

Abstract
The transcription factor 7-like 2 (TCF7L2) has been recently associated with diabetes risk, and it may exert its effect through metabolic syndrome (MetS)-related traits and be subjected to modification by environmental factors. We investigated the effect of single nucleotide polymorphisms (SNP), rs7903146 and rs12255372, within the TCF7L2 locus on postprandial lipemia and other MetS-related traits and their modulation by dietary fat. Data were collected from 1083 European Americans participating in the Genetics of Lipid Lowering Drugs and Diet Network Study. Carriers of the minor T allele at the C/T rs7903146 SNP had higher fasting plasma glucose (P = 0.012), lower homeostasis model assessment of β cell function (P = 0.041), higher plasma VLDL (P = 0.035), and lower large LDL particle (P = 0.007) concentrations and higher risk of MetS (P = 0.011) than CC individuals. Moreover, we identified significant interactions between this SNP and PUFA intake modulating fasting VLDL particle concentrations (P = 0.016) and postprandial triglycerides (TG) (P = 0.028), chylomicrons (P = 0.025), total VLDL (P = 0.026), and large VLDL (P = 0.018) concentrations. Thus, only T allele carriers with a PUFA intake ≥7.36% of energy had elevated fasting plasma VLDL concentrations and postprandial TG-rich lipoproteins. These variables did not differ in T allele carriers and noncarriers in the low-PUFA intake group. Moreover, these significant interactions were due exclusively to (n-6) PUFA intake. In summary, high (n-6) PUFA intakes (≥6.62% of energy intake) were associated with atherogenic dyslipidemia in carriers of the minor T allele at the TCF7L2 rs7903146 SNP and may predispose them to MetS, diabetes, and cardiovascular disease.
Introduction
Insulin resistance is associated with a cluster of glucose and lipid metabolism abnormalities and it is the hallmark of diabetes and metabolic syndrome (MetS),8 which are major cardiovascular disease (CVD) risk factors. Dyslipidemia associated with diabetes is characterized by increased plasma triglycerides (TG) and low concentrations of HDL cholesterol (HDL-C). Moreover, the availability of NMR lipoprotein profiling has led to an easier and more precise definition of the lipoprotein subclasses involved, providing support to the notion that the diabetic dyslipidemia is defined by elevation of fasting concentrations of small dense LDL, small HDL, and large VLDL particles as well as increased postprandial TG-rich lipoproteins (TRL) (1). Consistently, findings from 2 large cohort studies showed that elevated nonfasting TG levels were an independent predictor of CVD events and death and supported the relevant role of postprandial lipemia in CVD risk (2,3).
Type 2 diabetes shows considerable heritability and the identification of the specific loci involved has progressed since the use of genome-wide genetic screening in large populations. One of the novel genes identified in relation to diabetes risk is the transcription factor 7-like 2 gene (TCF7L2; formerly known as TCF4) located on chromosome 10 (10q25.3). More specifically, a microsatellite marker DG10S478 within intron 3 of TCF7L2 was associated with diabetes in 3 independent case-control studies, mainly in Caucasians (4). Subsequently, several studies replicated this finding in different ethnic groups (5–12), with the strongest and most consistent associations being reported for 2 single nucleotide polymorphisms (SNP), rs7903146C > T and rs12255372G > A. The TCF7L2 encodes for a Wnt signaling-associated transcription factor, TCF7L2, which is necessary for normal development of the pancreas and islets during embryonic growth (13) and plays a role in adipogenesis and adipocyte differentiation (14). The mechanisms by which common variants in the TCF7L2 gene increase risk of type 2 diabetes have been studied and work by Lissenko et al. (15) showed that one of the TCF7L2 variants (rs7903146) is associated with increased TCF7L2 mRNA expression in the pancreatic β cell, reducing insulin secretion and hence predisposing to diabetes. Moreover, TCF7L2 gene variants have been associated with other individual components of the MetS, including waist circumference, BMI, and lipid profiles (16–20), but these findings have been inconsistent. In Mexican and Finnish families with familial combined hyperlipidemia, the presence of the minor alleles at TCF7L2 rs7903146 and rs12255372 SNP was associated with higher plasma TG concentrations (20). In contrast, the minor T allele at the rs7903146 SNP was associated with a lesser atherogenic lipid profile, particularly lower plasma TG and higher plasma HDL-C concentrations in an elderly Italian cohort (18). Paradoxically, TCF7L2 SNP previously associated with increased diabetes risk have also been associated with reduced risk of obesity in some populations (16–19). To date, there is no information on the association between TCF7L2 gene variants and postprandial lipemia or whether dietary factors modulate the reported associations and could be one of the reasons for the reported disparities. We hypothesized that TCF7L2 variants were associated with MetS and other diabetes-related traits, including postprandial lipemia. Therefore, we investigated the associations of TCF7L2 SNP (rs7903146 and rs12255372) on diabetes-related traits and risk of MetS and their modification by dietary fat in a well-defined population of European Americans.
Materials and Methods
Participants and study design.
The Genetics of Lipid Lowering Drugs and Diet Network (GOLDN) study included 1200 individuals aged 17–92 y. The primary aim of the study was to characterize the genetic basis of TG responses to diet (high-fat test meal) and to a TG-lowering drug (fenofibrate). The majority of participants were re-recruited from the 3-generational pedigrees of 2 National Heart, Lung, and Blood Institute Family Heart Study field centers (Minneapolis, MN and Salt Lake City, UT) (21). Nearly all individuals were of European ancestry. Detailed design and methodology of the GOLDN study has been described (22,23). Individuals with inconsistent dietary data (total daily energy intake outside the range of 3.35–23.03 MJ in men or 2.51–18.84 MJ in women) were excluded, resulting in a sample size of 1083 individuals (515 men and 568 women). Moreover, some individuals did not complete the fat loading test and the analysis of postprandial response included 1011 participants. Baseline characteristics did not differ between any of the subgroups compared with the entire group. Briefly, exclusion criteria were: recent history (6 mo) of myocardial infarction; history of liver, kidney, pancreas, or gall bladder disease; history of malabsorption of nutrients; current use of insulin; abnormal renal or hepatic function; and pregnancy or nursing in women. Individuals who reported current use of lipid-lowering drugs or dietary supplements known to influence lipids were required to consult their physician for approval to discontinue these lipid-lowering agents for 4 wk before study participation.
During the fat loading test, participants were given a high-fat meal after 12 h of fasting. The meal, which participants were instructed to consume within 15 min, had 2.93 MJ/m body surface2 area (700 kcal/m2 body surface area), 3% of energy were derived from protein, 14% from carbohydrate, and 83% from fat sources. The cholesterol content and the ratio of polyunsaturated:saturated fat were 240 mg and 0.06, respectively (24). The average person consumed 175 mL heavy whipping cream (39.5% fat) and 7.5 mL of powdered, instant, nonfat, dry milk. The mixture was blended with water ice and 15 mL of chocolate- or strawberry-flavored syrup to increase palatability. Blood samples were drawn immediately before (time 0) and at 3.5 and 6 h after consuming this high-fat meal. For the duration of the 6-h study period, participants were instructed to take nothing by mouth except water, unsweetened coffee or tea, or sugarless soft drinks and to abstain from exercise and strenuous physical work until the final blood sample was obtained. The protocol was approved by the Institutional Review Boards at the University of Alabama, the University of Minnesota, the University of Utah, and Tufts University. Written informed consent was obtained from all participants.
Anthropometric and dietary assessments.
Anthropometric data including height, weight, and waist and hip circumferences were measured using a standard technique. BMI was calculated by dividing the weight (kg) by height (m2). The habitual dietary intake was estimated using the Dietary History Questionnaire developed by the National Cancer Institute (25). The questionnaire has been validated (26,27). We considered together the dietary intake of α-linolenic acid, eicosapentaenoic acid, docosahexaenoic acid, and docosapentaenoic acid as (n-3) PUFA. Linoleic acid and arachidonic acid were considered together as (n-6) PUFA. The percentages of dietary fat are more adequate to rank people than absolute intakes. Therefore, consumption of dietary fat was presented as percentage of total daily energy intake and was analyzed as both continuous and categorical variables. To construct categorical variables, the relative percent of dietary fat was dichotomized using the median value of the population (i.e. groups had intakes below and above the median of percentage dietary fat).
Biochemical analyses.
Venous blood was obtained from subjects after overnight fasting. All plasma samples were stored until the completion of their participation and then analyzed together. Measurements of plasma TG, total cholesterol, LDL-C and HDL-C, and postprandial lipemic response were determined as previously described (23). In the GOLDN study, NMR spectroscopy was used to quantify lipoprotein subclasses and average VLDL, LDL, and HDL particle diameters (nm) by using their difference signal amplitudes of each lipoprotein subclass and detail of this method (28,29). Conversion factors relating these signal amplitudes to subclass concentrations expressed in particle concentration units or lipid mass concentration units (cholesterol or TG). The particle concentrations (nmol/L for VLDL and LDL and μmol/L for HDL) were derived for each subclass standard by measuring the total concentration in core lipid (cholesterol ester plus TG) and dividing the volume occupied by the core volume per particle calculated from knowledge of the particle's diameter. Lipid mass concentrations of VLDL subclasses are given in mg/dL TG units and those of the LDL and HDL subclasses in mg/dL cholesterol units.
Moreover, the method was validated in this study population, suggesting that NMR is an alternative method for assessing TRL (30). For clinical purposes and to facilitate comparison with other publications using the NMR method, we present the fasting lipoprotein profiles in terms of molar levels of lipoprotein particles/L. However, the postprandial lipid responses are expressed as amount of lipid in lipoprotein by using lipid mass concentration rather than the molar level of lipoproteins.
Glucose was determined by a hexokinase-mediated reaction on the Hitachi 911 (Roche Diagnostics). Insulin was measured using RIA with a commercial kit (Linco Research). Insulin resistance and β cell function were quantified using homeostasis model assessment (HOMA-R and HOMA-B, respectively); HOMA-R = fasting insulin (pmol/L) × fasting glucose (mmol/L)/156.3 and HOMA-B = fasting insulin (pmol/L) × 2.88/[fasting glucose (mmol/L) − 3.5] (31). Blind duplicate samples from 5% of participants were sent to the laboratory to assess repeatability. For all lipid subfractions, the repeatability was >90%.
DNA isolation and TCF7L2 genotyping.
Genomic DNA was isolated from peripheral blood leukocytes using commercial Puregene DNA reagents (Gentra Systems). Two common variants in the TCF7L2 gene (rs7903146 and rs12255372) were genotyped using a Taqman assay with allele-specific probes on the ABI Prism 7900HT Sequence Detection system (Applied Biosystems) (32) according to routine laboratory protocols. The genotyping was successful in 1080 participants (99.7%) for rs7903146 and rs12255372 in the whole group.
Statistical analysis.
Statistical analyses were conducted using SAS for Windows version 9.1.3 (SAS Institute). Continuous variables were tested for normal distribution. Logarithmic or square root transformations were applied as appropriate to normalize the distribution of data. Data reported for those variables are back-transformed geometric means and 95% CI. As homogeneity of the genotype effect by sex was observed (P for interaction genotype × gender > 0.05) for any of the relevant traits, men and women were combined for subsequent analyses. Pearson χ2 and Fisher's exact tests were used to test TCF7L2 genotypes for departure from the Hardy-Weinberg equilibrium in unrelated subjects. Three genotype groups were first considered to identify different inherent models. The dominant models were better fit for these SNP concerning the interesting outcomes; thus, homozygous for the common allele was compared with carriers of the minor allele (heterozygous and homozygous for the minor allele) in the analysis.
The MetS was classified according to the 2005 National Cholesterol Education Program Adult Treatment Panel III guidelines (33). Diabetes was defined by either self-reported history of diabetes or a fasting plasma glucose concentration ≥ 7 mmol/L (≥126 mg/dL) (34). We used the generalized estimating equation linear and logistic regression with exchangeable correlation structure to adjust for the correlated observations due to familial relationship. Logistic regression method was applied to assess the association between TCF7L2 polymorphisms and disease outcomes (presence or absence of MetS). The presented results, OR, and 95% CI were the exponentiated β coefficient and 95% CI, which were computed at natural log-scale. To examine the postprandial lipemic response of TCF7L2 genotypes across 3 time points, we fitted a 3-level and individual growth mixed model using SAS Proc Mixed: level 1, individual measurements across 3 time points; level 2, individual nested within pedigree; and level 3, pedigree. To model the individual growth curve, we used an autoregressive, AR (1) error covariance matrix while treating the intercept and time as random effects. The individuals nested within pedigrees were modeled using a generalized linear mixed model (35). We also measured the postprandial lipemic response by calculating the area under the curve (AUC), which was defined as the area between the plasma concentration of the corresponding variable compared with the time curve and a line drawn parallel to the horizontal axis through the 0-h concentration. The logarithm of AUC was used as a dependent variable in the generalized estimating equation approach as implemented in the GENMOD procedure in SAS. The analyses were performed for the whole sample and for men and women separately to verify the homogeneity of genetic effect among men and women. The interactions between SNP and dietary fat intake (as continuous and as dichotomous variables) were tested in the multivariate interaction model. Covariates included age; gender; family relationship; smoking; alcohol consumption; BMI; physical activity; hormone treatment; drugs for diabetes, hypertension, and lowering cholesterol; and total energy intake. The pair-wise linkage disequilibrium between SNP was a scaled linkage disequilibrium estimate, D′ (D/Dmax, if D>0), and a correlation coefficient, r2, using the Helix Tree software package (Golden Helix). A 2-tailed P-value of <0.05 was considered significant.
Results
Genotype frequencies among unrelated participants did not deviate from the Hardy-Weinberg equilibrium. Allele frequencies of the variant alleles were 0.29 for rs7903146 and 0.31 for rs12255372. Consistent with previous reports, both SNP were in strong linkage disequilibrium (D' = 0.9; r2 = 0.9); therefore, we reported only those results pertaining to TCF7L2 rs7903146 C > T. The baseline clinical characteristics of study participants are shown (Table 1) and relevant metabolic traits for all participants according to the TCF7L2 rs7903146 genotype are presented (Tables 2 and 3). Under the dominant model, we found a significant difference in fasting plasma glucose concentration and β cell function assessed by HOMA-B between carriers of the minor rs7903146 T allele (CT+TT) and CC participants. Compared with CC participants, T allele carriers had a higher fasting plasma glucose level (6.35 vs. 6.24 mmol/L; P = 0.012) and lower β cell function (P = 0.041) after adjusting for age, gender, family relationship, BMI, smoking, alcohol consumption, physical activity, study sites, and hormone treatment, and diabetes, hypertension and hyperlipidemic medications (Table 2). We also observed that the T allele carriers had significantly higher plasma total VLDL (P = 0.035) and lower large LDL (P = 0.007) particle concentrations than did CC participants. The fasting plasma TG concentration tended (P = 0.057) to be greater in T allele carriers than in CC participants. In addition, T allele carriers tended to have smaller LDL size (P = 0.055) relative to CC participants (Table 3). In a multivariate adjusted model, the T allele carriers tended to have higher postprandial responses for TG (P for genotype = 0.096), VLDL (P for genotype = 0.062), and chylomicrons (P for genotype = 0.179). There were no significant genotype-related differences for insulin resistance and anthropometric variables. Furthermore, we used logistic regression to test the effect of the TCF7L2 SNP on the risk of diabetes and MetS-related components. In this population, 8 and 38% met the criteria for diabetes and MetS, respectively. Our study was not powered to replicate previous associations with diabetes risk, but we found that CC participants had a lower risk of MetS than carriers of the T allele (P = 0.011), primarily through lower risk of hypertriglyceridemia (P = 0.017) in a multivariate adjusted model (Table 4).
TABLE 1.
Baseline characteristics of the GOLDN participants1
| Men | Women | |
|---|---|---|
| n | 517 | 566 |
| Age, y | 49.4 ± 16.1 | 48.4 ± 16.2 |
| BMI, kg/m2 | 28.5 ± 4.9 | 28.0 ± 6.2 |
| Waist, cm | 101 ± 14.9 | 93 ± 17.2* |
| Plasma LDL-C, mmol/L | 3.19 ± 0.79 | 3.11 ± 0.83 |
| Plasma HDL-C, mmol/L | 1.08 ± 0.26 | 1.36 ± 0.36* |
| Plasma TG, mmol/L | 1.34 (0.90–2.11) | 1.14 (0.76–1.74)* |
| Fasting plasma glucose, mmol/L | 5.61 (5.33–5.99) | 5.27 (5.00–5.61)* |
| Plasma insulin, pmol/L | 62.5 (83.8–114.6) | 55.6 (76.4–107.6) |
| Current smoker, n (%) | 39 (7.5) | 42 (7.4) |
| Current drinker, n (%) | 256 (49.5) | 291 (51.4) |
| Hormonal treatment, n (%) | 0 (0) | 119 (21)* |
| Obesity,2n (%) | 169 (32.8) | 192 (33.8) |
| Diabetes,3n (%) | 50 (9.7) | 37 (6.5) |
Data are means ± SD or median (interquartile range). *Different from men, P < 0.05.
Obesity, BMI ≥30 kg/m2.
Diabetes, fasting plasma glucose ≥ 7 mmol/L or history of diabetes.
TABLE 2.
Anthropometric and insulin resistance variables according to TCF7L2 rs7903146 polymorphisms in the GOLDN study1
|
TCF7L2 rs7903146
|
|||
|---|---|---|---|
| CC | CT+TT | P2 | |
| n | 541 | 539 | |
| BMI, kg/m2 | 30.4 ± 0.8 | 30.2 ± 0.8 | 0.496 |
| Waist, cm | 102 ± 1.9 | 103 ± 2.0 | 0.358 |
| Hip, cm | 110 ± 1.5 | 110 ± 1.5 | 0.938 |
| Waist:hip ratio | 0.92 ± 0.01 | 0.93 ± 0.01 | 0.118 |
| Fasting plasma glucose, mmol/L | 6.24 (6.03–6.45) | 6.35 (6.13–6.57) | 0.012 |
| Fasting plasma insulin, pmol/L | 83.0 (92.3–102.5) | 83.0 (91.4–100.6) | 0.719 |
| HOMA-R | 3.68 (3.27–4.15) | 3.71 (3.32–4.15) | 0.779 |
| HOMA-B | 106 (94.9–118) | 102 (91.2–114) | 0.041 |
| Plasma cholesterol, mmol/L | 5.20 ± 0.11 | 5.22 ± 0.11 | 0.729 |
| Plasma LDL-C, mmol/L | 3.21 ± 0.10 | 3.16 ± 0.10 | 0.409 |
| Plasma HDL-C, mmol/L | 1.20 ± 0.03 | 1.19 ± 0.04 | 0.545 |
| Plasma TG, mmol/L | 1.84 (1.59–2.13) | 1.97 (1.70–2.28) | 0.057 |
Data are multivariate adjusted mean ± SEM or geometric mean (95% CI).
P-values were adjusted for age, gender, family relationship, smoking, alcohol consumption, study sites, physical activity, hormone treatment, and drugs for diabetes, hypertension and lowering cholesterol. BMI was additional adjusted in nonanthropometric variables.
TABLE 3.
Fasting plasma lipoprotein particle concentrations and diameters according to TCF7L2 rs7903146 polymorphisms in the GOLDN study1
|
TCF7L2 rs7903146
|
|||
|---|---|---|---|
| CC | CT+TT | P2 | |
| n | 541 | 539 | |
| Plasma lipoprotein particle concentrations | |||
| Total VLDL, nmol/L | 84.2 (73.9–95.1) | 91.6 (80.3–103.7) | 0.035 |
| Large VLDL, nmol/L | 2.90 (2.00–4.21) | 3.52 (2.47–5.02) | 0.060 |
| Medium VLDL, nmol/L | 35.2 (28.0–44.3) | 39.7 (31.9–49.4) | 0.056 |
| Small VLDL, nmol/L | 31.0 (26.1–36.4) | 30.6 (25.2–36.6) | 0.772 |
| Large LDL, nmol/L | 290 (222–367) | 250 (188–322) | 0.007 |
| Small LDL, nmol/L | 909 (765–1064) | 967 (812–1136) | 0.125 |
| Large HDL, μmol/L | 5.31 (4.59–6.08) | 4.96 (4.25–5.73) | 0.092 |
| Medium HDL, μmol/L | 1.89 (1.28–2.62) | 1.78 (1.21–2.45) | 0.491 |
| Small HDL, μmol/L | 22.0 (20.5–23.5) | 21.7 (20.3–23.2) | 0.470 |
| Particle size, nm | |||
| VLDL | 54.01 ± 1.12 | 53.83 ± 1.18 | 0.700 |
| LDL | 20.64 ± 0.10 | 20.53 ± 0.10 | 0.055 |
| HDL | 8.82 ± 0.05 | 8.79 ± 0.05 | 0.351 |
Data are multivariate adjusted mean ± SEM or geometric mean (95% CI).
P-values were adjusted for age, gender, family relationship, smoking, alcohol consumption, study sites, BMI, physical activity, hormone treatment, and drugs for diabetes, hypertension and lowering cholesterol.
The normal ranges of the lipoprotein subclasses in the U.S. population see (29).
TABLE 4.
OR (95% CI) for the prevalence of MetS components across TCF7L2 rs7903146 polymorphism in the GOLDN study1
|
TCF7L2 rs7903146
|
|||
|---|---|---|---|
| CC | CT+TT | P1 | |
| n | 541 | 539 | |
| Diabetes2 | 0.96 (0.61–1.52) | 1 | 0.856 |
| Obesity3 | 0.96 (0.72–1.28) | 1 | 0.764 |
| Impaired fasting glucose4 | 0.91 (0.67–1.24) | 1 | 0.555 |
| Abdominal obesity5 | 0.78 (0.60–1.02) | 1 | 0.076 |
| High blood pressure6 | 0.83 (0.58–1.17) | 1 | 0.279 |
| High TG7 | 0.68 (0.50–0.92) | 1 | 0.017 |
| Low HDL-C8 | 0.87 (0.66–1.15) | 1 | 0.315 |
| MetS9 | 0.68 (0.51–0.90) | 1 | 0.011 |
Data are OR (95% CI) or P-Values. Models were adjusted for age, gender, family relationship, smoking, alcohol consumption, study sites, physical activity, and hormone treatment.
Diabetes, fasting plasma glucose ≥ 7 mmol/L or history of diabetes.
Obesity, BMI ≥ 30 kg/m2.
Impaired fasting glucose, fasting plasma glucose ≥ 5.6 mmol/L or taking diabetic medication.
Abdominal obesity, waist circumference ≥ 102cm in men or ≥ 88 cm in women.
High blood pressure, systolic blood pressure ≥ 130 mm Hg or ≥ 85 mm Hg or taking antihypertensive drugs.
High TG, fasting plasma TG ≥ 1.7 mmol/L.
Low HDL-C, fasting plasma HDL-C < 1.03 mmol/L in men or < 1.29 mmol/L in women.
MetS by National Cholesterol Education Program criteria, any 3 or more of these components: impaired fasting glucose, abdominal obesity, high blood pressure, high TG, low HDL-C.
Because of the association with TRL variables and risk of hypertriglyceridemia, we further tested the hypothesis that dietary fat consumption modifies the association between TCF7L2 variants and lipid profiles (fasting and postprandial). At baseline, energy and macronutrient intakes did not differ between CC homozygotes and T allele carriers. We classified the reported percentage of dietary fat intakes as categorical variables according to their population median. The postprandial data represent the amount of lipid carried in particles of each subclass (e.g. mg/dL TG for VLDL particles and mg/dL cholesterol for LDL and HDL particles). Using a multivariate adjusted model, we identified significant interactions between rs7903146 and PUFA intake (<7.36 or ≥7.36% of energy intake) modulating fasting plasma VLDL (P = 0.016) concentrations. In participants classified within the lower one-half of PUFA intake, fasting plasma VLDL particle concentrations did not differ in either group, whereas T allele carriers in the upper one-half had higher plasma VLDL than CC participants (P = 0.011; P for interaction = 0.016). Likewise, we observed significant gene × PUFA interactions on postprandial responses for TG (P for interaction = 0.028), chylomicrons (P for interaction = 0.025), VLDL (P for interaction = 0.026), and large VLDL (P for interaction = 0.018) concentrations (Fig. 1A–F). Thus, carriers of the T allele who consumed PUFA ≥ 7.36% of energy exhibited higher postprandial TG (P = 0.008), chylomicrons (P = 0.006), VLDL (P = 0.013), and large VLDL (P = 0.009) concentrations than CC participants. On the other hand, in those with PUFA consumption < 7.36% of energy, there were no difference in the postprandial lipid response between T allele carriers and noncarriers. The gene × PUFA interactions did not observe for postprandial concentration of medium and small VLDL. Similarly, we did not find significant interactions between TCF7L2 rs7903146 SNP and dietary fat (total), SFA, or monounsaturated fatty acids on fasting and postprandial lipid profiles (data not shown). To examine whether the interaction between TCF7L2 rs7903146 and PUFA intake was dose dependent, we analyzed dietary PUFA as a continuous variable. The AUC lipid represented postprandial lipemic response. Consistently, we found significant gene × PUFA interactions for fasting plasma VLDL (P = 0.019) and postprandial TRL, represented by AUC TG (P = 0.004), chylomicrons (P = 0.004), VLDL (P = 0.002), and large VLDL (P = 0.005) (data not shown).
FIGURE 1 .

Plasma postprandial TG (A,B), chylomicrons (C,D), and VLDL (E,F) responses in participants classified by genetic variation at TCF7L2 rs7903146 and stratified by dietary PUFA intakes below (A,C,E) or above (B,D,F) the median. Values are geometric means adjusted for age, gender, family relationship, BMI, physical activity, smoking, alcohol consumption, study sites, hormone treatment, and drugs for diabetes, hypertension, and lipid lowering. PG denotes the multivariate genotype effect in the postprandial period for genotype and PI, the genotype × median dietary PUFA interaction. CC (n = 251), CT+TT (n = 253) below the PUFA intake median and CC (n = 255), CT+TT (n = 249) above the PUFA intake median. *Different from CC at that time, P < 0.05.
We further investigated whether the gene × PUFA interactions applied to the consumption of both (n-3) and (n-6) families. Using the same statistical models as those used above for total PUFA, we dichotomized (n-6) and (n-3) dietary PUFA intakes by their corresponding population medians (6.62% and 0.67% of energy, respectively). Our analyses showed that the significant interactions on fasting plasma VLDL particle concentrations and postprandial TG, chylomicrons, VLDL, and large VLDL concentrations were observed only for (n-6) PUFA intake (P for interaction = 0.012, 0.021, 0.031, 0.018, and 0.017, respectively) but not for the (n-3) family (P > 0.05). To investigate the possible dose-response relationship in these interactions, (n-6) and (n-3) PUFA intakes were considered as continuous variables. Similarly, we found a clear dosage effect on the variables listed above primarily for (n-6) PUFA (P for interaction = 0.015), which was not significant for (n-3) PUFA (P for interaction = 0.186) as for plasma total VLDL concentrations (Fig. 2A,B). The regression lines of the predicted plasma total VLDL levels derived from the regression models, including TCF7L2 rs7903146 and (n-6) and (n-3) PUFA intake (as continuous), are shown below.
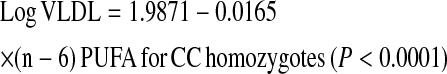 |
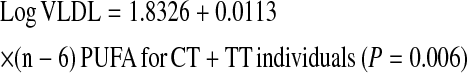 |
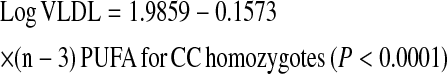 |
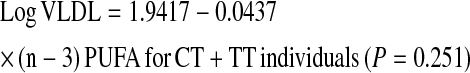 |
FIGURE 2 .

Scatterplots of the observed values and regression lines of predicted plasma total VLDL concentrations in participants classified by genetic variation at TCF7L2 rs7903146 and stratified by (n-6) (A) and (n-3) PUFA (B) intakes. Predicted values were calculated from the regression models containing PUFA intake, rs7903146 polymorphism, their interaction terms, and the potential confounders (age, gender, BMI, physical activity, smoking, alcohol consumption, study sites, hormone treatment, drugs for diabetes, hypertension, and lipid lowering, and energy intake). P-values for interaction terms were obtained from the multivariate adjusted model.
High (n-6) PUFA consumption was significantly associated with low plasma total VLDL concentrations in CC participants, whereas high (n-6) PUFA intake was significantly associated with high plasma total VLDL concentrations in the T allele carriers. With regard to (n-3) PUFA, high consumption was negatively correlated with plasma total VLDL concentrations in both genotype groups; however, the decrease in plasma VLDL concentrations was not significant in the T allele carriers (P = 0.251). These findings suggest that the interaction for total PUFA intake was primarily driven by (n-6) PUFA consumption.
Discussion
We found that TCF7L2 SNP was associated with higher fasting plasma glucose and lower HOMA-B, suggesting impaired β cell function without affecting HOMA-R and higher risk of MetS. However, given the small number of diabetics in this population, we could not demonstrate an association of this locus with diabetes. Moreover, we also observed the interaction between TCF7L2 rs7903146C > T and dietary PUFA primarily related to (n-6) PUFA in modulating fasting and postprandial lipid responses.
Our findings are in agreement with previous reports showing that the variant at TCF7L2 is associated with impaired insulin secretion rather than insulin resistance (15,19,36–39), supporting the idea that pancreatic β cell is the site of action for this gene product. Additionally, T allele carriers at TCF7L2 rs7903146 had significantly higher fasting VLDL particle concentrations and a trend toward higher fasting TG and postprandial lipemia and smaller LDL particle size, all of which are reported to be associated with increase CVD risk. In the Insulin Resistance Atherosclerosis Study, nondiabetic participants who subsequently developed diabetes after a mean follow-up of 5.2 y had 9.4% higher VLDL particle concentrations compared with participants who remained nondiabetic at follow-up (40). The difference in VLDL particle concentration was similar with our finding that VLDL particle concentrations in the T allele carriers were 8.8% higher than non-T allele carries. Furthermore, we found a significant association of this TCF7L2 variant with the risk of MetS, the strongest association being found with hypertriglyceridemia. This association was not found in the MONICA/KORA survey consisting of elderly German participants (41). The inconsistency may be related to the heterogeneity of MetS, with some components being more prevalent in one population over the others. The GOLDN study was conducted to ascertain the genetic basis of TG responses to a fat load and to a TG-lowering drug (fenofibrate). The study participants were re-recruited from the Family Heart study and may have been selected on the basis of higher TG and therefore, were at higher risk for CVD. MetS in the MONICA/KORA study may be more related to other factors rather than to hypertriglyceridemia, as was the case in our population.
The TCF7L2 transcription factor is highly expressed in most human tissues, including pancreatic β cells and adipocytes (42), and it is part of the Wnt/β-catenin signaling pathway. Moreover, an animal study revealed that modification of Wnt/β-catenin signaling in skeletal muscle induced weight loss and decreased insulin and TG levels in obese rats (43). Despite the paucity of information regarding the role of TCF7L2 in lipid metabolism, it has been shown that a mutation in LRP6, working through the same Wnt signaling pathway as TCF7L2, showed strong genetic linkage with early CAD, high LDL-C, high TG, hypertension, diabetes, and low bone mineral density (44). Few studies have investigated the association between TCF7L2 polymorphisms and fasting lipid traits (16–18,20). Consistent with our findings, the minor T alleles of TCF7L2 (both rs7903146 and rs12255372) were associated with elevated TG in participants with familial combined hyperlipidemia (20). Conversely, a report in elderly Italians showed that carriers of the minor allele at rs7903146 had a favorable lipid profile consisting of lower TG and higher HDL-C concentrations (18). Other studies did not find significant associations between TCF7L2 variants and lipid parameters (16,17). These inconsistencies could be due to the different experimental designs, including the age of the cohorts, that may introduce survival bias, but they could be also due to different environmental exposures with diet being a major factor. The Wnt/β-catenin signaling maintains the undifferentiated state of preadipocytes by inhibiting adipogenic gene expression, including PPARγ and CCAAT/enhancer binding protein α (14,45–47). In the meanwhile, dietary fatty acids, especially PUFA, are natural ligands for the PPAR family of transcription factors involved in regulation of energy and glucose and lipid metabolisms (48). Along these lines, we have reported the gene × PUFA interactions on TRL concentrations for the APOA5 gene in the Framingham Offspring study (49). The potential negative effects associated with increased TRL concentrations were reported in the APOA5-1131C carriers with high PUFA intake and more specifically (n-6) PUFA. In this regard, we further examine the hypothesis that PUFA intake may modulate the effect of TCF7L2 variant on TRL and postprandial lipemia. In agreement with our previous results, we observed novel findings pertaining to the modification of the associations between TCF7L2 variants and fasting and postprandial lipid parameters with reported PUFA consumption. Our findings suggest that the more atherogenic lipid profile associated with the minor T allele at the TCF7L2 rs7903146 SNP is expressed in conjunction with high consumption of dietary PUFA, consistent with the reported decrease in insulin secretion (assessed by glucose stimulated insulin secretion) associated with high PUFA intake (50). Moreover, we found that minor T allele carriers at the TCF7L2 rs7903146 SNP had lower β cell function. Therefore, we propose that the combination of the minor T allele and a high PUFA consumption act additively to reduce insulin secretion enough to impair homeostasis, resulting in increased FFA, hepatic VLDL secretion, and exaggerated fasting and postprandial hypertriglyceridemia, which may increase risk of atherosclerosis. These hypotheses need to be tested in experimental designs that include the measurement of serum FFA and lipoprotein lipase activity.
Given the differences in biological effects of (n-3) and (n-6) PUFA families, our data clearly support the idea that the reported gene × diet interactions were primarily driven by dietary (n-6) PUFA. Thus, elevated fasting VLDL and postprandial TRL concentrations were found in TCF7L2 rs7903146T allele carriers consuming high (n-6) PUFA. On the other hand, (n-3) PUFA decreased VLDL and postprandial TRL concentrations independently of the TCF7L2 rs7903146 polymorphism. These findings may explain the intraindividual responses in TG-related lipoprotein traits to dietary PUFA and the different effects of (n-3) and (n-6) PUFA.
In this study, we performed a number of statistical tests for different nutrients to assess interactions of lipid traits, potentially raising the problem of multiple comparisons. There is no consensus for selecting the most adequate strategy for correcting for multiple tests (51,52); therefore, we decided to present the unadjusted P-values. The findings of gene × diet interactions need to be interpreted with caution and maintained within the context of the specific circumstances of the population examined. In addition, taking into consideration the study design, we cannot provide evidence for the causality of this interaction or its mechanistic basis. Therefore, further work is required to replicate our findings and to determine the validity of the mechanisms suggested above. This includes intervention studies using specific fatty acids to confirm whether the gene × PUFA interaction regulating fasting and postprandial lipemia is supported by a higher level of evidence.
In summary, our findings support previous associations between genetic variants at the TCF7L2 locus and higher fasting plasma glucose, impaired β cell function, and higher risk of MetS. In addition, the current results underscore the role of TCF7L2 in lipid metabolism. Finally, we provide novel information about the modification of the associations between the variant allele at TCF7L2 and plasma lipids by (n-6) PUFA. These findings, if replicated, may help in the identification of a segment of the population that may benefit from restriction in their (n-6) PUFA intake to decrease their risk factors associated with MetS and CVD.
Supported by the National Heart, Lung and Blood Institute grants U01 HL72524, Genetic and Environmental Determinants of Triglycerides and HL-54776 and the National Institute of Diabetes and Digestive and Kidney grant DK075030, and by contracts 53-K06-5-10 and 58-1950-9-001 from the USDA, Agricultural Research Service.
Author disclosures: D. Warodomwichit, D. K. Arnett, E. K. Kabagambe, M. Y. Tsai, J. E. Hixson, R. J. Straka, M. Province, P. An, C. Lai, I. Borecki, and J. M. Ordovas, no conflicts of interest.
Abbreviations used: AUC, area under the curve; CVD, cardiovascular disease; GOLDN, Genetics of Lipid Lowering Drugs and Diet Network; HOMA-B, homeostasis model assessment of β cell function; HOMA-R, homeostasis model assessment of insulin resistance; MetS, metabolic syndrome; SNP, single nucleotide polymorphism; TG, triglyceride; TRL, triglyceride-rich lipoprotein.
References
- 1.Taskinen MR. Diabetic dyslipidaemia: from basic research to clinical practice. Diabetologia. 2003;46:733–49. [DOI] [PubMed] [Google Scholar]
- 2.Bansal S, Buring JE, Rifai N, Mora S, Sacks FM, Ridker PM. Fasting compared with nonfasting triglycerides and risk of cardiovascular events in women. JAMA. 2007;298:309–16. [DOI] [PubMed] [Google Scholar]
- 3.Nordestgaard BG, Benn M, Schnohr P, Tybjaerg-Hansen A. Nonfasting triglycerides and risk of myocardial infarction, ischemic heart disease, and death in men and women. JAMA. 2007;298:299–308. [DOI] [PubMed] [Google Scholar]
- 4.Grant SFA, Thorleifsson G, Reynisdottir I, Benediktsson R, Manolescu A, Sainz J, Helgason A, Stefansson H, Emilsson V, et al. Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nat Genet. 2006;38:320–3. [DOI] [PubMed] [Google Scholar]
- 5.Cauchi S, El Achhab Y, Choquet H, Dina C, Krempler F, Weitgasser R, Nejjari C, Patsch W, Chikri M, et al. TCF7L2 is reproducibly associated with type 2 diabetes in various ethnic groups: a global meta-analysis. J Mol Med. 2007;85:777–82. [DOI] [PubMed] [Google Scholar]
- 6.Chandak GR, Janipalli C, Bhaskar S, Kulkarni S, Mohankrishna P, Hattersley A, Frayling T, Yajnik C. Common variants in the TCF7L2 gene are strongly associated with type 2 diabetes mellitus in the Indian population. Diabetologia. 2007;50:63–7. [DOI] [PubMed] [Google Scholar]
- 7.Hayashi T, Iwamoto Y, Kaku K, Hirose H, Maeda S. Replication study for the association of TCF7L2 with susceptibility to type 2 diabetes in a Japanese population. Diabetologia. 2007;50:980–4. [DOI] [PubMed] [Google Scholar]
- 8.Scott LJ, Bonnycastle LL, Willer CJ, Sprau AG, Jackson AU, Narisu N, Duren WL, Chines PS, Stringham HM, et al. Association of transcription factor 7-like 2 (TCF7L2) variants with type 2 diabetes in a Finnish sample. Diabetes. 2006;55:2649–53. [DOI] [PubMed] [Google Scholar]
- 9.Elbein SC, Chu W, Das S, Yao-Borengasser A, Hasstedt S, Wang H, Rasouli N, Kern P. Transcription factor 7-like 2 polymorphisms and type 2 diabetes, glucose homeostasis traits and gene expression in US participants of European and African descent. Diabetologia. 2007;50:1621–30. [DOI] [PubMed] [Google Scholar]
- 10.Groves CJ, Zeggini E, Minton J, Frayling TM, Weedon MN, Rayner NW, Hitman GA, Walker M, Wiltshire S, et al. Association analysis of 6,736 U.K. TCF7L2 as a subjects provides replication and confirms type 2 diabetes susceptibility gene with a substantial effect on individual risk. Diabetes. 2006;55:2640–4. [DOI] [PubMed] [Google Scholar]
- 11.Humphries SE, Gable D, Cooper J, Ireland H, Stephens J, Hurel S, Li K, Palmen J, Miller M, et al. Common variants in the TCF7L2 gene and predisposition to type 2 diabetes in UK European Whites, Indian Asians and Afro-Caribbean men and women. J Mol Med. 2006;84:1005–14. [DOI] [PubMed] [Google Scholar]
- 12.Zhang C, Qi L, Hunter DJ, Meigs JB, Manson JE, van Dam RM, Hu FB. Variant of transcription factor 7-Like 2 (TCF7L2) gene and the risk of type 2 diabetes in large cohorts of U.S. women and men. Diabetes. 2006;55:2645–8. [DOI] [PubMed] [Google Scholar]
- 13.Papadopoulou S, Edlund H. Attenuated Wnt signaling perturbs pancreatic growth but not pancreatic function. Diabetes. 2005;54:2844–51. [DOI] [PubMed] [Google Scholar]
- 14.Ross SE, Hemati N, Longo KA, Bennett CN, Lucas PC, Erickson RL, MacDougald OA. Inhibition of adipogenesis by Wnt signaling. Science. 2000;289:950–3. [DOI] [PubMed] [Google Scholar]
- 15.Lyssenko V, Lupi R, Marchetti P, Del Guerra S, Orho-Melander M, Almgren P, Sjogren M, Ling C, Eriksson KF, et al. Mechanisms by which common variants in the TCF7L2 gene increase risk of type 2 diabetes. J Clin Invest. 2007;117:2155–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cauchi S, Meyre D, Choquet H, Dina C, Born C, Marre M, Balkau B, Froguel P. TCF7L2 variation predicts hyperglycemia incidence in a French general population: the data from an epidemiological study on the Insulin Resistance Syndrome (DESIR) study. Diabetes. 2006;55:3189–92. [DOI] [PubMed] [Google Scholar]
- 17.Kimber CH, Doney A, Pearson E, McCarthy M, Hattersley A, Leese G, Morris A, Palmer C. TCF7L2 in the Go-DARTS study: evidence for a gene dose effect on both diabetes susceptibility and control of glucose levels. Diabetologia. 2007;50:1186–91. [DOI] [PubMed] [Google Scholar]
- 18.Melzer D, Murray A, Hurst AJ, Weedon MN, Bandinelli S, Corsi AM, Ferrucci L, Paolisso G, Guralnik JM, et al. Effects of the diabetes linked TCF7L2 polymorphism in a representative older population. BMC Med. 2006;4:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Florez JC, Jablonski KA, Bayley N, Pollin TI, de Bakker PIW, Shuldiner AR, Knowler WC, Nathan DM, Altshuler D. TCF7L2 polymorphisms and progression to diabetes in the diabetes prevention program. N Engl J Med. 2006;355:241–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huertas-Vazquez A, Plaisier C, Weissglas-Volkov D, Sinsheimer J, Canizales-Quinteros S, Cruz-Bautista I, Nikkola E, Herrera-Hernandez M, Davila-Cervantes A, et al. TCF7L2 is associated with high serum triacylglycerol and differentially expressed in adipose tissue in families with familial combined hyperlipidaemia. Diabetologia. 2008;51:62–9. [DOI] [PubMed] [Google Scholar]
- 21.Higgins M, Province M, Heiss G, Eckfeldt J, Ellison RC, Folsom AR, Rao DC, Sprafka JM, Williams R. NHLBI Family Heart Study: objectives and design. Am J Epidemiol. 1996;143:1219–28. [DOI] [PubMed] [Google Scholar]
- 22.Corella D, Arnett DK, Tsai MY, Kabagambe EK, Peacock JM, Hixson JE, Straka RJ, Province M, Lai C-Q, et al. The -256T>C polymorphism in the apolipoprotein A-II gene promoter is associated with body mass index and food intake in the Genetics of Lipid Lowering Drugs and Diet Network Study. Clin Chem. 2007;53:1144–52. [DOI] [PubMed] [Google Scholar]
- 23.Lai C-Q, Arnett DK, Corella D, Straka RJ, Tsai MY, Peacock JM, Adiconis X, Parnell LD, Hixson JE, et al. Fenofibrate effect on triglyceride and postprandial response of apolipoprotein a5 variants: the GOLDN Study. Arterioscler Thromb Vasc Biol. 2007;27:1417–25. [DOI] [PubMed] [Google Scholar]
- 24.Patsch JR, Karlin JB, Scott LW, Smith LC, Gotto AM. Inverse relationship between blood levels of high density lipoprotein subfraction 2 and magnitude of postprandial lipemia. Proc Natl Acad Sci USA. 1983. March 1983;80:1449–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.National Cancer Institute. Risk factor monitoring and methods 2007. Jun 25 [cited 2008 Feb 12]. Available from: http://riskfactor.cancer.gov/DHQ/.
- 26.Thompson FE, Subar AF, Brown CC, Smith AF, Sharbaugh CO, Jobe JB, Mittl B, Gibson JT, Ziegler RG. Cognitive research enhances accuracy of food frequency questionnaire reports: results of an experimental validation study. J Am Diet Assoc. 2002;102:212–25. [DOI] [PubMed] [Google Scholar]
- 27.Millen AE, Midthune D, Thompson FE, Kipnis V, Subar AF. The National Cancer Institute Diet History Questionnaire: validation of pyramid food servings. Am J Epidemiol. 2006;163:279–88. [DOI] [PubMed] [Google Scholar]
- 28.Otvos JD, Jeyarajah EJ, Bennett DW, Krauss RM. Development of a proton nuclear magnetic resonance spectroscopic method for determining plasma lipoprotein concentrations and subspecies distributions from a single, rapid measurement. Clin Chem. 1992;38:1632–8. [PubMed] [Google Scholar]
- 29.Jeyarajah EJ, Cromwell WC, Otvos JD. Lipoprotein particle analysis by nuclear magnetic resonance spectroscopy. Clin Lab Med. 2006;26:847–70. [DOI] [PubMed] [Google Scholar]
- 30.Tsai MY, Georgopoulos A, Otvos JD, Ordovas JM, Hanson NQ, Peacock JM, Arnett DK. Comparison of ultracentrifugation and nuclear magnetic resonance spectroscopy in the quantification of triglyceride-rich lipoproteins after an oral fat load. Clin Chem. 2004;50:1201–4. [DOI] [PubMed] [Google Scholar]
- 31.Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28:412–9. [DOI] [PubMed] [Google Scholar]
- 32.Livak KJ. Allelic discrimination using fluorogenic probes and the 5′ nuclease assay. Genet Anal. 1999;14:143–9. [DOI] [PubMed] [Google Scholar]
- 33.Grundy SM, Cleeman JI, Daniels SR, Donato KA, Eckel RH, Franklin BA, Gordon DJ, Krauss RM, Savage PJ, et al. Diagnosis and management of the metabolic syndrome: an American Heart Association/National Heart, Lung, and Blood Institute Scientific Statement. Circulation. 2005;112:2735–52. [DOI] [PubMed] [Google Scholar]
- 34.Kuzuya T, Nakagawa S, Satoh J, Kanazawa Y, Iwamoto Y, Kobayashi M, Nanjo K, Sasaki A, Seino Y, et al. Report of the Committee on the Classification and Diagnostic Criteria of Diabetes Mellitus. Diabetes Res Clin Pract. 2002;55:65–85. [DOI] [PubMed] [Google Scholar]
- 35.Singer JD. Using SAS PROC MIXED to fit multilevel models, hierarchical models, and individual growth models. J Educ Behav Stat. 1998;24:323–55. [Google Scholar]
- 36.Loos RJ, Franks PW, Francis RW, Barroso I, Gribble FM, Savage DB, Ong KK, O'Rahilly S, Wareham NJ. TCF7L2 polymorphisms modulate proinsulin levels and beta-cell function in a British Europid population. Diabetes. 2007;56:1943–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Saxena R, Gianniny L, Burtt NP, Lyssenko V, Giuducci C, Sjogren M, Florez JC, Almgren P, Isomaa B, et al. Common single nucleotide polymorphisms in TCF7L2 are reproducibly associated with type 2 diabetes and reduce the insulin response to glucose in nondiabetic individuals. Diabetes. 2006;55:2890–5. [DOI] [PubMed] [Google Scholar]
- 38.Munoz J, Lok KH, Gower BA, Fernandez JR, Hunter GR, Lara-Castro C, De Luca M, Garvey WT. Polymorphism in the transcription factor 7-like 2 (TCF7L2) gene is associated with reduced insulin secretion in nondiabetic women. Diabetes. 2006;55:3630–4. [DOI] [PubMed] [Google Scholar]
- 39.Wang J, Kuusisto J, Vänttinen M, Kuulasmaa T, Lindström J, Tuomilehto J, Uusitupa M, Laakso M. Variants of transcription factor 7-like 2 (TCF7L2) gene predict conversion to type 2 diabetes in the Finnish Diabetes Prevention Study and are associated with impaired glucose regulation and impaired insulin secretion. Diabetologia. 2007;50:1192–200. [DOI] [PubMed] [Google Scholar]
- 40.Festa A, Williams K, Hanley AJG, Otvos JD, Goff DC, Wagenknecht LE, Haffner SM. Nuclear magnetic resonance lipoprotein abnormalities in prediabetic subjects in the Insulin Resistance Atherosclerosis Study. Circulation. 2005;111:3465–72. [DOI] [PubMed] [Google Scholar]
- 41.Marzi C, Huth C, Kolz M, Grallert H, Meisinger C, Wichmann HE, Rathmann W, Herder C, Illig T. Variants of the transcription factor 7-like 2 gene (TCF7L2) are strongly associated with type 2 diabetes but not with the metabolic syndrome in the MONICA/KORA surveys. Horm Metab Res. 2007;39:46–52. [DOI] [PubMed] [Google Scholar]
- 42.Cauchi S, Meyre D, Dina C, Choquet H, Samson C, Gallina S, Balkau B, Charpentier G, Pattou F, et al. Transcription factor TCF7L2 genetic study in the French population: expression in human {beta}-cells and adipose tissue and strong association with type 2 diabetes. Diabetes. 2006;55:2903–8. [DOI] [PubMed] [Google Scholar]
- 43.Aslanidi G, Kroutov V, Philipsberg G, Lamb K, Campbell-Thompson M, Walter GA, Kurenov S, Ignacio Aguirre J, Keller P, et al. Ectopic expression of Wnt10b decreases adiposity and improves glucose homeostasis in obese rats. Am J Physiol Endocrinol Metab. 2007;293:E726–36. [DOI] [PubMed] [Google Scholar]
- 44.Mani A, Radhakrishnan J, Wang H, Mani A, Mani MA, Nelson-Williams C, Carew KS, Mane S, Najmabadi H, et al. LRP6 mutation in a family with early coronary disease and metabolic risk factors. Science. 2007;315:1278–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu J, Farmer SR. Regulating the balance between peroxisome proliferator-activated receptor {gamma} and {beta}-catenin signaling during adipogenesis: a glycogen synthase kinase 3{beta} phosphorylation-defective mutant of {beta}-catenin inhibits expression of a subset of adipogenic genes. J Biol Chem. 2004;279:45020–7. [DOI] [PubMed] [Google Scholar]
- 46.Moldes M, Zuo Y, Morrison RF, Silva D, Park BH, Liu J, Farmer SR. Peroxisome-proliferator-activated receptor gamma suppresses Wnt/beta-catenin signalling during adipogenesis. Biochem J. 2003;376:607–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kawai M, Mushiake S, Bessho K, Murakami M, Namba N, Kokubu C, Michigami T, Ozono K. Wnt/Lrp/[beta]-catenin signaling suppresses adipogenesis by inhibiting mutual activation of PPAR[gamma] and C/EBP. Biochem Biophys Res Commun. 2007;363:276–82. [DOI] [PubMed] [Google Scholar]
- 48.Bragt MC, Popeijus HE. Peroxisome proliferator-activated receptors and the metabolic syndrome. Physiol Behav. 2008;94:187–97. [DOI] [PubMed] [Google Scholar]
- 49.Lai C-Q, Corella D, Demissie S, Cupples LA, Adiconis X, Zhu Y, Parnell LD, Tucker KL, Ordovas JM. Dietary intake of n-6 fatty acids modulates effect of apolipoprotein A5 gene on plasma fasting triglycerides, remnant lipoprotein concentrations, and lipoprotein particle size: the Framingham Heart Study. Circulation. 2006;113:2062–70. [DOI] [PubMed] [Google Scholar]
- 50.Xiao C, Giacca A, Carpentier A, Lewis G. Differential effects of monounsaturated, polyunsaturated and saturated fat ingestion on glucose-stimulated insulin secretion, sensitivity and clearance in overweight and obese, non-diabetic humans. Diabetologia. 2006;49:1371–9. [DOI] [PubMed] [Google Scholar]
- 51.Rothman KJ. No adjustments are needed for multiple comparisons. Epidemiology. 1990;1:43–6. [PubMed] [Google Scholar]
- 52.Feise RJ. Do multiple outcome measures require p-value adjustment? BMC Med Res Methodol. 2002;2:8. [DOI] [PMC free article] [PubMed] [Google Scholar]