

Abstract
Protein-protein interactions sequester enzymes close to their substrates. Protein kinase C (PKC) is one example of a ubiquitous signaling molecule with effects that are dependent upon localization. Short peptides derived from interaction sites between each PKC isozyme and its receptor for activated C kinase act as highly specific inhibitors and have become available as selective drugs in basic research and animal models of human diseases, such as myocardial infarction and hyperglycemia. Whereas the earlier inhibitory peptides are highly specific, we believe that peptides targeting additional interactions between PKC and selective substrates will generate even more selective tools that regulate different functions of individual isozymes. Here, we discuss the methodologies and applications for identifying selective regulators of PKC.
The evolution of rational drug design
The process of drug discovery has evolved from a series of serendipitous findings to a more systematic search accompanied by rationally designed molecules. The evolution of this process is best illustrated by the discovery of aspirin. The benefit of willow leaves in reducing pain and inflammation was first described by the Babylonians nearly 4000 years ago and later prescribed as a medication by Hippo-crates [1]. Two millennia later, salicylic acid was purified to reduce side-effects associated with other components in the plant extract, and by the end of the 19th century, this substance was sold by Bayer as acetylsalicylic acid under the name aspirin [1]. The mechanism of action of aspirin and the targets of aspirin, the cyclo-oxygenase enzymes (see Glossary), were discovered 80 years later [2], and the crystal structure of this enzyme was solved as recently as 1994 [3,4]. The ability to use automated high-throughput screens of thousands of molecules and the development of more advanced nuclear magnetic resonance (NMR), X-ray crystallography and molecular dynamic simulation techniques resulted in more systematic searches of new drugs and directed small molecule design based on three-dimensional information of proteins bound with their ligands. These two seemingly incongruent approaches (unbiased search vs rational design) are in fact complementary, leading to the current approach to drug discovery.
Glossary.
- β sandwich
a motif composed of eight β strands creating two flat sheets connected with a short helix ‘hinge’. The C2 domain can bind 2-3 calcium ions, thus increasing interaction with negatively charged phospholipids (i.e. phosphatidylserine) in membranes. Calcium and lipid binding is a common feature of C2 domains.
- Cyclo-oxygenase enzymes
a family of enzymes responsible for the conversion of arachidonic acid to prostaglandins. They are the targets of non-steroidal anti-inflammatory agents, which can reduce symptoms of inflammation and pain.
- Diacylglycerol
a second messenger signaling molecule, which anchors PKC to membranes, resulting in its activation. It is generated from the phospholipase-C2-dependent cleavage of phosphatidyl inositol bisphosphate and is mimicked by phorbol esters, such as phorbol 12-myristate 13-acetate.
- First-generation peptide regulators
short peptides (6-10 amino acids) that either disrupt or enhance PKC isozyme binding to their respective RACKs.
- Second-generation peptide regulators
short peptides that can regulate PKC subdomain interactions with target substrates independent of RACK binding, therefore conferring even greater specificity.
- Isozymes
homologous enzymes that are products of different genes or alternatively spliced mRNA of the same gene and, thus, belong to the same family of enzymes.
- Constraints (local and global)
as applied here, constraints introduced within a peptide substantially reduce the theoretically possible conformational states of a linear peptide and are powerful tools for studying interactions in biological systems. Local constraints can be enforced by using unnatural or natural amino acids, and global constraints can be induced by cyclization of the peptide.
- Molecular dynamic simulation
computer simulation that enables modeling of the three-dimensional position of atoms within a molecule to understand dynamic changes in their structure and interactions.
- Phospholipase C
an enzyme that catalyzes the hydrolysis of phospholipids such as phosphatidyl inositol bisphosphate to diacylglycerol and the remaining lipid head group, inositol trisphosphate. Similar to PKC, phospholipase C contains a C2 domain, which enables its anchoring to different proteins.
- Phospholipase D
an enzyme that catalyzes the hydrolysis of phosphatidylcholine to phosphatidic acid that can be further converted to diacylglycerol. Similar to PKC, phospholipase D contains a C2 domain, enabling its anchoring to different proteins.
- Pseudo RACKs
regions on PKC that are homologous to the RACK protein. The pseudo-RACK site in PKC binds to the RACK-binding site in an intramolecular interaction, which stabilizes the isozyme in its inactive state. Peptides corresponding to this sequence interfere with the intramolecular interaction and, thus, activate PKC.
- Phorbol 12-myristate 13-acetate
a tumor promoter that mimics diacylglycerol binding to and activation of PKC.
- RACKs
proteins in the cellular particulate fraction that bind activated PKC in a saturable and specific manner but are themselves not substrates of PKC.
- Synaptotagmin
a protein involved in synaptic vesicle discharge. The protein has no catalytic activity, but it contains two repeats of the C2 domain and, like PKC, these domains enable its anchoring to membranes.
Structural information on drug-protein interactions identifies binding pockets for small molecules in the protein targets and provides measurements of the forces that govern the binding of these small molecules to their protein. This has facilitated the rational design of drugs that mimic or compete with these interactions. However, many crucial signaling events in the cell occur in multiprotein complexes and involve multiple protein-protein interactions. Identification of drugs that interfere with these protein-protein interactions has proven difficult because the interaction sites between two proteins constitute large, flat surfaces rather than small hydrophobic pockets. Nevertheless, protein-protein interactions are crucial in signal transduction [5-7], and inhibitors of such interactions are potentially useful drugs.
Over the past 20 years, focus has been on identifying means to regulate the function of individual members of the protein kinase C (PKC) family. This was a challenge because of the presence of multiple isozymes, which have different activation requirements, within the PKC family (Box 1). PKC isozymes have distinct regulatory and catalytic domains (Figure 1). The C2 domain has been identified as a region within the regulatory domain of these enzymes that mediates protein-protein interactions between individual PKC isozymes and their anchoring proteins, receptors for activated C kinase (RACKs) (Figure 2). Using a rational approach, we successfully generated short peptides derived from this C2 domain and other PKC domains that bind to the specific RACKs and disrupt the anchoring and function of the corresponding PKC isozymes (Figure 2a). These PKC-regulating peptides mimic the interaction site on PKC or its RACK, thereby stimulating or inhibiting the resulting signaling pathways in a highly specific manner, which is necessary to generate effective therapeutic agents.
Box 1. Modes of PKC activation.
PKC is a family of highly homologous kinases that differ in their activation requirements and substrate specificities. The main mechanism of activation includes stimulation of G-protein coupled receptors (GPCRs), leading to activation of phospholipase C (PLC). The classical isozymes (α, βI, βII and γ) are activated by diacylglycerol (DAG), a lipid-derived second messenger generated through the PLC-dependent hydrolysis of phosphatidyl inositol bisphosphate (PIP2) and elevated cellular calcium, which is increased by inositol trisphosphate, another PIP2 hydrolysis product. The novel isozymes (ε, δ, η and θ) are insensitive to calcium because of the lack of crucial residues in their C2 domain, and the atypical isozymes (ζ and λ/ι) are not sensitive to either DAG or calcium (Figure 1). GPCR-independent mechanisms of activation include oxidation of key regulatory residues in PKC [120], peroxide-mediated phosphorylation [121] and direct binding of steroid hormones to the C2 domain [122]. Because of the ubiquitous nature of these isozymes and their wide tissue distribution, the signaling pathways they are involved in are extensive.
Figure 1.
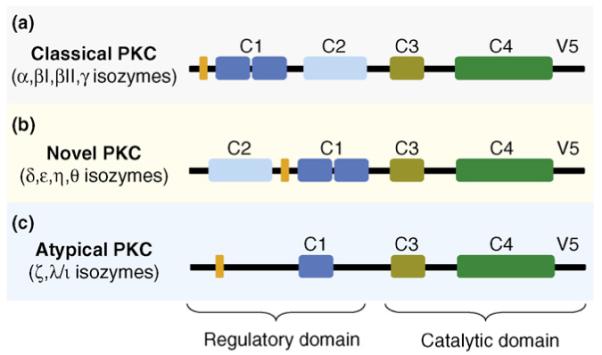
PKC isozymes and domains. PKC isozymes are classified based upon their activation requirements, which are the result of differences in domains within the regulatory region. (a) The classical isozymes (α, βI, βII and γ) bind calcium at the C2 domain, increasing the affinity of the C1 domain for diacylglycerol (DAG). (b) The novel isozymes (ε, δ, η and θ) are not sensitive to calcium but can be activated by binding DAG. (c) The atypical isozymes (ζ and λ/ι) lack a C2 domain and a functional C1 domain and, therefore, are not sensitive to either calcium or DAG. The catalytic domains are highly conserved throughout all of the PKC isozymes, and individual isozymes contain pseudosubstrate (orange) domains, which in the absence of stimuli maintain the isozyme in an inactive conformation.
Figure 2.
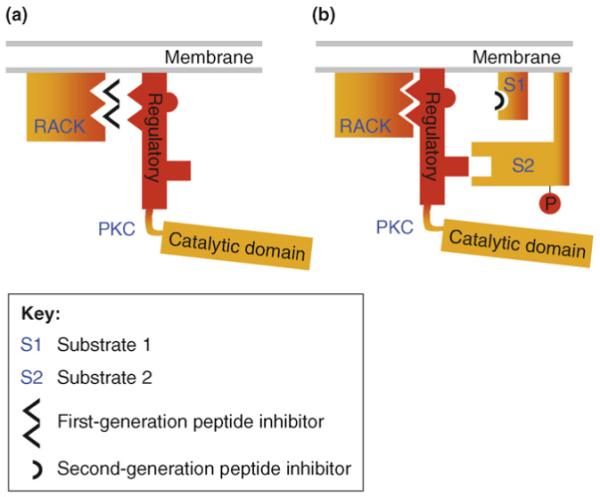
Mechanisms of peptide inhibition of PKC function. (a) Short peptides corresponding to a region within the C2 domain that mediates the binding of PKC to its anchoring protein, receptor for activated C kinase (RACK), act as competitive inhibitors; they bind to RACK, thereby preventing its binding to PKC. These first-generation peptides specifically block anchoring of PKC to its RACK and, therefore, the activity of all the functions that are mediated by that PKC isozyme at each of its subcellular locations. (b) Second-generation PKC-regulating peptides can be derived from regions within the C2 domain or other domains in PKC. Each of these peptides inhibits the binding of the kinase to a specific substrate at a particular subcellular location. These second-generation peptides are expected to interfere with binding of PKC to an individual substrate and, thus, block the function related to phosphorylation of that substrate (e.g. S1), without affecting PKC translocation, binding to its RACK or phosphorylation of another substrate (e.g. S2).
The role of PKC in endocrine signaling has been known for many years and has been reviewed extensively. PKC is involved in prostaglandin synthesis in the corpus luteum [8], fuel metabolism in pancreatic β cells [9], GLUT4 exocytosis [10], prolactin signaling [11], thyroid-stimulating hormone signaling [12] and pituitary secretion of ACTH [13], in addition to diabetic kidney disease [14], diabetic nephropathy [15] and thyroid dysfunction [16]. Therefore, PKC-regulating peptides might be useful for the treatment of different diseases, including endocrine and metabolic disorders. In this review, we discuss the techniques that were used to generate such peptide inhibitors of protein-protein interactions in PKC signaling and propose an approach to identify the next generation of peptide inhibitors with potentially greater selectivity to alter specific functions of individual PKC isozymes.
Mechanisms that govern selectivity of members of the PKC family
PKC is a family of homologous serine/threonine-related isozymes that are involved in many signaling events in normal and disease states. There are three different sub-families of isozymes within the PKC family that are further classified according to their mode of activation and regulatory domain homology (see Box 1 and Figure 1). These include the classic PKCs α, βI, βII and γ isozymes; the novel PKCs δ, ε, η, and θ isozymes; and the atypical PKCs ζ and ι/λ isozymes. Many studies have demonstrated that individual PKC isozymes have unique and at times, even opposing roles in the heart (reviewed in Refs [17,18]), brain (reviewed in Ref. [19]), liver (reviewed in Ref. [20]), pancreas (reviewed in Ref. [9]), vasculature (reviewed in Ref. [21]) and other organs, illustrating the need for isozyme-selective inhibitors and activators.
Elevations in the second messenger diacylglycerol lead to translocation and anchoring of the activated isozymes to select subcellular compartments near specific cellular substrates [22,23]. For example, βIIPKC is found in the cytosolic compartment in non-stimulated cells (e.g. heart muscle cells in culture), and within seconds of cell stimulation, activated βIIPKC translocates to the plasma membrane as well as to the perinucleus [24], where it binds RACK1 [25]. RACK1 anchors βIIPKC near the L-type calcium channels in these cells where the activated βIIPKC phosphorylates and inhibits the channel [26,27]. (Note that regulation of L-type calcium channels is crucial in a variety of other physiological responses, e.g. for insulin release from pancreatic β-islet cells [28].) RACK1 binds βIIPKC via a region within the C2 domain [25] and an additional region in the V5 domain [29] (Figure 1). The RACK1 structure is composed of seven ∼40-amino-acid repeats, called the tryptophan-aspartic acid (WD40) motif, that are arranged in a propeller-blade-like structure (reviewed in Ref. [30]). This structure enables it to bind different proteins simultaneously [31,32] and, thus, RACK1 acts as a scaffold, bringing activated PKC into contact with its various substrates [31,33]. Although only one additional RACK has been cloned (εRACK) [34], it is likely that RACKs of other activated PKC isozymes are also scaffolds for multiple proteins [32].
Binding of activated PKCs to their RACKs provides access to a subset of substrates and, therefore, is required for PKC function [35]. This finding led to our approach of generating inhibitors of PKC signaling that selectively interfere with the binding of a specific PKC isozyme with its RACK without affecting the binding and function of other isozymes. A series of rational approaches enabled the identification of the interaction site of each cognate PKC for its RACK. The technology for identifying peptide regulators of PKC isozymes has been reviewed extensively elsewhere [21,36] and, therefore, is discussed here only briefly. Peptides corresponding to these sites bind to and selectively inhibit each PKC isozyme from binding to its RACK [36] (Figure 2a). Because PKC is a multidomain protein [37] and the interactions between the domains are dynamic [38,39], there are multiple intramolecular protein-protein interactions within the enzyme that keep the enzyme in the inactive state. The first such inhibitory intramolecular interaction was identified by Kemp and collaborators, who found that the N terminus of the enzyme (in the regulatory domain; see Figure 1, orange domain) contains a substrate-like sequence that binds the catalytic site in domain C4 when PKC is inactive [40]. We, therefore, developed peptides that interfered with these intramolecular interactions within PKC [36]. These short peptides induce activation and translocation of the corresponding isozyme by mimicking the action of the RACK on the isozyme and, therefore, are termed ‘pseudo RACKs’ (ΨRACK) [41].
PKC-regulating peptides have been used in different models of human diseases. For example, we found that activators of εPKC and inhibitors of δPKC diminish injury associated with myocardial infarction [42-44], diminish graft coronary artery disease associated with cardiac transplantation [45] and increase the number of surviving β-islet cells for pancreatic cell transplantation [46]. Other isozyme-specific peptide inhibitors designed to prevent cardiac hypertrophy [29] have also shown therapeutic potential in animal models of diabetes [46,47], pain [48,49], ischemic brain injury [50,51] and cancer [52]. Furthermore, these peptides have helped elucidate the roles of PKC isozymes in glucose-signaling pathways in pancreatic β cells [53].
Inhibiting protein-protein interactions; emerging rules for rational drug design
Protein-protein interactions are central to all cellular processes, from cellular proliferation to programmed cell death, rendering them a large and important class of targets for drug development. Understanding the mechanisms by which proteins interact enables the development of molecules to modulate these interactions. The first model for ligand-protein interactions was suggested in 1894 by Emil Fisher, who proposed that the intimate contact between two interacting molecules is analogous to a lock and key fit whereby the interaction is only possible if the two molecules have a similar geometric configuration [54] (Figure 3a). This model laid the groundwork for understanding protein substrate interactions but did not take into account the highly dynamic nature of these interactions. Later models addressed this discrepancy, proposing that protein substrate interactions were not as rigid as once thought. The ‘induced fit’ model proposed by Daniel E. Koshland, Jr. [55,56] postulated that a local change occurs in the conformation of an enzyme caused by binding of its substrate (Figure 3b). Another model suggested that the conformational complementarity in which both ligand and enzyme exist is an ensemble of conformational equilibria; therefore, binding itself shifts this equilibria to the stabilized bound conformation [57] (Figure 3c).
Figure 3.
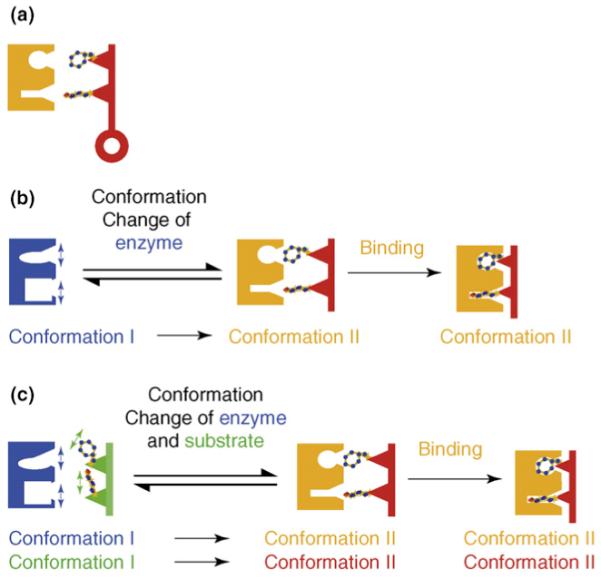
Three proposed models for describing protein-protein interactions. (a) The ‘lock and key’ model proposed by Emil Fisher in 1894 states that ligand-protein interactions are only possible if the enzyme and substrate have a similar geometric configuration. Amino acid residues on the enzyme will tightly interact with residues in the substrate [55]. (b) The ‘induced fit’ model postulated by Daniel E. Koshland, Jr. in 1958 suggests that local conformational changes in the enzyme accommodate binding of a substrate that makes it catalytically active [56]. (c) The ‘conformational complementarity’ model postulates that both the enzyme and ligand exist in an ensemble of conformational equilibria and the binding shifts these equilibria to the stabilized bound conformation [57].
Disruption of protein-protein interactions is the target of rational drug design. However, protein-protein interactions constitute large surface interfaces of approximately 750-1500 Å in each partner, and structural studies of protein-protein pairs have not shown many small binding pockets that enable the fitting of small-molecule inhibitors [58]. Rather, the selective interaction is probably the sum of many low-affinity interactions between these large interfaces. Furthermore, interactions between two large surfaces involve a considerable degree of flexibility and adaptive conformational changes. Therefore, generating an inhibitor of protein-protein interaction poses a challenge. Many of the successful regulators of protein-protein interactions are peptides that have been designed to mimic the structure of one surface of the interacting proteins. Such regulating peptides serve as decoys for one of the proteins, preventing the binding of that protein to the target protein. These flexible, naturally selected peptides can interact with the many interaction sites within the binding domain of a protein much better than rigid small molecules, thereby increasing efficacy and diminishing non-specific interactions.
In the past 20 years, rational approaches have been used to develop peptides that regulate the function of PKCs, starting with the first peptide, which was a cataly-tic-site-binding peptide derived from the pseudosubstrate site in PKC [40]. The subsequent development of peptides that disrupt protein-protein interactions between PKC and its RACK revealed a set of rules that can facilitate rational approaches to identify peptide regulators of other protein-protein interactions. These rules are described briefly here, and references of the studies that used these approaches are provided. First, enzymes can interact with multiple unrelated proteins. Sequences shared by these non-related proteins might represent at least part of the binding site for that enzyme. Therefore, peptides corresponding to these sequences might interfere with these interactions, thereby affecting the function of the enzyme [59,60]. Second, non-related enzymes might contain homologous domains that serve a similar yet unique function. For example, the C2 domains mediate anchoring of phospholipases C, D, synaptotagmin and PKC to different anchoring proteins [25,61,62]. Non-homologous sequences within these homologous domains in each enzyme are likely to represent the unique interaction site for the corresponding partner proteins. Therefore, peptides corresponding to these short unique sequences can specifically interfere with the function of the enzyme from which they were derived [25,63]. Third, peptide regulators of protein-protein interactions can be generated by identifying evolutionarily conserved sequences (e.g. conserved sequences between εPKCs from the evolutionarily distant sea snail Aplisia Californica and rat) [64]. Peptides derived from these conserved sequences might be selective regulators of protein-protein interactions [64]. Fourth, it is possible to identify sequences that are involved in dynamic and regulatable intramolecular interactions (i.e. auto-regulatory sites). Intramolecular interactions within a variety of enzymes between the catalytic domain and the regulatory domain maintain the enzyme in an inactive state. Peptides mimicking these intramolecular interactions can disrupt the inhibitory action of the regulatory domain and, thus, activate the enzyme [65,66].
Second-generation regulators of PKC function
The PKC peptides that specifically regulate the binding of each PKC isozyme to its RACK (first-generation peptide regulators, described previously) represent the use of a rational design of peptide regulators of protein-protein interactions [36]. One example of where such design has been useful is the C2 domain of PKC, which mediates the interaction of PKC with its RACK [25,61,64,67-69,27,70,71] (Figure 4a). In a recent study, a series of short peptides was derived from regions that span the length of the C2 domain of εPKC [68] (Figure 4b). The biological activities of the peptides were tested in four assays: activation or inhibition of translocation in primary skeletal muscle cells; activation or inhibition of MARCKS phosphorylation, a known PKC substrate involved in actin crosslinking using both wild-type and εPKC-knockout cells; increase or decrease of infarct size in a model of myocardial infarction (εPKC activation decreases infarct size [72]); and effect on PKC translocation in vivo [68]. Some were found to be selective for only εPKC, whereas other peptides affected several PKC isozymes. Furthermore, some peptides exerted inhibitory activity, whereas others activated εPKC [68].
Figure 4.

Interaction cultures in the C2 domain of εPKC. (a) Space-filled surface mapping of the C2 domain of εPKC. The peptides derived from this domain and their activities are mapped back to the C2 domain of εPKC; they are colored in blue to represent PKC activators (not selective for εPKC), green to represent εPKC-selective activators, yellow to represent inconclusive results about their selectivity or red to represent εPKC inhibitors. εPKC-selective activators are peptides that cause phosphorylation of MARCKS in WT cells but not in εKO-cells and protect cardiac tissue from ischemia and reperfusion damage. Three of the four PKC-inhibiting peptides cluster to one area on the surface of the C2 domain (red regions). However, the selectivity of these inhibitory peptides for εPKC has not yet been determined. (b) Multiple sequence alignment of the C2 domain of five PKC isozymes identifies potential new peptides with similar biological activities derived from other isozymes. The alignment is based on structure (β strands are aligned) and sequence. Sequences corresponding to peptides with different biological activities are colored (red to represent inhibitors, green to represent selective activators or blue to represent isozyme non-selective activators). Peptides (indicated in color) from other isozymes are as follows: βPKC agonist ΨβRACK (SVEIWD) activates all the classical PKC isozymes [25], and βPKC antagonists βC2-4 (SLNPEWNET) and βC2-2 (MDPNGLSDPYVKL) are inhibitors of all the classical PKC isozymes [25]; δPKC agonist (δΨRACK, MRAAEDPM) and antagonist (δV1-1, SFNSYELGSL) are selective for δPKC [70], ηPKC agonist (ηΨRACK, HETPLGYD) and antagonist (ηV-2, EAVGLQPT) [67]. Sequence alignment of C2 domains of several PKC isozymes predicts regions from which potential peptide regulators (selective and nonselective) for other PKC isozymes can be generated (color, boxed areas). Reproduced, with permission, from Ref. [68].
In addition to PKC, the C2 domain is found in over 60 proteins [73], and NMR and crystal structures of a variety of these C2 domains show a similar fold of a β sandwich made of four antiparallel β strands [69,73-77]. Examination of the structure of the C2 domain from εPKC revealed that peptides derived from this domain that exert similar biological activities are clustered into distinct regions within the domain [68] (Figure 4a). We hypothesize that these regions in the C2 domain represent discrete protein-protein interaction surfaces within εPKC (between the C2 and other domains of the enzyme) and/or between εPKC and other partner proteins and, thus, might represent different functionalities of individual PKC isozymes. This indicates that in addition to isozyme-specific regulators, it might be possible to generate inhibitors and activators of PKC signaling that selectively interfere with a subset of protein-protein interactions for each isozyme and, thereby, might confer an even higher degree of specificity.
Within the same cells, individual PKC isozymes are found at multiple subcellular sites [5,78-81]. For example, in cardiac myocytes, activated εPKC is found on cross-striated structures, on the plasma membrane and at the intercalated discs [24,81]. Therefore, it can be hypothesized that different protein substrates and downstream effectors would be found in each of these subcellular locations. Furthermore, although RACKs anchor the activated PKCs at these subcellular sites, additional unique protein-protein interactions between PKC and its substrates might provide further anchoring and specificity at these subcellular sites (e.g. a myofilament-binding site in the C2 domain [82], a Golgi-binding site [34], an intra-sarcoplasmic reticulum calsequestrin-binding site [83], a neurocytoskeletal elements-binding site [84] and a unique actin-binding site within the C1 domain of εPKC [85] were identified). These unique interactions enable different signaling events that are dependent upon the region of the individual PKC isozyme that is available for binding. Thus, we hypothesize that regulators of each interaction should interfere with only one of the functions that are mediated by that isozyme. Such second-generation peptides will not only regulate specific PKC isozymes (i.e. εPKC vs δPKC) but also block or induce a subset of subcellular and substrate-specific PKC interactions (e.g. permit εPKC phosphorylation of substrate 1 but block εPKC phosphorylation of substrate 2) (Figure 2b). Furthermore, although there is limited sequence homology between the C2 domains of different PKC isozymes, because of the structural homology between the isozymes, we suggest that peptides derived from similar regions within the C2 domain of other isozymes will interfere with the function of the isozyme of interest. These second-generation peptides can be used to determine the role of each particular isozyme at its sub-cellular location and, accordingly, which of these should be regulated to obtain optimal protection from tissue injury and which should be left unaltered to limit undesired side-effects of intervention.
Challenges in using peptides as drugs
Peptides are not considered useful drugs because of their limited bioavailability, their inability to cross biological membranes, their short half-life and their multiple conformations, only a few of which are capable of interacting with the target. Recent efforts in multiple laboratories have addressed these potential limitations. First, the biological activity of peptide regulators of protein-protein interactions can be increased by modifying the natural peptides to restrict their conformation via local or global modifications. Methods to induce local constraints include incorporation of D-configuration amino acids, N-alkylated amino acids and non-natural amino acids [86]. Incorporation of global constraints can be done through cyclization of the peptide [87]. Peptide resistance to degradation by proteases and peptidases can be increased by blocking their free N- and C-termini [88] and by using constraints to restrict the peptide conformation [89-91]. Finally, several methods have been developed to enable peptide penetration across cell membranes, including addition of arginine-rich peptides that can carry the peptides by micropinocytosis [92,93]. The feasibility of restricting the peptides’ conformation has been demonstrated with several naturally occurring peptides, including substance P [94,95], somatostatin [96], pheromone-biosynthesis-activating neuropeptide [97] and gonadotropin-releasing hormone [98], resulting in the development of receptor-selective and metabolically stable peptides. Two properties of the natural protein must be preserved in these modified peptides: the conformation that enables optimal binding to the target protein, and a certain degree of conformational flexibility to enable induced fit of the peptide to its target [99].
An early example of a peptide regulator that had limited success in the clinic for treating HIV is Enfuvirtide. Enfuvirtide was rationally designed to bind to viral proteins to prevent fusion of the capsid with the plasma membrane [100,101] and was shown to be effective at reducing viral load, while having a favorable toxicity profile [102]. Recent studies using peptides and peptide mimetics other than PKC-regulating peptides have demonstrated their potential therapeutic applications in chronic diseases such as cardiovascular diseases [103,104], cancer [105], sepsis [106], diabetes mellitus (a highly prevalent chronic disease) [107], chronic hepatitis C viral infection [108] and others. In addition, regulating peptides might be used to block not only protein-protein interactions but also other interactions, such as protein-nucleic-acid and protein-peptide interactions [109-111]. We suggest that the advantages (which include selectivity, bioavailability, low molecular weight, low production cost, stability and lack of antigenicity) of peptide regulators make them useful drugs.
The targets of regulatory peptides are often ubiquitously expressed throughout the body. Therefore, tissue specificity is another challenge when using peptides as therapeutic agents. Many peptides are rendered cell permeable (through linking with cell-permeable peptides such as TAT [46-56]); once they reach the systemic circulation, the possibility of off-target effects increases greatly. Two ways to diminish this are through localized delivery and through controlled activation. Upon entry of TAT-conjugated peptides into the cell, the linking disulfide bonds are cleaved and the peptide cargo might be trapped [112]. Therefore, when peptides are delivered locally to the desired site of action, most of the therapeutic agent will be taken up into the cell, decreasing the concentration available for other non-target tissues. The delivery of light-activatable peptides also represents a way to deliver peptides in a controlled manner. By shining light directly onto the therapeutic target, peptides that contain a linked chemical group (i.e. azobenzene) that is susceptible to light-mediated chemical changes will become activated in that area only and not in other non-illuminated tissues [113].
Concluding remarks
Inhibiting protein-protein interactions using small molecules is a challenge because these sites constitute many low-affinity interactions spread on a large surface on each of the protein partners. This review highlights several simple approaches in which short peptides derived from these interacting surfaces were used to interfere selectively and effectively with individual PKC isozymes and other proteins or with intramolecular inhibitory interactions. These rationally designed peptides are 6-10 amino acids long and derived from one of the interacting domains and thereby act as mimetics to block the association of two proteins or two domains. Although these peptides represent only part of the interaction surface, they were found to be highly selective and effective and, thus, useful agents for basic research, as well as potential drugs for human diseases. One of these peptides, a selective inhibitor of δPKC, has been tested in patients with acute myocardial infarction in phase IIa clinical trials, showing promise as a novel therapeutic for diminishing the associated injury to the heart [114]. This mechanism proceeds through the peptide inhibiting the following: δPKC-mediated increases in the apoptotic response [115], inactivation of pyruvate dehydrogenase (through direct phosphorylation and inhibition of the inactivating kinase [116]), improvements in ATP regeneration [43] and diminished cAbl-dependent endoplasmic-reticulum-induced apoptosis [117]. The role of PKC in these metabolism-associated fundamental processes indicates that regulation of PKC will be a therapeutic target for other endocrinology-related disorders. Indeed, the peptide inhibitor of δPKC (δV1-1) and activator of εPKC (ΨεRACK) diminished respective hyperglycemic and streptazotocininduced apoptotic signaling and oxidative stress [47,118], decreased insulin release from islet β cells [53] and increased survival of isolated islet β cells [46]. Additionally, studies show that glucose stimulation of islet β cells causes subcellular translocation of many of the PKC isozymes, indicating many regulatory points in insulin secretion pathways for the isozyme-specific peptides [53,119].
Because protein-protein interactions provide novel sets of targets to regulate signal transduction in disease states, there is increased effort in identifying regulators that interfere with these interactions. These targets are traditionally regarded as hard to regulate because of the attempts to use small molecules - the more traditional approach of drug discovery. Several studies show that peptides can be used effectively in vivo and are well tolerated. We found that in chronic treatments with different PKC peptide regulators, there was no apparent toxicity, there was no apparent activation of compensatory pathways, they did not elicit an immune response and there was no tolerance to the effect of the peptides [112]. Although traditionally, peptides have not been commonly used as drugs, the increased interest of the pharmaceutical industry in peptides that act on surface receptors is encouraging and indicates that the use of peptides to target intracellular signaling events will follow. We believe that because natural short peptides derived from the interaction sites of important signaling proteins can be rationally designed (thus saving tremendous amounts of time, effort and cost), we will see such peptides developed as drugs for treating human diseases.
Acknowledgements
The research for this article was supported by the National Institutes of Health Grants NIH AA11147, HL52141 and HL76670 to D.M.-R.
Footnotes
Disclaimer statement
D.M.-R. is the founder of KAI Pharmaceuticals, a company that plans to bring PKC regulators to the clinic. However, none of the work in her lab is supported by the company.
References
- 1.Mahdi JG, et al. The historical analysis of aspirin discovery, its relation to the willow tree and antiproliferative and anticancer potential. Cell Prolif. 2006;39:147–155. doi: 10.1111/j.1365-2184.2006.00377.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vane JR, Botting RM. The mechanism of action of aspirin. Thromb. Res. 2003;110:255–258. doi: 10.1016/s0049-3848(03)00379-7. [DOI] [PubMed] [Google Scholar]
- 3.Garavito RM, Mulichak AM. The structure of mammalian cyclooxygenases. Annu. Rev. Biophys. Biomol. Struct. 2003;32:183–206. doi: 10.1146/annurev.biophys.32.110601.141906. [DOI] [PubMed] [Google Scholar]
- 4.Picot D, et al. The X-ray crystal structure of the membrane protein prostaglandin H2 synthase-1. Nature. 1994;367:243–249. doi: 10.1038/367243a0. [DOI] [PubMed] [Google Scholar]
- 5.Mochly-Rosen D. Localization of protein kinases by anchoring proteins: a theme in signal transduction. Science. 1995;268:247–251. doi: 10.1126/science.7716516. [DOI] [PubMed] [Google Scholar]
- 6.Fry DC. Drug-like inhibitors of protein-protein interactions: a structural examination of effective protein mimicry. Curr. Protein Pept. Sci. 2008;9:240–247. doi: 10.2174/138920308784533989. [DOI] [PubMed] [Google Scholar]
- 7.Liu Y, et al. Protein interaction predictions from diverse sources. Drug Discov. Today. 2008;13:409–416. doi: 10.1016/j.drudis.2008.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Diaz FJ, et al. Regulation of progesterone and prostaglandin F2α production in the CL. Mol. Cell. Endocrinol. 2002;191:65–80. doi: 10.1016/s0303-7207(02)00056-4. [DOI] [PubMed] [Google Scholar]
- 9.Nesher R, et al. β-cell protein kinases and the dynamics of the insulin response to glucose. Diabetes. 2002;51(Suppl 1):S68–S73. doi: 10.2337/diabetes.51.2007.s68. [DOI] [PubMed] [Google Scholar]
- 10.Ishiki M, Klip A. Minireview: recent developments in the regulation of glucose transporter-4 traffic: new signals, locations, and partners. Endocrinology. 2005;146:5071–5078. doi: 10.1210/en.2005-0850. [DOI] [PubMed] [Google Scholar]
- 11.Goffin V, et al. Prolactin: a hormone at the crossroads of neuroimmunoendocrinology. Ann. N. Y. Acad. Sci. 1998;840:498–509. doi: 10.1111/j.1749-6632.1998.tb09588.x. [DOI] [PubMed] [Google Scholar]
- 12.Ginsberg J. Protein kinase C as a mediator of TSH and thyroid autoantibody action. Autoimmunity. 1992;13:51–59. doi: 10.3109/08916939209014635. [DOI] [PubMed] [Google Scholar]
- 13.Volpi S, et al. Vasopressinergic regulation of the hypothalamic pituitary adrenal axis and stress adaptation. Stress. 2004;7:75–83. doi: 10.1080/10253890410001733535. [DOI] [PubMed] [Google Scholar]
- 14.Flyvbjerg A, et al. The involvement of growth hormone (GH), insulin-like growth factors (IGFs) and vascular endothelial growth factor (VEGF) in diabetic kidney disease. Curr. Pharm. Des. 2004;10:3385–3394. doi: 10.2174/1381612043383106. [DOI] [PubMed] [Google Scholar]
- 15.Noh H, King GL. The role of protein kinase C activation in diabetic nephropathy. Kidney Int. 2007;106:S49–S53. doi: 10.1038/sj.ki.5002386. [DOI] [PubMed] [Google Scholar]
- 16.Knauf JA, et al. Involvement of protein kinase Cε (PKCε) in thyroid cell death. A truncated chimeric PKCε cloned from a thyroid cancer cell line protects thyroid cells from apoptosis. J. Biol. Chem. 1999;274:23414–23425. doi: 10.1074/jbc.274.33.23414. [DOI] [PubMed] [Google Scholar]
- 17.Churchill EN, Mochly-Rosen D. The roles of PKCδ and ε isoenzymes in the regulation of myocardial ischaemia/reperfusion injury. Biochem. Soc. Trans. 2007;35:1040–1042. doi: 10.1042/BST0351040. [DOI] [PubMed] [Google Scholar]
- 18.Budas GR, et al. Cardioprotective mechanisms of PKC isozyme-selective activators and inhibitors in the treatment of ischemia-reperfusion injury. Pharmacol. Res. 2007;55:523–536. doi: 10.1016/j.phrs.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 19.Bright R, Mochly-Rosen D. The role of protein kinase C in cerebral ischemic and reperfusion injury. Stroke. 2005;36:2781–2790. doi: 10.1161/01.STR.0000189996.71237.f7. [DOI] [PubMed] [Google Scholar]
- 20.Nitti M, et al. PKC signaling in oxidative hepatic damage. Mol. Aspects Med. 2008;29:36–42. doi: 10.1016/j.mam.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 21.Churchill E, et al. PKC isozymes in chronic cardiac disease: possible therapeutic targets? Annu. Rev. Pharmacol. Toxicol. 2008;48:569–599. doi: 10.1146/annurev.pharmtox.48.121806.154902. [DOI] [PubMed] [Google Scholar]
- 22.Kraft AS, Anderson WB. Phorbol esters increase the amount of Ca2+, phospholipid-dependent protein kinase associated with plasma membrane. Nature. 1983;301:621–623. doi: 10.1038/301621a0. [DOI] [PubMed] [Google Scholar]
- 23.Mochly-Rosen D, et al. A protein kinase C isozyme is translocated to cytoskeletal elements on activation. Cell Regul. 1990;1:693–706. doi: 10.1091/mbc.1.9.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Disatnik MH, et al. Localization of protein kinase C isozymes in cardiac myocytes. Exp. Cell Res. 1994;210:287–297. doi: 10.1006/excr.1994.1041. [DOI] [PubMed] [Google Scholar]
- 25.Ron D, et al. C2 region-derived peptides inhibit translocation and function of β protein kinase C in vivo. J. Biol. Chem. 1995;270:24180–24187. doi: 10.1074/jbc.270.41.24180. [DOI] [PubMed] [Google Scholar]
- 26.McHugh D, et al. Inhibition of cardiac L-type calcium channels by protein kinase C phosphorylation of two sites in the N-terminal domain. Proc. Natl. Acad. Sci. U. S. A. 2000;97:12334–12338. doi: 10.1073/pnas.210384297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang ZH, et al. C2 region-derived peptides of β-protein kinase C regulate cardiac Ca2+ channels. Circ. Res. 1997;80:720–729. doi: 10.1161/01.res.80.5.720. [DOI] [PubMed] [Google Scholar]
- 28.Yang SN, Berggren PO. The role of voltage-gated calcium channels in pancreatic β-cell physiology and pathophysiology. Endocr. Rev. 2006;27:621–676. doi: 10.1210/er.2005-0888. [DOI] [PubMed] [Google Scholar]
- 29.Stebbins EG, Mochly-Rosen D. Binding specificity for RACK1 resides in the V5 region of β II protein kinase C. J. Biol. Chem. 2001;276:29644–29650. doi: 10.1074/jbc.M101044200. [DOI] [PubMed] [Google Scholar]
- 30.Smith TF, et al. The WD repeat: a common architecture for diverse functions. Trends Biochem. Sci. 1999;24:181–185. doi: 10.1016/s0968-0004(99)01384-5. [DOI] [PubMed] [Google Scholar]
- 31.Birikh KR, et al. Interaction of “readthrough” acetylcholinesterase with RACK1 and PKCβ II correlates with intensified fear-induced conflict behavior. Proc. Natl. Acad. Sci. U. S. A. 2003;100:283–288. doi: 10.1073/pnas.0135647100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schechtman D, Mochly-Rosen D. Adaptor proteins in protein kinase C-mediated signal transduction. Oncogene. 2001;20:6339–6347. doi: 10.1038/sj.onc.1204778. [DOI] [PubMed] [Google Scholar]
- 33.Patterson RL, et al. RACK1 binds to inositol 1,4, 5-trisphosphate receptors and mediates Ca2+ release. Proc. Natl. Acad. Sci. U. S. A. 2004;101:2328–2332. doi: 10.1073/pnas.0308567100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Csukai M, et al. The coatomer protein β’-COP, a selective binding protein (RACK) for protein kinase Cε. J. Biol. Chem. 1997;272:29200–29206. doi: 10.1074/jbc.272.46.29200. [DOI] [PubMed] [Google Scholar]
- 35.Smith BL, Mochly-Rosen D. Inhibition of protein kinase C function by injection of intracellular receptors for the enzyme. Biochem. Biophys. Res. Commun. 1992;188:1235–1240. doi: 10.1016/0006-291x(92)91363-u. [DOI] [PubMed] [Google Scholar]
- 36.Souroujon MC, Mochly-Rosen D. Peptide modulators of protein-protein interactions in intracellular signaling. Nat. Biotechnol. 1998;16:919–924. doi: 10.1038/nbt1098-919. [DOI] [PubMed] [Google Scholar]
- 37.Ohno S, Nishizuka Y. Protein kinase C isotypes and their specific functions: prologue. J. Biochem. 2002;132:509–511. doi: 10.1093/oxfordjournals.jbchem.a003249. [DOI] [PubMed] [Google Scholar]
- 38.Gallegos LL, Newton AC. Spatiotemporal dynamics of lipid signaling: protein kinase C as a paradigm. IUBMB Life. 2008;60:782–789. doi: 10.1002/iub.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Newton AC. Regulation of the ABC kinases by phosphorylation: protein kinase C as a paradigm. Biochem. J. 2003;370:361–371. doi: 10.1042/BJ20021626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.House C, Kemp BE. Protein kinase C contains a pseudosubstrate prototope in its regulatory domain. Science. 1987;238:1726–1728. doi: 10.1126/science.3686012. [DOI] [PubMed] [Google Scholar]
- 41.Dorn GW, II, et al. Sustained in vivo cardiac protection by a rationally designed peptide that causes varε protein kinase C translocation. Proc. Natl. Acad. Sci. U. S. A. 1999;96:12798–12803. doi: 10.1073/pnas.96.22.12798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu GS, et al. Protein kinase C-ε is responsible for the protection of preconditioning in rabbit cardiomyocytes. J. Mol. Cell. Cardiol. 1999;31:1937–1948. doi: 10.1006/jmcc.1999.1026. [DOI] [PubMed] [Google Scholar]
- 43.Inagaki K, et al. Inhibition of δ-protein kinase C protects against reperfusion injury of the ischemic heart in vivo. Circulation. 2003;108:2304–2307. doi: 10.1161/01.CIR.0000101682.24138.36. [DOI] [PubMed] [Google Scholar]
- 44.Inagaki K, et al. Additive protection of the ischemic heart ex vivo by combined treatment with δ-protein kinase C inhibitor and ε-protein kinase C activator. Circulation. 2003;108:869–875. doi: 10.1161/01.CIR.0000081943.93653.73. [DOI] [PubMed] [Google Scholar]
- 45.Tanaka M, et al. Suppression of graft coronary artery disease by a brief treatment with a selective εPKC activator and a δPKC inhibitor in murine cardiac allografts. Circulation. 2004;110:II194–II199. doi: 10.1161/01.CIR.0000138389.22905.62. [DOI] [PubMed] [Google Scholar]
- 46.Kvezereli M, et al. Islet cell survival during isolation improved through protein kinase C epsilon activation. Transplant. Proc. 2008;40:375–378. doi: 10.1016/j.transproceed.2008.01.014. [DOI] [PubMed] [Google Scholar]
- 47.Malhotra A, et al. PKC-ε-dependent survival signals in diabetic hearts. Am. J. Physiol. 2005;289:H1343–H1350. doi: 10.1152/ajpheart.01200.2004. [DOI] [PubMed] [Google Scholar]
- 48.Sweitzer SM, et al. Protein kinase C ε and γ: involvement in formalin-induced nociception in neonatal rats. J. Pharmacol. Exp. Ther. 2004;309:616–625. doi: 10.1124/jpet.103.060350. [DOI] [PubMed] [Google Scholar]
- 49.Sweitzer SM, et al. Exaggerated nociceptive responses on morphine withdrawal: roles of protein kinase C ε and γ. Pain. 2004;110:281–289. doi: 10.1016/j.pain.2004.04.004. [DOI] [PubMed] [Google Scholar]
- 50.Bright R, et al. Protein kinase C δ mediates cerebral reperfusion injury in vivo. J. Neurosci. 2004;24:6880–6888. doi: 10.1523/JNEUROSCI.4474-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bright R, et al. varεPKC confers acute tolerance to cerebral ischemic reperfusion injury. Neurosci. Lett. 2008;441:120–124. doi: 10.1016/j.neulet.2008.05.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim J, et al. Centrosomal PKCβII and pericentrin are critical for human prostate cancer growth and angiogenesis. Cancer Res. 2008;68:6831–6839. doi: 10.1158/0008-5472.CAN-07-6195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yedovitzky M, et al. Translocation inhibitors define specificity of protein kinase C isoenzymes in pancreatic β-cells. J. Biol. Chem. 1997;272:1417–1420. doi: 10.1074/jbc.272.3.1417. [DOI] [PubMed] [Google Scholar]
- 54.Fischer E. Einfluss der Configuration auf die Wirkung der Enzyme. Ber. Dtsch. Chem. Ges. 1894;27:2985–2993. [Google Scholar]
- 55.Pauling L. A theory of the structure and process of formation of antibodies. J. Am. Chem. Soc. 1940;62:2643–2657. [Google Scholar]
- 56.Koshland DE. Application of a theory of enzyme specificity to protein synthesis. Proc. Natl. Acad. Sci. U. S. A. 1958;44:98–104. doi: 10.1073/pnas.44.2.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Monod J, et al. On the nature of allosteric transitions: a plausible model. J. Mol. Biol. 1965;12:88–118. doi: 10.1016/s0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- 58.Arkin MR, Wells JA. Small-molecule inhibitors of protein-protein interactions: progressing towards the dream. Nat. Rev. Drug Discov. 2004;3:301–317. doi: 10.1038/nrd1343. [DOI] [PubMed] [Google Scholar]
- 59.Carr DW, et al. Association of the type II cAMP-dependent protein kinase with a human thyroid RII-anchoring protein. Cloning and characterization of the RII-binding domain. J. Biol. Chem. 1992;267:13376–13382. [PubMed] [Google Scholar]
- 60.Aitken A, et al. Kinase and neurotransmitters. Nature. 1990;344:594. doi: 10.1038/344594a0. [DOI] [PubMed] [Google Scholar]
- 61.Mochly-Rosen D, et al. p65 fragments, homologous to the C2 region of protein kinase C, bind to the intracellular receptors for protein kinase C. Biochemistry. 1992;31:8120–8124. doi: 10.1021/bi00150a003. [DOI] [PubMed] [Google Scholar]
- 62.Disatnik MH, et al. Phospholipase C-γ 1 binding to intracellular receptors for activated protein kinase C. Proc. Natl. Acad. Sci. U. S. A. 1994;91:559–563. doi: 10.1073/pnas.91.2.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bommert K, et al. Inhibition of neurotransmitter release by C2-domain peptides implicates synaptotagmin in exocytosis. Nature. 1993;363:163–165. doi: 10.1038/363163a0. [DOI] [PubMed] [Google Scholar]
- 64.Johnson JA, et al. A protein kinase C translocation inhibitor as an isozyme-selective antagonist of cardiac function. J. Biol. Chem. 1996;271:24962–24966. doi: 10.1074/jbc.271.40.24962. [DOI] [PubMed] [Google Scholar]
- 65.Ron D, Mochly-Rosen D. An autoregulatory region in protein kinase C: the pseudoanchoring site. Proc. Natl. Acad. Sci. U. S. A. 1995;92:492–496. doi: 10.1073/pnas.92.2.492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kobe B, et al. Intrasteric regulation of protein kinases. Adv. Second Messenger Phosphoprotein Res. 1997;31:29–40. doi: 10.1016/s1040-7952(97)80006-7. [DOI] [PubMed] [Google Scholar]
- 67.Gray MO, et al. A selective ε-protein kinase C antagonist inhibits protection of cardiac myocytes from hypoxia-induced cell death. J. Biol. Chem. 1997;272:30945–30951. doi: 10.1074/jbc.272.49.30945. [DOI] [PubMed] [Google Scholar]
- 68.Brandman R, et al. Peptides derived from the C2 domain of protein kinase C ε (εPKC) modulate εPKC activity and identify potential protein-protein interaction surfaces. J. Biol. Chem. 2007;282:4113–4123. doi: 10.1074/jbc.M608521200. [DOI] [PubMed] [Google Scholar]
- 69.Banci L, et al. Molecular dynamics characterization of the C2 domain of protein kinase Cβ. J. Biol. Chem. 2002;277:12988–12997. doi: 10.1074/jbc.M106875200. [DOI] [PubMed] [Google Scholar]
- 70.Chen L, et al. Opposing cardioprotective actions and parallel hypertrophic effects of δ PKC and ε PKC. Proc. Natl. Acad. Sci. U. S. A. 2001;98:11114–11119. doi: 10.1073/pnas.191369098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chen L, et al. Molecular transporters for peptides: delivery of a cardioprotective εPKC agonist peptide into cells and intact ischemic heart using a transport system, R7. Chem. Biol. 2001;8:1123–1129. doi: 10.1016/s1074-5521(01)00076-x. [DOI] [PubMed] [Google Scholar]
- 72.Inagaki K, et al. Cardioprotection by ε-protein kinase C activation from ischemia: continuous delivery and antiarrhythmic effect of an ε-protein kinase C-activating peptide. Circulation. 2005;111:44–50. doi: 10.1161/01.CIR.0000151614.22282.F1. [DOI] [PubMed] [Google Scholar]
- 73.Rizo J, Sudhof TC. C2-domains, structure and function of a universal Ca2+-binding domain. J. Biol. Chem. 1998;273:15879–15882. doi: 10.1074/jbc.273.26.15879. [DOI] [PubMed] [Google Scholar]
- 74.Sutton RB, Sprang SR. Structure of the protein kinase Cβ phospholipid-binding C2 domain complexed with Ca2+ Structure. 1998;6:1395–1405. doi: 10.1016/s0969-2126(98)00139-7. [DOI] [PubMed] [Google Scholar]
- 75.Pappa H, et al. Crystal structure of the C2 domain from protein kinase C-δ. Structure. 1998;6:885–894. doi: 10.1016/s0969-2126(98)00090-2. [DOI] [PubMed] [Google Scholar]
- 76.Nalefski EA, et al. C2 domains from different Ca2+ signaling pathways display functional and mechanistic diversity. Biochemistry. 2001;40:3089–3100. doi: 10.1021/bi001968a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ochoa WF, et al. Structure of the C2 domain from novel protein kinase Cε. A membrane binding model for Ca2+-independent C2 domains. J. Mol. Biol. 2001;311:837–849. doi: 10.1006/jmbi.2001.4910. [DOI] [PubMed] [Google Scholar]
- 78.Gallegos LL, et al. Targeting protein kinase C activity reporter to discrete intracellular regions reveals spatiotemporal differences in agonist-dependent signaling. J. Biol. Chem. 2006;281:30947–30956. doi: 10.1074/jbc.M603741200. [DOI] [PubMed] [Google Scholar]
- 79.Rybin VO, et al. Stimulus-specific differences in protein kinase Cδ localization and activation mechanisms in cardiomyocytes. J. Biol. Chem. 2004;279:19350–19361. doi: 10.1074/jbc.M311096200. [DOI] [PubMed] [Google Scholar]
- 80.Disatnik MH, et al. Stimulus-dependent subcellular localization of activated protein kinase C; a study with acidic fibroblast growth factor and transforming growth factor-β 1 in cardiac myocytes. J. Mol. Cell. Cardiol. 1995;27:2473–2481. doi: 10.1006/jmcc.1995.0235. [DOI] [PubMed] [Google Scholar]
- 81.Disatnik MH, et al. Distinct responses of protein kinase C isozymes to c-erbB-2 activation in SKBR-3 human breast carcinoma cells. Cell Growth Differ. 1994;5:873–880. [PubMed] [Google Scholar]
- 82.Huang X, Walker JW. Myofilament anchoring of protein kinase C-ε in cardiac myocytes. J. Cell Sci. 2004;117:1971–1978. doi: 10.1242/jcs.01044. [DOI] [PubMed] [Google Scholar]
- 83.Rodriguez MM, et al. Characterization of the binding and phosphorylation of cardiac calsequestrin by ε protein kinase C. FEBS Lett. 1999;454:240–246. doi: 10.1016/s0014-5793(99)00697-3. [DOI] [PubMed] [Google Scholar]
- 84.Zeidman R, et al. PKCε, via its regulatory domain and independently of its catalytic domain, induces neurite-like processes in neuroblastoma cells. J. Cell Biol. 1999;145:713–726. doi: 10.1083/jcb.145.4.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Prekeris R, et al. Identification and localization of an actin-binding motif that is unique to the epsilon isoform of protein kinase C and participates in the regulation of synaptic function. J. Cell Biol. 1996;132:77–90. doi: 10.1083/jcb.132.1.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Feng JA, et al. Chimeric protein engineering. Int. J. Pept. Res. Ther. 2007;13:151–160. [Google Scholar]
- 87.Kessler H. Peptide Conformations. 19. Conformation and biological-activity of cyclic-peptides. Angew. Chem. Int. Ed. Engl. 1982;21:512–523. [Google Scholar]
- 88.Adessi C, et al. Pharmacological profiles of peptide drug candidates for the treatment of Alzheimer’s disease. J. Biol. Chem. 2003;278:13905–13911. doi: 10.1074/jbc.M211976200. [DOI] [PubMed] [Google Scholar]
- 89.Tugyi R, et al. Partial D-amino acid substitution: Improved enzymatic stability and preserved Ab recognition of a MUC2 epitope peptide. Proc. Natl. Acad. Sci. U. S. A. 2005;102:413–418. doi: 10.1073/pnas.0407677102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Briand JP, et al. A retro-inverso peptide corresponding to the GH loop of foot-and-mouth disease virus elicits high levels of long-lasting protective neutralizing antibodies. Proc. Natl. Acad. Sci. U. S. A. 1997;94:12545–12550. doi: 10.1073/pnas.94.23.12545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hamamoto K, et al. Antimicrobial activity and stability to proteolysis of small linear cationic peptides with D-amino acid substitutions. Microbiol. Immunol. 2002;46:741–749. doi: 10.1111/j.1348-0421.2002.tb02759.x. [DOI] [PubMed] [Google Scholar]
- 92.Kaplan IM, et al. Cationic TAT peptide transduction domain enters cells by macropinocytosis. J. Control. Release. 2005;102:247–253. doi: 10.1016/j.jconrel.2004.10.018. [DOI] [PubMed] [Google Scholar]
- 93.Friedler A, et al. Backbone cyclic peptide, which mimics the nuclear localization signal of human immunodeficiency virus type 1 matrix protein, inhibits nuclear import and virus production in nondividing cells. Biochemistry. 1998;37:5616–5622. doi: 10.1021/bi972878h. [DOI] [PubMed] [Google Scholar]
- 94.Byk G, et al. Synthesis and biological activity of NK-1 selective, N-backbone cyclic analogs of the C-terminal hexapeptide of substance P. J. Med. Chem. 1996;39:3174–3178. doi: 10.1021/jm960154i. [DOI] [PubMed] [Google Scholar]
- 95.Bitan G, et al. Synthesis and biological activity of novel backbone-bicyclic substance-P analogs containing lactam and disulfide bridges. J. Pept. Res. 1997;49:421–426. doi: 10.1111/j.1399-3011.1997.tb00894.x. [DOI] [PubMed] [Google Scholar]
- 96.Gilon C, et al. A backbone-cyclic, receptor 5-selective somatostatin analogue: synthesis, bioactivity, and nuclear magnetic resonance conformational analysis. J. Med. Chem. 1998;41:919–929. doi: 10.1021/jm970633x. [DOI] [PubMed] [Google Scholar]
- 97.Altstein M, et al. Backbone cyclic peptide antagonists, derived from the insect pheromone biosynthesis activating neuropeptide, inhibit sex pheromone biosynthesis in moths. J. Biol. Chem. 1999;274:17573–17579. doi: 10.1074/jbc.274.25.17573. [DOI] [PubMed] [Google Scholar]
- 98.Barda Y, et al. Backbone metal cyclization: novel Tc-99m labeled GnRH analog as potential SPECT molecular imaging agent in cancer. Nucl. Med. Biol. 2004;31:921–933. doi: 10.1016/j.nucmedbio.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 99.Friedler A, et al. Development of a functional backbone cyclic mimetic of the HIV-1 Tat arginine-rich motif. J. Biol. Chem. 2000;275:23783–23789. doi: 10.1074/jbc.M002200200. [DOI] [PubMed] [Google Scholar]
- 100.Derdeyn CA, et al. Sensitivity of human immunodeficiency virus type 1 to the fusion inhibitor T-20 is modulated by coreceptor specificity defined by the V3 loop of gp120. J. Virol. 2000;74:8358–8367. doi: 10.1128/jvi.74.18.8358-8367.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.O’Brien WA, et al. Anti-human immunodeficiency virus type 1 activity of an oligocationic compound mediated via gp120 V3 interactions. J. Virol. 1996;70:2825–2831. doi: 10.1128/jvi.70.5.2825-2831.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lalezari JP, et al. A phase II clinical study of the long-term safety and antiviral activity of enfuvirtide-based antiretroviral therapy. AIDS. 2003;17:691–698. doi: 10.1097/00002030-200303280-00007. [DOI] [PubMed] [Google Scholar]
- 103.Lal H, et al. Integrins: novel therapeutic targets for cardiovascular diseases. Cardiovasc. Hematol. Agents Med. Chem. 2007;5:109–132. doi: 10.2174/187152507780363223. [DOI] [PubMed] [Google Scholar]
- 104.Andronati SA, et al. Peptidomimetics - antagonists of the fibrinogen receptors: molecular design, structures, properties and therapeutic applications. Curr. Med. Chem. 2004;11:1183–1211. doi: 10.2174/0929867043365314. [DOI] [PubMed] [Google Scholar]
- 105.Sawyer TK. Cancer metastasis therapeutic targets and drug discovery: emerging small-molecule protein kinase inhibitors. Expert Opin. Investig. Drugs. 2004;13:1–19. doi: 10.1517/13543784.13.1.1. [DOI] [PubMed] [Google Scholar]
- 106.Ayala A, et al. Blockade of apoptosis as a rational therapeutic strategy for the treatment of sepsis. Novartis Found Symp. 2007;280:37–49. doi: 10.1002/9780470059593.ch4. discussion 49-52, 160-164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Jain R, Chawrai S. Advancements in the anti-diabetes chemotherapeutics based on amino acids, peptides, and peptidomimetics. Mini Rev. Med. Chem. 2005;5:469–477. doi: 10.2174/1389557053765583. [DOI] [PubMed] [Google Scholar]
- 108.Walker MP, et al. Promising candidates for the treatment of chronic hepatitis C. Expert Opin. Investig. Drugs. 2003;12:1269–1280. doi: 10.1517/13543784.12.8.1269. [DOI] [PubMed] [Google Scholar]
- 109.Braisted AC, Wells JA. Minimizing a binding domain from protein A. Proc. Natl. Acad. Sci. U. S. A. 1996;93:5688–5692. doi: 10.1073/pnas.93.12.5688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wrighton NC, et al. Small peptides as potent mimetics of the protein hormone erythropoietin. Science. 1996;273:458–464. doi: 10.1126/science.273.5274.458. [DOI] [PubMed] [Google Scholar]
- 111.Yanofsky SD, et al. High affinity type I interleukin 1 receptor antagonists discovered by screening recombinant peptide libraries. Proc. Natl. Acad. Sci. U. S. A. 1996;93:7381–7386. doi: 10.1073/pnas.93.14.7381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Begley R, et al. Biodistribution of intracellularly acting peptides conjugated reversibly to Tat. Biochem. Biophys. Res. Commun. 2004;318:949–954. doi: 10.1016/j.bbrc.2004.04.121. [DOI] [PubMed] [Google Scholar]
- 113.Bose M, et al. The incorporation of a photoisomerizable amino acid into proteins in E. coli. J. Am. Chem. Soc. 2006;128:388–389. doi: 10.1021/ja055467u. [DOI] [PubMed] [Google Scholar]
- 114.Bates E, et al. Intracoronary KAI-9803 as an adjunct to primary percutaneous coronary intervention for acute ST-segment elevation myocardial infarction. Circulation. 2008;117:886–896. doi: 10.1161/CIRCULATIONAHA.107.759167. [DOI] [PubMed] [Google Scholar]
- 115.Murriel CL, et al. Protein kinase Cδ activation induces apoptosis in response to cardiac ischemia and reperfusion damage: a mechanism involving BAD and the mitochondria. J. Biol. Chem. 2004;279:47985–47991. doi: 10.1074/jbc.M405071200. [DOI] [PubMed] [Google Scholar]
- 116.Churchill EN, et al. Reperfusion-induced translocation of δPKC to cardiac mitochondria prevents pyruvate dehydrogenase reactivation. Circ. Res. 2005;97:78–85. doi: 10.1161/01.RES.0000173896.32522.6e. [DOI] [PubMed] [Google Scholar]
- 117.Qi X, et al. δPKC participates in the endoplasmic reticulum stress-induced response in cultured cardiac myocytes and ischemic heart. J. Mol. Cell. Cardiol. 2007;43:420–428. doi: 10.1016/j.yjmcc.2007.07.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Shizukuda Y, et al. Protein kinase C-δ modulates apoptosis induced by hyperglycemia in adult ventricular myocytes. Am. J. Physiol. 2002;282:H1625–H1634. doi: 10.1152/ajpheart.00783.2001. [DOI] [PubMed] [Google Scholar]
- 119.Warwar N, et al. Dynamics of glucose-induced localization of PKC isoenzymes in pancreatic β-cells: diabetes-related changes in the GK rat. Diabetes. 2006;55:590–599. doi: 10.2337/diabetes.55.03.06.db05-0001. [DOI] [PubMed] [Google Scholar]
- 120.Chu F, et al. PKC isozyme S-cysteinylation by cystine stimulates the pro-apoptotic isozyme PKCδ and inactivates the oncogenic isozyme PKCε. Carcinogenesis. 2003;24:317–325. doi: 10.1093/carcin/24.2.317. [DOI] [PubMed] [Google Scholar]
- 121.Konishi H, et al. Activation of protein kinase C by tyrosine phosphorylation in response to H2O2. Proc. Natl. Acad. Sci. U. S. A. 1997;94:11233–11237. doi: 10.1073/pnas.94.21.11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Alzamora R, et al. Direct binding and activation of protein kinase C isoforms by aldosterone and 17β-estradiol. Molecular Endocrinology. 2007;21:2637–2650. doi: 10.1210/me.2006-0559. [DOI] [PubMed] [Google Scholar]