

Abstract
TNFAIP3 encodes the ubiquitin modifying enzyme, A20, a key regulator of inflammatory signaling pathways. We previously reported association between TNFAIP3 variants and systemic lupus erythematosus (SLE). In order to further localize the risk variant(s), we performed a meta-analysis using genetic data available from two Caucasian case/control datasets (1453 total cases, 3381 total controls) and 713 SLE trio families. The best result was found at rs5029939 (P = 1.67 × 10−14, OR = 2.09, 95% CI 1.68–2.60). We then imputed SNPs from the CEU Phase II HapMap using genotypes from 431 SLE cases and 2155 controls. Imputation identified eleven SNPs in addition to three observed SNPs, which together, defined a 109 kb SLE risk segment surrounding TNFAIP3. When evaluating whether the rs5029939 risk allele was associated with SLE clinical manifestations, we observed that heterozygous carriers of the TNFAIP3 risk allele at rs5029939 have a two-fold increased risk of developing renal or hematologic manifestations compared to homozygous non-risk subjects. In summary, our study strengthens the genetic evidence that variants in the region of TNFAIP3 influence risk for SLE, particularly in patients with renal and hematologic manifestations, and narrows the risk effect to a 109 kb DNA segment that spans the TNFAIP3 gene.
Keywords: systemic lupus erythematosus, TNFAIP3, imputation, meta-analysis
Introduction
Systemic lupus erythematosus (SLE) is an autoimmune disease characterized by dysregulated interferon responses and loss of tolerance to self-antigens. Deposition of immune complexes in tissues results in local and systemic inflammation, often progressing to organ dysfunction and failure. Recent genome-wide association studies (GWAS) in SLE subjects of European ancestry have identified several new and confirmed risk loci 1–4. Our group reported association of genetic variants in the region of TNFAIP3 with human SLE and identified two independent genetic risk effects 2. One effect, identified by rs6920220 is located approximately 185 kb upstream of TNFAIP3 and is also associated with risk for rheumatoid arthritis (RA) 2; 5–7. The second effect, spanning the TNFAIP3 locus, is comprised of a low frequency haplotype (~2% in European ancestry) marked by rs10499197, rs5029939, rs7749323 and the non-synonymous variant rs2230926 2; 8.
TNFAIP3 encodes A20, a zinc-finger protein required for efficient termination of the NF-κB signaling axis downstream of TNFR, TLR, IL1R and NOD2 9–13. A20 is a unique dual function ubiquitin editing enzyme that catalyzes the deubiquitylation of several NF-κB pathway proteins, including TRAF6, RIP1, RIP2 and IKKγ/NEMO 9; 11; 12; 14; 15 through an amino-terminal ovarian tumor domain. The carboxy terminal zinc finger domain of A20 functions as an E3 ubiquitin ligase catalyzing K48-linked ubiquitylation of substrate proteins, which targets them for proteosome degradation 12. The importance of A20 in attenuating NF-κB is evident in mice engineered to lack expression of A20, which die in the first 6 weeks of life due to uncontrolled systemic organ inflammation 11.
In order to further characterize and determine the magnitude of the association signals in the region of TNFAIP3, we performed a meta-analysis of existing genotype data from two independent case/control datasets and one trio family dataset. In addition, we tested for evidence of genetic association in silico by imputing genotypes from the Phase II HapMap over a 5 MB window spanning TNFAIP3, using our previously published GWAS dataset as the source of observed genotypes. We then tested whether the TNFAIP3 SLE risk haplotype defined by risk and non-risk alleles at rs5029939 were associated with specific SLE sub-phenotypes.
Results
Meta-analysis was performed with genotype data from four SNPs in a sample set comprised of 1453 independent SLE cases, 3381 independent controls and 713 independent trio families. The results of the meta-analysis demonstrated significant association approaching or exceeding criteria for genome-wide significance (P < 1 × 10−8) for all SNPs (Table 1). SNP rs5029939 located in the second intron of TNFAIP3 and originally identified in our GWAS, produced a convincing meta-analysis P-value of 1.67 × 10−14 and a combined odds ratio of 2.09 (95% CI: 1.68–2.60) in the case/control datasets (Table 1). In our previous study we reported an OR equal to 2.28 (95% CI: 1.71–3.06) for marker rs5029939 2. Note that while the meta-analysis OR at rs5029939 decreased to 2.09, the 95% confidence interval around this OR was reduced, indicating an improvement in the precision of the estimate, a primary goal of meta-analysis. In SLE cases of European ancestry, HLA-DR3 and HLA-DR2 alleles are the only risk alleles to consistently demonstrate odds ratios near 2.0 or higher 16. These results suggest that the genetic effect size marked by rs5029939 in the TNFAIP3 gene is similar to that of the HLA and larger than any of the recently identified SLE risk genes including IRF5 17, STAT4 18, BLK 3 and ITGAM 19. Given the relatively low frequency of the risk allele at rs5029939, this effect aligns with the common disease, rare variant hypothesis of complex genetic disease.
Table 1.
Study Specific and Meta-analysis Association Results for Four SNPs in the Region of TNFAIP3
| SNP | Associated Allele | GWAS 431 cases 2155 controls |
BE2 1022 cases 1226 controls |
CMH |
TRIO N=713 |
Fisher P-value | |
|---|---|---|---|---|---|---|---|
| OR (95% CI) | P-value | ||||||
|
| |||||||
| rs10499197 | G | 1.23E-05 | NA | 1.00E-04 | 2.65E-08 | ||
| case freq | 0.0603 | T:69 | |||||
| control freq | 0.0302 | U:31 | |||||
|
| |||||||
| rs5029939 | G | 7.85E-08 | 1.03E-05 | 2.09 (1.68–2.60) | 5.18E-12 | 8.86E-05 | 1.67E-14 |
| case freq | 0.0696 | 0.0580 | T:85 | ||||
| control freq | 0.0314 | 0.0307 | U:41 | ||||
|
| |||||||
| rs2230926 | G | NA | 1.42E-05 | 1.10E-03 | 2.96E-07 | ||
| case freq | 0.0575 | T:79 | |||||
| control freq | 0.0307 | U43 | |||||
|
| |||||||
| rs7749323 | A | 4.64E-06 | NA | 3.81E-05 | 4.15E-09 | ||
| case freq | 0.0615 | T:73 | |||||
| control freq | 0.0300 | U:31 | |||||
CMH = Cochran Mantel Haenszel analysis of case/control data
Fisher P-value - was obtained by combining P-values from Trio family dataset with study specific or combined CMH P-value using Fisher’s method
OR = Odds Ratio
CI = Confidence Intervals
T = Transmitted allele count
U = Untransmitted allele count
freq = frequency
Genotypes were imputed to determine the contribution of untyped variants to the genetic association in the region of TNFAIP3 and to further define the boundaries of the SLE risk haplotype. Imputation was performed over a 5 Mb (135–140 Mb) interval centered on TNFAIP3 from marker rs4896151 to marker rs1977772 on chromosome 6q using our previously published GWAS dataset as the source of observed genotypes and the Phase II HapMap as the source of imputed genotypes 20. In addition to TNFAIP3, this interval contains at least 20 genes, some with a possible role in the immune system such as interleukin 20 receptor alpha (IL20RA), interleukin 22 receptor alpha (IL22RA), interferon gamma receptor 1 (INFGR1) and mitogen-activated protein kinase kinase kinase 5 (MAP3K5). Also included was the region upstream of TNFAIP3 associated with risk for RA 6; 7 and the region downstream of TNFAIP3 near PERP, recently reported to be associated with SLE 8. Imputation expanded the number of SNPs in the 5 MB region from 390 observed SNPs to 3,670 total SNPs. Following exclusion of imputed SNPs based on quality control measures (information scores < 0.7 and/or ≤ 2 proxy SNPs used to impute any give SNP (NPRX)), 2497 SNPs remained in the final imputed dataset (Figure 1).
Figure 1.
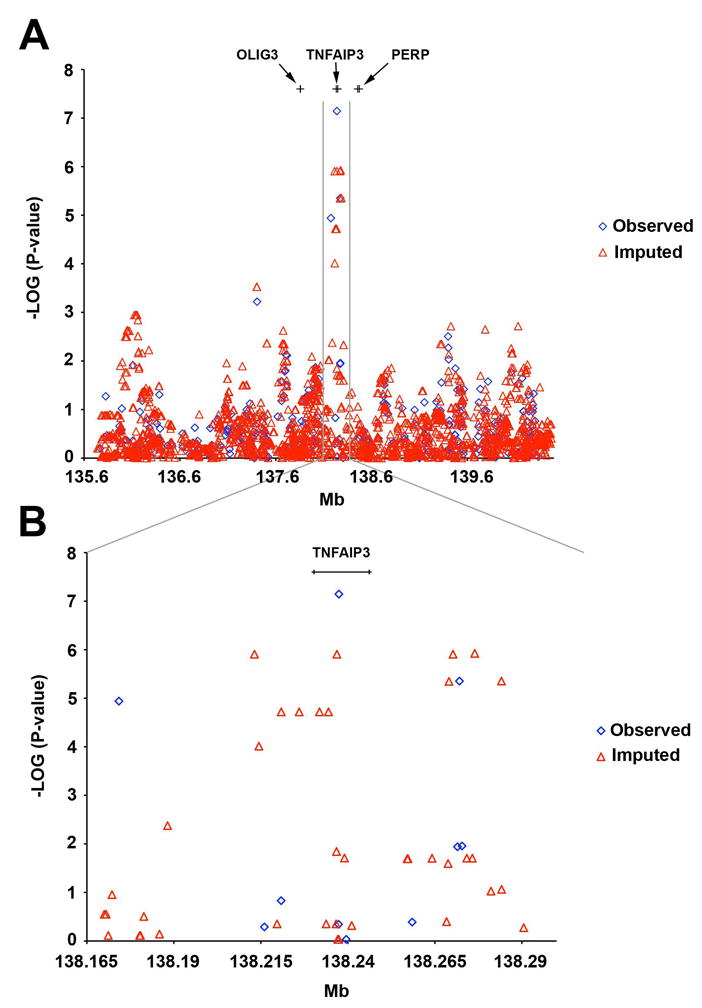
Results of Imputation Across a 5MB Region Centered on TNFAIP3. A. Results showing full 5MB imputation interval. Imputed SNPs are in red and observed SNPs in blue. Locations of genes flanking TNFAIP3 are indicated at the top. B. Expanded view of region surrounding TNFAIP3. Eleven imputed SNPs that demonstrated association with SLE (P-value < 1 × 10−4) are represented as red triangles. The observed SNPs, rs10499197, rs5029939, and rs7749323 that demonstrate significant association are represented as blue diamonds.
The strongest association signals were detected in the vicinity of TNFAIP3 (Figure 1) with both observed and imputed SNPs. No other region in the 5MB interval reached significance at P < 1.0 × 10−4, including variants in the region 185 kb upstream associated with RA and SLE or the region near PERP (Supplemental Table 1). In contrast, eleven imputed SNPs spanning the TNFAIP3 locus demonstrated association with SLE at P < 1.0 × 10−4 (Table 2). Imputation accuracy for all eleven SNPs was greater than 99%. The concordance rates between observed genotypes and imputed genotypes for the three observed SNPs (rs10499197, rs5029939, rs7749323) exceeded 99%, indicating robust imputation over this region. SNP rs5029939 was the most statistically significant variant among all observed and imputed SNPs (Table 2). The exon three missense SNP, rs2230926, was not imputed as it was not present in either the GWAS or HapMap datasets, however, rs5029939 is in strong LD (r2=0.99) and may indeed incorporate the effect at rs2230926 2.
Table 2.
Eleven imputed SNPs and three observed SNPs are associated with SLE in the region of TNFAIP3
| SNP | BP | NPRX | INFO | A1 | A2 | F_A | F_U | CHISQ | P | OR |
|---|---|---|---|---|---|---|---|---|---|---|
| rs10499197 | 138174209 | 3 | 1.01 | C | A | 0.0603 | 0.0302 | 19.25 | 1.15E-05 | 2.064 |
| rs9494883 | 138213159 | 4 | 1.00 | G | A | 0.0634 | 0.0298 | 23.52 | 1.24E-06 | 2.203 |
| rs9494885 | 138214441 | 4 | 0.74 | C | T | 0.1275 | 0.0837 | 15.19 | 9.72E-05 | 1.600 |
| rs11970411 | 138220854 | 5 | 1.02 | C | G | 0.1221 | 0.0774 | 18.28 | 1.91E-05 | 1.657 |
| rs9494886 | 138226023 | 5 | 1.02 | G | C | 0.1221 | 0.0774 | 18.28 | 1.91E-05 | 1.657 |
| rs3757173 | 138231847 | 5 | 1.02 | G | A | 0.1221 | 0.0774 | 18.28 | 1.91E-05 | 1.657 |
| rs719149 | 138234438 | 5 | 1.02 | A | G | 0.1221 | 0.0774 | 18.28 | 1.91E-05 | 1.657 |
| rs5029937 | 138236844 | 4 | 1.00 | T | G | 0.0634 | 0.0298 | 23.52 | 1.24E-06 | 2.203 |
| rs5029939 | 138237416 | 4 | 0.86 | C | G | 0.0696 | 0.0313 | 29.02 | 7.17E-08 | 2.314 |
| rs7752903 | 138269057 | 3 | 1.01 | G | T | 0.0615 | 0.0299 | 21.04 | 4.50E-06 | 2.122 |
| rs9494894 | 138270213 | 4 | 1.00 | C | T | 0.0634 | 0.0298 | 23.52 | 1.24E-06 | 2.203 |
| rs7749323 | 138272082 | 3 | 1.00 | T | C | 0.0615 | 0.0299 | 21.07 | 4.44E-06 | 2.123 |
| rs9494895 | 138276471 | 5 | 0.98 | T | C | 0.0612 | 0.0283 | 23.59 | 1.19E-06 | 2.241 |
| rs6932056 | 138284130 | 3 | 1.01 | C | T | 0.0615 | 0.0299 | 21.07 | 4.44E-06 | 2.123 |
Observed SNPs are shown in italicized font
BP = base pair position on chromosome 6, build 36
NPRX - number of proxy SNPs used to impute
INFO - score of accuracy of imputation
A1/A2 - allele 1 or 2
F_A/F_U - allele frequency in affected/unaffected
Imputation also defined the length of the associated TNFAIP3 risk haplotype. Before imputation, association with SNPs on the 3′ end extended as far as rs7749323 and following imputation, additional SNPs extended the risk haplotype approximately 12 kb downstream to marker rs6932056, resulting in risk segment approximately 109 kb in length.
The distribution of allele frequencies and odds ratios for the imputed SNPs was consistent with the presence of more than one haplotype. Therefore, we evaluated the haplotypic and LD relationships for the observed and imputed SNPs listed in Table 2. Three haplotypes with frequency greater than 1% were identified (Figure 2). Conditional logistic regression analysis implemented in PLINK 21 was used to determine if the haplotypes contributed independent genetic risk for SLE using haplotype 1 as the reference haplotype. The omnibus likelihood ratio test (LRT) yielded a P-value = 4.0 × 10−4, consistent with the fact that variants in the region of TNFAIP3 influence risk for SLE. The analysis demonstrated an independent effect for haplotype 3 (LRT P = 1.0 × 10−4) but not haplotype 2 (LRT P = 0.55) (Figure 2). Conditioning on haplotype 3 in comparison to the reference haplotype, resulted in no evidence of association (LRT P=0.42). In contrast, significant evidence of association was seen when the reference haplotype was conditioned on haplotype 2 (LRT P=9.7×10−5) (Figure 2). These results support the conclusion that genetic variation carried on haplotype 3 is responsible for the association with SLE. As was seen in the meta-analysis, SNP alleles carried exclusively on haplotype 3 produced OR ≥ 2.0.
Figure 2.
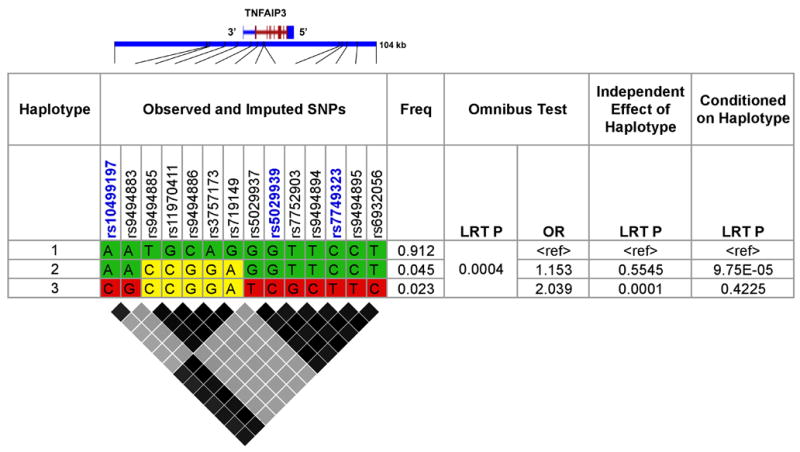
Conditional haplotype analyses for the imputed TNFAIP3 risk haplotype. Three haplotypes are shown with frequencies > 1%. Imputed SNPs are in black font and observed SNPs are in blue font. LD relationships (r2) are shown in the figure below the table with black diamonds corresponding to high LD (r2 = 0.75–1.0) and gray diamonds corresponding to low (r2 < 0.5). LRT = likelihood ratio test.
Clinical data were available for 1351 female SLE cases of European descent and was used to define SLE sub-phenotypes based on revised ACR criteria (malar rash, discoid rash, photosensitivity, oral ulcers, arthritis, serositis, nephritis, neurologic disorder, hematologic disorder, antinuclear antibody, and immunologic disorder) 22; 23 and presence of anti-Ro/SSA and anti-La/SSB autoantibodies (Table 3). Case subsets were compared to a group of 1172 female controls without a personal or family history of autoimmune disease. For comparison of anti-Ro/SSA and anti-La/SSB antibodies the control group consisted of 348 subjects that were negative for these autoantibodies by serologic testing. Association analysis was performed by comparing of the frequencies of the risk (C) and non-risk (G) genotypes at rs5029939, which tags the SLE risk haplotype. There were 144 SLE cases (Freq. = 0.057) and 71 controls (Freq. = 0.031) that carried the CG genotype. Frequencies for the CC genotype in cases and controls were low (CC Cases = 0.006, CC Controls = 0) precluding analysis of the CC genotype.
Table 3.
Association of alleles at rs5029939 with SLE sub-phenotypes compared to healthy controls.
| CG | GG | Attributable Risk | OR | ||||
|---|---|---|---|---|---|---|---|
|
|
|||||||
| Trait | Case:Cont | Case:Cont | CG GG | 95% CI | CG GG | 95% CI | P-value |
| SLE | 144:71 | 1198:1101 | 0.15 | 0.08–0.21 | 1.86 | 1.39–2.51 | 3.75E-05 |
| Anti-Ro/SSA | 22:25 | 173:347 | 0.14 | −0.01–0.27 | 1.77 | 0.97–3.22 | 0.065 |
| Anti-La/SSB | 9:25 | 61:348 | 0.16 | −0.02–0.31 | 2.05 | 0.91–4.62 | 0.082 |
| Nephritis | 49:71 | 330:1101 | 0.19 | 0.11–0.27 | 2.30 | 1.57–3.38 | 2.27E-05 |
| Malar Rash | 80:71 | 703:1101 | 0.14 | 0.06–0.21 | 1.77 | 1.26–2.46 | 9.0E-04 |
| Discoid Rash | 4:71 | 77:1101 | −0.04 | −0.22–0.14 | 0.81 | 0.29–2.27 | 0.68 |
| Photosensitivity | 89:71 | 748:1101 | 0.15 | 0.07–0.22 | 1.85 | 1.33–2.56 | 2.0E-04 |
| Arthritis | 112:71 | 961:1101 | 0.14 | 0.07–0.21 | 1.81 | 1.33–2.56 | 2.0E-04 |
| Oral Ulcers | 59:71 | 548:1101 | 0.13 | 0.04–0.21 | 1.46 | 0.90–2.36 | 0.12 |
| Serositis | 64:71 | 538:1101 | 0.15 | 0.06–0.23 | 1.85 | 1.30–2.63 | 7.0E-04 |
| Neurologic | 13:71 | 171:1101 | 0.04 | −0.10–0.17 | 1.18 | 0.64–2.18 | 0.60 |
| Hematologic | 88:71 | 661:1101 | 0.18 | 0.10–0.24 | 2.06 | 1.49–2.86 | 1.58E-05 |
| Immunologic | 97:71 | 809:1101 | 0.15 | 0.07–0.22 | 1.86 | 1.35–2.56 | 1.0E-0-4 |
| ANA | 148:14 | 1277:228 | 0.13 | 0.02–0.23 | 1.89 | 1.07–3.33 | 0.028 |
ANA = anti-nuclear antibodies
Cont = Control
Of the sub-phenotypes evaluated, nephritis and hematologic disorder demonstrated lower P-values and higher attributable risk and odds ratios compared to the SLE phenotype, even though only 28% and 56% of the cases, respectively, were used in the analyses of these phenotypes (Table 3). Note that the analysis of the SLE phenotype without the nephritis cases or the hematologic cases resulted in an increase in the P-value from 3.75×10−5 to 0.0012 and 0.01, respectively (Table 4). Excluding both the nephritis and hematologic sub-phenotypes from the SLE phenotype resulted in a non-significant association with the CG genotype of rs5029939 (P-value = 0.053). Taken together, these results suggest that SLE patients with the CG genotype at rs5029939 are over two fold more likely to develop lupus nephritis and/or hematologic manifestations compared to SLE patients with the GG genotype.
Table 4.
Conditional analysis of clinical traits
| CG | GG | ||||
|---|---|---|---|---|---|
|
|
|||||
| SLE Conditioned on: | Case:Cont | Case:Cont | OR | 95% C.I. | P-value |
| Nephritisa | 95:71 | 868:1101 | 1.70 | 1.23–2.34 | 0.0012 |
| Hematologyb | 56:71 | 537:1101 | 1.62 | 1.12–2.33 | 0.01 |
| Nephritis or Hematologyc | 40:71 | 416:1101 | 1.49 | 0.99–2.23 | 0.0525 |
Nephritis cases were excluded from analysis
Hematology cases were excluded from analsyis
Cases with either nephritis and/or hematology were excluded from analysis
We then performed an analysis of SLE cases only, stratified by SLE sub-phenotypes. This analysis failed to produce any statistically significant associations for any of the sub-phenotypes (Supplementary Table 2). This is likely due to reduced statistical power due to the smaller sample sizes that result when using only SLE cases. Considering the nephritis sub-phenotype for example, power analyses suggest that we would need approximately 560 cases (SLE with nephritis) to detect an effect size similar to the case-control results, whereas our data included 379 lupus nephritis subjects (Supplementary Table 2).
Next we evaluated if clusters of clinical sub-phenotypes were associated with the risk allele at rs5029939. To define the clusters we used a principle components approach which produced five clusters from 10 of the 11 ACR criteria evaluated, the first three of which explained 56.5% of the total variation. Anti-nuclear antibodies were present in 98% of the case subjects, which precluded clustering of this sub-phenotype. Overall, the sub-phenotypes within each of the five clusters (Cluster 1 - malar rash, photosensitivity, and oral ulcers; Cluster 2 - renal, immunologic and hematologic manifestations; Cluster 3 - arthritis and serositis; Cluster 4 - neurologic disorder; Cluster 5 - discoid rash) were moderately correlated (0.33 – 0.57), but no correlation was observed with variables outside their respective clusters (Supplementary Table 3). A component score was then estimated for each cluster and each case subject using the principle components derived from the clustering procedure, generating five new covariates for each case subject. Logistic regression analysis was performed using rs5029939 (omitting homozygous risk individuals) as the dependent variable and SLE and the cluster component score as independent variables (Table 5). Only the results for the first three clusters are reported as clusters 4 and 5 explained only 2.2% of the total variation and sub-phenotypes within these clusters were not associated with rs5029939. In line with our analysis using individual sub-phenotypes, cluster 2 (renal, hematologic and immunologic manifestations) demonstrated a better fit to the model when compared to cluster 1, cluster 3, or SLE (Table 5). Importantly, when SLE was adjusted for cluster 2, association with rs5029939 was insignificant (Wald P = 0.81), yet association with SLE remained when adjusting for cluster 1 (P = 0.04) or cluster 3 (P = 0.06). These results suggest the association between rs5029939 and subjects with renal, hematologic and immunologic manifestations is not due to confounding with SLE but rather represents a sub-phenotype specific genetic effect.
Table 5.
Logistic regression of rs5029939 with SLE and cluster component scores.
| Model | Wald P-value | LRT Model Fit |
|---|---|---|
| SLE | 0.0015 | 0.0016 |
| Cluster 1 | 0.0102 | 0.0124 |
| Cluster 2 | 0.0003 | 0.0002 |
| Cluster 3 | 0.0062 | 0.0066 |
| SLE + Cluster 1 | 0.0435 | 0.0067 |
| SLE + Cluster 2 | 0.8099 | 0.0016 |
| SLE + Cluster 3 | 0.0631 | 0.0061 |
Cluster 1 – malar rash, photosensitivity, oral ulcers
Cluster 2 – renal, hematologic, immunologic manifestations
Cluster 3 – arthritis, serositis
LRT – likelihood ratio test
Discussion
Genome-wide association scans in human SLE have been successful in identifying novel risk loci 1−4. Our group recently identified association between SLE and variants in the region of TNFAIP3, the gene encoding the ubiquitin modifying enzyme A20 2. Genetic association in the region of TNFAIP3 has also been described for other autoimmune diseases including RA and Crohn’s disease 5–7. For SLE and RA, TNFAIP3 is a genetically complex locus. The region 185 kb upstream of TNFAIP3, confers risk for both RA and SLE 2; 5–8. A nearby independent effect that confers protection for RA has been inconsistently observed in SLE 2; 5–8. Directly surrounding TNFAIP3 we previously reported an independent haplotype associated with SLE defined by a highly correlated (r2>0.98) set of SNPs (rs10499197, rs5029939, rs2230926 and rs7749323), an effect that has been replicated in an independent SLE cohort 2; 8. Whether this haplotype confers risk for RA is unknown. Finally, another association was reported in the region near PERP located 240 kb downstream of TNFAIP3 8; this effect awaits replication in either SLE or RA.
The meta-analysis and imputation results presented here further support the association between SLE and variants within and flanking the TNFAIP3 gene. Specifically, the evidence for association was strengthened for four SNPs through meta-analysis of 1453 SLE cases, 3381 controls and 713 independent SLE trio families. The strongest association was located at marker rs5029939 in intron 2 of TNFAIP3. Marker rs5029939 is a proxy for rs2230926, which results in a phenylalanine to cysteine substitution at position 127 of A20. Preliminary data suggest that the 127C allele may be less efficient in attenuating NF-κB signaling 8, however, additional work is necessary to determine if this effect is seen in cells that carry the specific genotypes. Most importantly, the meta-analysis improves our confidence in the estimate of the OR for the rs5029939 risk allele compared with our previous study. The OR for this marker and others in linkage disequilibrium with it is approximately 2.0, thus approaching the magnitude seen only in HLA-DR3 and DR2 alleles in SLE patients of European ancestry.
Our imputation results provide support for 11 new SNPs that together with the three observed SNPs from our GWAS form a 109 kb SLE risk haplotype. All the SNPs on this haplotype are highly correlated thus it is not possible with the current dataset to determine if the functional allele is among the fourteen identified SNPs or remains undiscovered. Preliminary bioinformatic analysis (not shown) does not support an obvious functional role for any of the observed or imputed SNPs with the exception of rs2230926 described above.
Apart from the association at rs2230926, which our data supports through proxy SNP rs5029939, our imputation fails to support the other associations with SLE described by Musone, et al. 8. In the region 185 kb upstream of TNFAIP3, Musone et al. reported association with two protective variants, rs13192841 and rs12527282 8. These SNPs are proxies for rs10499194, which was associated with a protective effect in RA 7; an effect that was absent in our previously published SLE study 2. Similarly, the results from our imputation failed to reveal evidence for association at any of these markers (Supplemental Table 1). Therefore, we believe the presence of a protective association 185 kb upstream of TNFAIP3 is still in question for SLE. The previously reported association in the region of PERP marked by rs6922466 was also not significant in our imputation analysis (Supplemental Table 1). We acknowledge that our imputed dataset is likely underpowered to detect an association that was not detected in our observed GWAS dataset. In addition, locus and/or genetic heterogeneity in these regions may lead to association signals that are not reproducibly observed in independent SLE sample collections. Genotyping these and additional variants in larger SLE cohorts will be necessary to further characterize these associations.
Our analysis of SLE clinical sub-phenotypes shows that subjects heterozygous for the TNFAIP3 risk (CG) genotype at rs5029939 were over two times more likely to experience lupus nephritis (OR = 2.3) and/or hematologic manifestations (OR = 2.06) than GG homozygotes. This observation, combined with the lack of association when the SLE cases with nephritis or hematologic manifestations were removed from the analysis, suggests that the SLE risk haplotype at TNFAIP3 influences the development of these SLE sub-phenotypes. To more precisely determine if the TNFAIP3 risk haplotype directly influences risk for developing nephritis and SLE associated hematologic disorders, an analysis of a larger number of SLE case subjects stratified by presence or absence of nephritis and hematologic manifestations would be needed. The current case-only data set was underpowered for this type of analysis and did not produce any statistically significant findings (Supplemental Table 2). We estimate that to validate an association between lupus nephritis and the CG genotype at rs5029939 that produces an odds ratio of 2.3 as reported here using a case only approach would require approximately 560 SLE cases.
In summary, genetic variation in the region of TNFAIP3 has been shown to influence risk for human SLE. Our results support a potent genetic effect (OR≥2) located on a ~109 kb DNA segment in the region of TNFAIP3. Lupus nephritis and hematologic disorders are among the most severe manifestations observed in the clinical management of SLE patients. Our observation that the TNFAIP3 risk haplotype may influence the development of these complications suggests that TNFAIP3 plays an important role in SLE pathogenesis. Further characterization of the role of TNFAIP3 in SLE will be aided by the identification of the precise functional variant(s) responsible for the association with human SLE.
Methods
Datasets
For the meta-analysis in the region of TNFAIP3, we used genotype data from subjects of self-described European ancestry from our GWAS study of 431 SLE cases, 2155 controls and 740 trio families 2 and an independent case-control dataset, referred to as BE2. The BE2 dataset was comprised of 1313 SLE cases and 1226 controls selected from the Lupus Family Registry and Repository (LFRR) and the University of Minnesota SLE collection 24. All data sets were evaluated for subjects genotyped in more than one study. We removed 291 subjects from the BE2 set resulting in 1453 independent SLE cases and 3381 independent controls for meta-analysis.
Genotyping
Genotyping methods for the GWAS and Trio sample sets have been described previously 2. Genotyping in the BE2 study were performed on the BeadXpress Platform (Illumina) using the GoldenGate Chemistry at OMRF. SNPs were discarded if they failed to pass any of the following quality control metrics: a minimum Gencall score of 0.4, 10% Gencall score >0.8, call rate > 90%, and Hardy-Weinberg proportions greater than 0.01. In addition, we manually evaluated each SNP to ensure that the cluster characteristics were robust.
Meta-analysis and Imputation Methods
SNPs were chosen for inclusion in the meta-analysis if they were genotyped in a minimum of two datasets and were significantly associated with SLE in at least one dataset. PLINK was used to merge the genotype data from our GWAS and BE2 studies. Meta-analysis statistics were then generated using the Cochran Mantel-Haenszel (CMH) method implemented in SAS v. 9.1 (SAS Inst., Inc., Cary, N.C.). The four SNPs genotyped in the trio families were combined with the CMH derived case/control P-values using Fisher’s method 25.
Imputation was performed by merging the GWAS genotype data from the 5MB interval flanking TNFAIP3 with HapMap Phase II data from the same region using PLINK 21. This process generated a list of SNPs for which differences in strand orientation prohibited further merging of the data. The strand orientation of these SNPs was “flipped” in the HapMap genotype file to match the strand orientation for the GWAS data file. SNPs with A/T or G/C alleles cannot be detected by PLINK and were strand corrected manually. Once the merged dataset was assembled, we imputed the genotype data using the “proxy_impute” PLINK command. As a quality control measure, SNPs with an information score < 0.7 and/or NPRX ≤ 2 (n = 1173 SNPs) were removed, resulting in 2497 SNPs imputed SNPs. As an independent test for the quality of the imputation we also used the IMPUTE package to generate an imputed dataset with similar results (data not shown) 26.
SLE Sub-Phenotype Analysis
SLE sub-phenotypes included the 11 criteria used for classification from the American College of Rheumatology, as revised 22; 23, and the presence of anti-Ro/SSA and anti-La/SSB autoantibodies. For the 11 ACR criteria, cases were given a score of 0, 1, 2, or 3 based on the degree of confidence that a particular manifestation was present via review of the medical records. Cases denoted with 3 were considered positive for the manifestation, whereas individuals given a 0 were considered negative. Cases receiving a 1 or 2 were given a missing value for the condition. The presence of autoantibodies (anti-Ro, anti-La, and anti-nuclear antibodies) were evaluated at the CLIA approved OMRF Clinical Immunology Laboratory, Oklahoma City, OK, USA. Controls were assumed to be normal, healthy individuals and did not have a family history of SLE. Association of rs5029939 genotypes with clinical sub-phenotypes was assessed using SAS to model a generalized linear model assuming a binomial distribution for the dependent variable and using the logit and probit link functions to estimate odds ratio and attributable risk, respectively. Probability values were not corrected for multiple testing.
SLE criteria were grouped using the VARCLUS procedure in SAS, an oblique principle component analysis procedure that divides a group of variables into a set of distinct clusters. Within each cluster the VARCLUS procedure attempts to maximize the variation in the data accounted for by the fewest principle components, therefore grouping variables within a cluster that are correlated and separate variables into distinct clusters that are uncorrelated. Individuals included in the VARCLUS cluster analysis were all SLE cases. Cases were scored on a scale of 0–3 as described above and a component score for each cluster was estimated using the scoring coefficient from the cluster analysis obtained using the SCORE procedure in SAS. Therefore five new variables were created for each observation, with each new variable representing a cluster. Regression analysis was performed using the LOGISTIC procedure in SAS, specifying the rs5029939 genotype (omitting homozygous risk individuals due to low sample size) as the dependent variable and SLE and Cluster component score as independent variables.
Supplementary Material
Acknowledgments
We thank Joshua Ojwang, Joshua Cavett, Billy Herring and Adam Adler for their technical assistance. The authors would like to thank all of the patients and family members who participated in this study, as well as the many referring physicians. Portions of the samples used in this study were obtained from the Lupus Family Registry and Repository (AR62277) (see http://lupus.omrf.org).
This study makes use of data generated by the Wellcome Trust Case-Control Consortium. A full list of the investigators who contributed to the generation of the data is available from www.wtccc.org.uk. Funding for the project was provided by the Wellcome Trust under award 076113 (see Ref.
Control subjects from the National Institute of Mental Health Schizophrenia Genetics Initiative (NIMH-GI), data and biomaterials are being collected by the “Molecular Genetics of Schizophrenia II” (MGS-2) collaboration. The investigators and co-investigators are: ENH/Northwestern University, Evanston, IL, MH059571, Pablo V. Gejman, M.D. (Collaboration Coordinator; PI), Alan R. Sanders, M.D.; Emory University School of Medicine, Atlanta, GA,MH59587, Farooq Amin, M.D. (PI); Louisiana State University Health Sciences Center; New Orleans, Louisiana, MH067257, Nancy Buccola APRN, BC, MSN (PI); University of California-Irvine, Irvine, CA,MH60870, William Byerley, M.D. (PI); Washington University, St. Louis, MO,U01, MH060879, C. Robert Cloninger, M.D. (PI); University of Iowa, Iowa, IA,MH59566, Raymond Crowe, M.D. (PI), Donald Black, M.D.; University of Colorado, Denver, CO, MH059565, Robert Freedman, M.D. (PI); University of Pennsylvania, Philadelphia, PA, MH061675, Douglas Levinson M.D. (PI); University of Queensland, Queensland, Australia, MH059588, Bryan Mowry, M.D. (PI); Mt. Sinai School of Medicine, New York, NY,MH59586, Jeremy Silverman, Ph.D. (PI). The samples were collected by V L Nimgaonkar’s group at the University of Pittsburgh, as part of a multi-institutional collaborative research project with J Smoller, MD DSc and P Sklar, MD PhD (Massachusetts General Hospital) (grant MH 63420).
Grant Support: This work was supported through grants from the United States National Institutes of Health (AI063274 (P.M.G.), AR052125 (P.M.G.), AR043247 (K.L.M.), AI024717 (J.B.H.), AR062277 (J.B.H.), AR042460 (J.B.H.), AR049084 (J.B.H.)), the Mary Kirkland Scholarship (J.B.H.), the Alliance for Lupus Research (J.B.H.), the U.S. Department of Veterans Affairs (J.B.H.), Lupus Foundation of Minnesota (P.M.G., K.L.M.), National Arthritis Foundation (K.L.M.) and Arthritis Foundation, Oklahoma Chapter (P.M.G.).
References
- 1.Harley JB, Alarcon-Riquelme ME, Criswell LA, Jacob CO, Kimberly RP, Moser KL, et al. Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat Genet. 2008;40(2):204–210. doi: 10.1038/ng.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Graham RR, Cotsapas C, Davies L, Hackett R, Lessard CJ, Leon JM, et al. Genetic variants near TNFAIP3 on 6q23 are associated with systemic lupus erythematosus. Nat Genet. 2008 doi: 10.1038/ng.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hom G, Graham RR, Modrek B, Taylor KE, Ortmann W, Garnier S, et al. Association of systemic lupus erythematosus with C8orf13-BLK and ITGAM-ITGAX. N Engl J Med. 2008;358(9):900–909. doi: 10.1056/NEJMoa0707865. [DOI] [PubMed] [Google Scholar]
- 4.Kozyrev SV, Abelson AK, Wojcik J, Zaghlool A, Linga Reddy MV, Sanchez E, et al. Functional variants in the B-cell gene BANK1 are associated with systemic lupus erythematosus. Nat Genet. 2008;40(2):211–216. doi: 10.1038/ng.79. [DOI] [PubMed] [Google Scholar]
- 5.Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447(7145):661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thomson W, Barton A, Ke X, Eyre S, Hinks A, Bowes J, et al. Rheumatoid arthritis association at 6q23. Nat Genet. 2007;39(12):1431–1433. doi: 10.1038/ng.2007.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Plenge RM, Cotsapas C, Davies L, Price AL, de Bakker PI, Maller J, et al. Two independent alleles at 6q23 associated with risk of rheumatoid arthritis. Nat Genet. 2007;39(12):1477–1482. doi: 10.1038/ng.2007.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Musone SL, Taylor KE, Lu TT, Nititham J, Ferreira RC, Ortmann W, et al. Multiple polymorphisms in the TNFAIP3 region are independently associated with systemic lupus erythematosus. Nat Genet. 2008 doi: 10.1038/ng.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hitotsumatsu O, Ahmad RC, Tavares R, Wang M, Philpott D, Turer EE, et al. The ubiquitin-editing enzyme A20 restricts nucleotide-binding oligomerization domain containing 2-triggered signals. Immunity. 2008;28(3):381–390. doi: 10.1016/j.immuni.2008.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee EG, Boone DL, Chai S, Libby SL, Chien M, Lodolce JP, et al. Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science. 2000;289(5488):2350–2354. doi: 10.1126/science.289.5488.2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boone DL, Turer EE, Lee EG, Ahmad RC, Wheeler MT, Tsui C, et al. The ubiquitin-modifying enzyme A20 is required for termination of Toll-like receptor responses. Nat Immunol. 2004;5(10):1052–1060. doi: 10.1038/ni1110. [DOI] [PubMed] [Google Scholar]
- 12.Wertz IE, O’Rourke KM, Zhou H, Eby M, Aravind L, Seshagiri S, et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature. 2004;430(7000):694–699. doi: 10.1038/nature02794. [DOI] [PubMed] [Google Scholar]
- 13.Jaattela M, Mouritzen H, Elling F, Bastholm L. A20 zinc finger protein inhibits TNF and IL-1 signaling. J Immunol. 1996;156(3):1166–1173. [PubMed] [Google Scholar]
- 14.Evans PC, Ovaa H, Hamon M, Kilshaw PJ, Hamm S, Bauer S, et al. Zinc-finger protein A20, a regulator of inflammation and cell survival, has de-ubiquitinating activity. Biochem J. 2004;378(Pt 3):727–734. doi: 10.1042/BJ20031377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mauro C, Pacifico F, Lavorgna A, Mellone S, Iannetti A, Acquaviva R, et al. ABIN-1 binds to NEMO/IKKgamma and co-operates with A20 in inhibiting NF-kappaB. J Biol Chem. 2006;281(27):18482–18488. doi: 10.1074/jbc.M601502200. [DOI] [PubMed] [Google Scholar]
- 16.Fernando MM, Stevens CR, Walsh EC, De Jager PL, Goyette P, Plenge RM, et al. Defining the role of the MHC in autoimmunity: a review and pooled analysis. PLoS Genet. 2008;4(4):e1000024. doi: 10.1371/journal.pgen.1000024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Graham RR, Kozyrev SV, Baechler EC, Reddy MV, Plenge RM, Bauer JW, et al. A common haplotype of interferon regulatory factor 5 (IRF5) regulates splicing and expression and is associated with increased risk of systemic lupus erythematosus. Nat Genet. 2006;38(5):550–555. doi: 10.1038/ng1782. [DOI] [PubMed] [Google Scholar]
- 18.Remmers EF, Plenge RM, Lee AT, Graham RR, Hom G, Behrens TW, et al. STAT4 and the risk of rheumatoid arthritis and systemic lupus erythematosus. N Engl J Med. 2007;357(10):977–986. doi: 10.1056/NEJMoa073003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nath SK, Han S, Kim-Howard X, Kelly JA, Viswanathan P, Gilkeson GS, et al. A nonsynonymous functional variant in integrin-alpha(M) (encoded by ITGAM) is associated with systemic lupus erythematosus. Nat Genet. 2008;40(2):152–154. doi: 10.1038/ng.71. [DOI] [PubMed] [Google Scholar]
- 20.de Bakker PI, McVean G, Sabeti PC, Miretti MM, Green T, Marchini J, et al. A high-resolution HLA and SNP haplotype map for disease association studies in the extended human MHC. Nat Genet. 2006;38(10):1166–1172. doi: 10.1038/ng1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. American journal of human genetics. 2007;81(3):559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997;40 (9):1725. doi: 10.1002/art.1780400928. [DOI] [PubMed] [Google Scholar]
- 23.Tan EM, Cohen AS, Fries JF, Masi AT, McShane DJ, Rothfield NF, et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1982;25(11):1271–1277. doi: 10.1002/art.1780251101. [DOI] [PubMed] [Google Scholar]
- 24.Gaffney PM, Langefeld CD, Graham RR, Ortmann WA, Williams AH, Rodine PR, et al. Fine-mapping chromosome 20 in 230 systemic lupus erythematosus sib pair and multiplex families: evidence for genetic epistasis with chromosome 16q12. American journal of human genetics. 2006;78(5):747–758. doi: 10.1086/503686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fisher RA. Statistical Methods for Research Workers. 13. Oliver and Lloyd, Ltd; London: 1925. [Google Scholar]
- 26.Marchini J, Howie B, Myers S, McVean G, Donnelly P. A new multipoint method for genome-wide association studies by imputation of genotypes. Nat Genet. 2007;39(7):906–913. doi: 10.1038/ng2088. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.