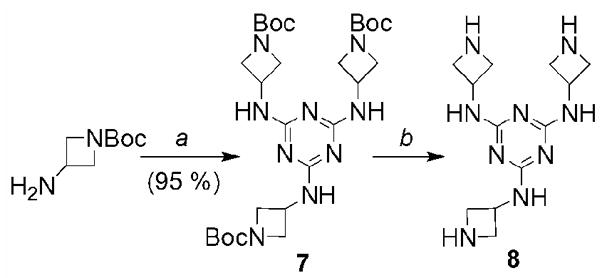
Synthesis of the core, 8. a) THF, cyanuric chloride, Hunig’s Base (DIPEA), 70 °C, 7d. b) (1:1) TFA:DCM, rt, overnight.
This strategy affords a highly reactive core possessing three azetidine groups which yield dendrimer, 1, after reaction with a large excess of 6 (Scheme 4). The reported yield of 36% represents the amount of material obtained in pure form after extensive chromatography, and is not a reflection of an unsuccessful reaction. Indeed, we estimate conversion to product occurred in >80%.
Scheme 4.

Synthesis of 1. e) THF, BEMP resin, 70 °C, 7d.
In concluding, we foresee that each linker offers interesting features that could be exploited in future work. Aminoazetidine (L1) offers a highly reactive and sterically unencumbered amine that might find use in situations where piperidine-type amines are unreactive or sluggish. Aminopyrrolidine (L2) offers opportunities to explore chiral environments in these dendrimers. Aminopiperidine (L3) offers an inexpensive linker that aligns with our current reliance on aminomethylpiperidine groups. In addition, these linkers convey spectroscopically unique signatures to different regions of the dendrimer architecture; an effect only rarely observed in related architectures.24–29
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (NIGMS 65460). KXM thanks the National Science Foundation for a predoctoral fellowship through the NSF GK-12 program.
Footnotes
Supplementary Data
Supplementary data associated with this article can be found, in the online version, …
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Steffensen MB, Simanek EE. Org Lett. 2003;5:2359. doi: 10.1021/ol0347491. [DOI] [PubMed] [Google Scholar]
- 2.Steffensen MB, Simanek EE. Angew Chem Int Ed. 2004;43:5177. doi: 10.1002/anie.200460031. [DOI] [PubMed] [Google Scholar]
- 3.Zhang W, Simanek EE. Org Lett. 2000;2:843. doi: 10.1021/ol005585g. [DOI] [PubMed] [Google Scholar]
- 4.Zhang W, Simanek EE. Tet Lett. 2001;42:5355. [Google Scholar]
- 5.Zhang W, Nowlan DT, Thomson LM, Lackowski WM, Simanek EE. J Am Chem Soc. 2001;123:8914. doi: 10.1021/ja0041369. [DOI] [PubMed] [Google Scholar]
- 6.Zhang W, Gonzalez SO, Simanek EE. Macromolecules. 2002;35:9015. [Google Scholar]
- 7.Zhang W, Tichy SE, Perez LM, Maria GC, Lindahl PA, Simanek EE. J Am Chem Soc. 2003;125:5086. doi: 10.1021/ja0210906. [DOI] [PubMed] [Google Scholar]
- 8.Bell SA, McLean ME, Oh SK, Tichy SE, Zhang W, Corn RM, Crooks RM, Simanek EE. Bioconjugate Chem. 2003;14:488. doi: 10.1021/bc020075n. [DOI] [PubMed] [Google Scholar]
- 9.Umali AP, Simanek EE. Org Lett. 2003;5:1245. doi: 10.1021/ol034161u. [DOI] [PubMed] [Google Scholar]
- 10.Zhang W, Jiang J, Qin C, Perez LM, Parrish AR, Safe SF, Simanek EE. Supramolecular Chemistry. 2003;15:607. [Google Scholar]
- 11.Neerman MF, Chen HT, Parrish AR, Simanek EE. Molecular Pharmaceutics. 2004;1:390. doi: 10.1021/mp049957p. [DOI] [PubMed] [Google Scholar]
- 12.Neerman MF, Zhang W, Parrish AR, Simanek EE. Int J Pharm. 2004;281:129. doi: 10.1016/j.ijpharm.2004.04.023. [DOI] [PubMed] [Google Scholar]
- 13.Chen HT, Neerman MF, Parrish AR, Simanek EE. J Am Chem Soc. 2004;126:10044. doi: 10.1021/ja048548j. [DOI] [PubMed] [Google Scholar]
- 14.Acosta EJ, Gonzalez SO, Simanek EE. J Poly Sci, Part A: Polym Chem. 2005;43:168. [Google Scholar]
- 15.Hollink E, Simanek EE. Org Lett. 2006;8:2293. doi: 10.1021/ol060559p. [DOI] [PubMed] [Google Scholar]
- 16.Yoo S, Lunn JD, Gonzalez SO, Ristich JA, Simanek EE, Shantz DF. Chem Mater. 2006;18:2935. [Google Scholar]
- 17.Simanek, E. E.; Hollink, E. US Patent 041933.
- 18.Crampton HL, Hollink E, Perez LM, Simanek EE. New J Chem. 2007;7:1283. doi: 10.1039/b617875h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Acosta EJ, Carr CS, Simanek EE, Shantz DF. Adv Mater. 2004;16:985. [Google Scholar]
- 20.Lim J, Simanek EE. Molecular Pharmaceutics. 2005;2:273. doi: 10.1021/mp050030e. [DOI] [PubMed] [Google Scholar]
- 21.Caswell LR, Goldsmith ME. J Org Chem. 1989;54:5101. [Google Scholar]
- 22.Searles S, Tamres M, Block F, Quarterman LA. J Am Chem Soc. 1956;78:4917. [Google Scholar]
- 23.Frenna V, Vivona N, Consiglio G, Spinelli D. J Chem Soc, Perkin Trans II: Phys Org Chem. 1985;12:1865. [Google Scholar]
- 24.Ihre H, Hult A, Soderlind E. J Am Chem Soc. 1996;118:6388. [Google Scholar]
- 25.Gorman CB, Hager MW, Parkhurst BL, Smith JC. Macromolecules. 1998;31:815. [Google Scholar]
- 26.Lellek V, Stibor I. J Mater Chem. 2000;10:1061. [Google Scholar]
- 27.Murer PK, Lapierre JM, Greiveldinger G, Seebach D. Helv Chim Acta. 1997;80:1648. [Google Scholar]
- 28.Chai M, Niu Y, Youngs WJ, Rinaldi PL. J Am Chem Soc. 2001;123:4670. doi: 10.1021/ja002824m. [DOI] [PubMed] [Google Scholar]
- 29.van Genderen MHP, Baars MWPL, van Hest JCM, de Brabander - van den Berg EMM, Meijer EW. Recl Trav Chim Pays – Bas. 1994;113:573. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.