

Abstract
Cancer cells can overcome the ability of polyamine biosynthesis inhibitors from completely depleting their internal polyamines by the importation polyamines from external sources. We have developed a group of lipophilic polyamine analogs that potently inhibit the cellular polyamine uptake system and greatly increase the effectiveness of polyamine depletion when used in combination with DFMO, a well-studied polyamine biosynthesis inhibitor. By the attachment of an length-optimized C16 lipophilic substituent to the epsilon-nitrogen atom of our earlier lead compound, d-Lys-Spm (5), we have produced an analog, d-Lys(C16acyl)-Spm (11) with several orders of magnitude more potent cell growth inhibition on a variety of cultured cancer cell types including breast (MDA-MB-231), prostate (PC-3), melanoma (A375) and ovarian (SK-OV-3), among others. We discuss these results in the context of a possible membrane-catalyzed interaction with the extracellular polyamine transport apparatus. The resulting novel two-drug combination therapy targeting cellular polyamine metabolism has shown exceptional efficacy against cutaneous squamous cell carcinomas (SCC) in a transgenic ornithine decarboxylase (ODC) mouse model of skin cancer. A majority (88%) of large, aggressive SCCs exhibited complete or near-complete remission to this combination therapy, while responses to each agent alone were poor. The availability of a potent polyamine transport inhibitor allows, for the first time, for a real test of the hypothesis that starving cells of polyamines will lead to objective clinical response.
Keywords: Polyamine depletion, polyamine transport inhibitor, putrescine, spermidine, spermine, polyamine uptake, cytostatic, MDA-MB-231 breast, A375 melanoma, PC-3 prostate, SK-OV-3 ovarian, cell growth inhibitor, transporter, molecular targeting, Schwyzer effect, membrane-catalyzed, squamous cell carcinoma
Introduction
Humanity’s ability to use chemotherapeutic agents to interrupt cellular metabolic processes constitutes a significant achievement and has supported many advances in medical treatment over the last half century. As one of the first rationally designed chemotherapeutics, α-difluoromethylornithine (DFMO) once held great promise in the fight against cancer. Despite early results achieved against cancer cell lines grown in culture, the use of this mechanism-based inhibitor of the first step in the biosynthesis of the polyamines failed to translate to success in the clinic. Extensive research now points to the fact that proliferating cells treated with DFMO can overcome this metabolic blockage by importing their required polyamines from extracellular sources. By compensating for the loss of one avenue for obtaining polyamines, the cell utilizes an alternative biochemical mechanism to obtain the molecules necessary for survival and continued growth.
The biological association between increased polyamine concentration and tumor growth is well established.1;2 Numerous multidisciplinary studies have shown that intracellular concentrations of polyamines are highly regulated at many steps in their biosynthesis, catabolism and transport (Figure 1). The fact that the cell contains such a complex system for the tight control of the levels of these molecules indicates that specific concentrations are required depending on the dynamics of cell growth, differentiation and cycling. Ornithine decarboxylase (ODC), the rate-limiting enzyme in polyamine biosynthesis, catalyzes the conversion of ornithine to putrescine 1; which is then converted to the tri-and tetra-amines spermidine 2 and spermine 3. An increase in the activity of ODC has been associated with tumor growth.3–5 Inhibition of polyamine biosynthesis in cells in culture by α-difluoromethylornithine (DFMO), a well-studied mechanism-based inhibitor of ODC, causes a substantial depletion of intracellular putrescine and spermidine resulting in cell growth inhibition. Upon supplementing the culture media with exogenous polyamines, this depletion causes transport activity to rise several-fold,6;7 allowing the cells to return to their original hyperproliferative rate of growth.
Figure 1.

Cellular polyamine depletion therapy: Synthesis, recycling and transport must be considered.
Cutaneous squamous cell carcinoma (SCC) is an epidermoid carcinoma of the skin, composing 20% of dermatological malignancies.8 Together with basal cell carcinoma, it is classified as a nonmelanoma skin cancer, which is the most common type of cancer in the Caucasian population. Incidence has reached epidemic proportions with 400,000 cases in the U.S. in 1980, 600,000 cases in 1990 and presently over 1 million new cases diagnosed annually.9 Although most cases are easily cured if detected early, if the tumor is allowed to progress and metastasize then treatment becomes much more complicated and less successful.10
We have extended our earlier discovery of amino acid-spermine conjugates11 by attaching lipophilic substituents to the ε-amino group of the lysine portion of our earlier lead compound d-Lys-Spm 5. These agents were characterized by their ability to inhibit cell growth in combination with DFMO even in the presence of extracellular spermidine (EC50 values). These analogs did not have pronounced cytotoxic effects on cells when used alone (IC50 value). Their ability to block uptake of radiolabeled spermidine (Ki values) was also measured and determined to be in the nanomolar range. Measurement of these analogs’ inability to rescue cells from the growth inhibitory effects of DFMO in the absence of extracellular polyamines showed that these analogs do not supply the cell with their polyamine requirements. Under these culture conditions, depletion of the intracellular levels of polyamines was demonstrated. This paper reports the optimization of lipophilic polyamine analogs produced by N-acylation or N-alkylation of the ε-amine group of the lysine portion of the Lys-spm conjugates. The dramatic improvement in the potency of these agents was demonstrated on multiple cell lines and translated to a murine model of SCC. Oral delivery of DFMO and an optimized polyamine transport inhibitor resulted in tumor growth inhibition demonstrating animal proof-of-concept for polyamine depletion therapy. Insights into these results are discussed in the context of prior examples of greatly improved potency of molecules directed towards membrane-associated targets, as described by use of a membrane-catalyzed binding mechanism.
Results
To optimize our previously described polyamine transport inhibitor (PTI), d-Lys-Spm (5), we functionalized the ε-position of the lysine moiety in order to overcome anticipated difficulties with this class of molecule’s oral uptake (Fig. 2). Upon addition of a C16-acyl group to the l-Lys-spm analog 4, we observed a dramatic increase in the resulting molecule’s (10) ability to inhibit cell growth in conjugation with DFMO when tested against several types of cancer cell lines in culture (Table 1). The cellular assay included 0.5 µM spermidine added to the media in order to mimic the conditions anticipated in vivo where an excess of extracellular polyamines are expected. By performing these cell culture experiments for a six-day time period we theorize that complete depletion of cellular polyamine levels occurs, allowing for the interruption of some critical cellular function; shorter culture lengths showed reduced levels of growth inhibition. Compound 10 showed greater than 100-fold improvement in its ability to inhibit cell growth when compared to its un-substituted stereo-partner 4 (in MDA-MB-231 cells, EC50 values of 0.06 µM vs. 7.0 µM for 10 and 4, respectively). Representative growth inhibition curves for combination therapy with unacylated d-Lys-spm analog 5 and the C16 acylated L-Lys-spm analog 10 in A375 human melanoma cells are shown in Fig. 3. The inhibition of cell proliferation is particularly significant since it occurred even when exogenous spermidine (0.5 µM) was present in the culture media. This improvement was matched by the d-Lys-Spm stereo-pair 11 and 5 where the C16-acylated version showed an EC50 value of 0.076 µM (compared to an EC50 value of 2.7 µM for 5 using MDA-MB-231 cells, respectively). As demonstrated by these data, we conclude that there is no significant difference between the activities of the l- or d-stereoisomers. This lack of difference was repeated when the ability of these analogs to inhibit the uptake of radiolabelled spermidine into MB-MDA-231 cells (Ki values for the L-/D- stereopairs 4/5 were 32 vs. 28 nM and 10/11 were 7.5 vs. 10.5 nM).
Figure 2.

Design of analogs with higher lipophilicity.
Table 1.
6-Day EC50 values for ε-acyl-substituted lipophilic lysine-spermine conjugates.a
 | |||||||
|---|---|---|---|---|---|---|---|
| EC50 with DFMOb (µM) | |||||||
| Compound | R Group | X | Stereo-> chemistry |
Breast (MDA-231) |
Prostate (PC-3) |
Melanoma (A375) |
Ovarian (SK-OV-3) |
| 4 | - | - | L | 7 | 4 | 1.5 | 0.51 |
| 5 | - | - | D | 2.7 | 3 | 1.8 | 2 |
| 6 | -CH(CH3)2 | C | D | 4.7 | 6.4 | 2.6 | 20.5 |
| 7 | -C(CH3)3 | C | D | 11.4 | 1.9 | 6.3 | 13.6 |
| 8 | -CH2(CH2)5CH3 | C | L | 1.1 | 1.1 | 2.0 | 30 |
| 9 | -CH2(CH2)11CH3 | C | L | 0.21 | 0.19 | 0.025 | 4.6 |
| 10 | -CH2(CH2)13CH3 | C | L | 0.06 | 0.05 | 0.02 | 0.2 |
| 11 | -CH2(CH2)13CH3 | C | D | 0.076 | 0.12 | 0.031 | 0.17 |
| 12 | -CH2(CH2)14CH3 | S | L | 0.15 | 0.044 | 0.012 | 0.09 |
| 13 | -CH2(CH2)14CH3 | C | L | 0.025 | 0.022 | 0.01 | 0.13 |
| 14 | -CH2(CH2)15CH3 | C | L | 0.013 | 0.012 | 0.002 | 0.024 |
| 15 | -CH2(CH2)15CH3 | C | D | 0.028 | 0.091 | 0.016 | 0.20 |
| 16 | -CH2(CH2)17CH3 | C | L | 0.010 | 0.0095 | 0.005 | 0.052 |
| 17 |  |
C | D | 17.8 | 39.3 | 18.3 | 65 |
| 18 | 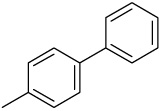 |
C | D | 0.77 | 0.63 | 0.37 | 2.1 |
| 19 |  |
C | L | 0.029 | 0.022 | 0.01 | 0.076 |
| 20 |  |
C | L | 0.039 | 0.056 | 0.014 | 0.13 |
Data obtained following 6 day incubations in the presence of 5.0 mM DFMO, 0.5 µM spermidine and 1.0 mM aminoguanidine.
All EC50 values are the average of at least two independent determinations and were calculated from growth inhibition curves generated from the mean of three determinations per concentration (eight drug concentrations), with the variation between individual values being <15%.
Figure 3.

Comparison of growth-inhibitory activities against A375 cells between unsubstituted (5) and lipophilic Lys-spm (10) conjugates in combination with DFMO.a
aA375 cells cultured for 6 days in combination with 5.0 mM DFMO, 0.5 µM spermidine and 1.0 mM aminoguanidine together with the amount of Lys-spm conjugates shown.
These observations were extended by the synthesis and biological testing of a series of ε-acyl and alkyl substituted Lys-spm analogs. As demonstrated in Table 1 and Table 2, the analogs with long chain lipophilic substituents consistently inhibited cell growth in conjunction with DFMO in the low nanomolar concentration range. With an EC50 value of 10 nM in MDA-MB-231 breast cancer cells, the C20-acyl analog 16 demonstrated the best potency of the acylated analogs. Analog 27, the C16-alkylated version, showed the best activity in the alkylated series (EC50 value of 19 nM in MDA-MB-231 cells). Throughout these series, it appears that incremental improvements in potency are obtained upon addition of carbon atoms to the ε-nitrogen position of lysine in the conjugates. This trend provides options to overcome unforeseen toxicities in the longer chain analogs since moderately lengthened analogs such as 8, with a C8-acyl chain, still retain substantial activity. No rounded or floating cells were observed with any of the Lys-spm conjugates described here. This supports the lack of a surfactant-like, cytotoxic effect of the compounds and might further suggest that the therapy may be working through a cytostatic mechanism.
Table 2.
6-Day EC50 values for ε-alkyl-substituted lipophilic lysine-spermine conjugates.a
 | |||||||
|---|---|---|---|---|---|---|---|
| EC50 with DFMOb (µM) | |||||||
| Compound | R Group | Stereo- chemistry |
Breast (MDA-231) |
Prostate (PC-3) |
Melanoma (A375) |
Ovarian (SK-OV-3) |
|
| 4 | - | L | 7 | 4 | 1.5 | 0.15 | |
| 5 | - | D | 2.7 | 3 | 1.8 | 2 | |
| 21 | -CH2CH(CH3)2 | D | 1.7 | 1.6 | 0.67 | 3.3 | |
| 22 | -CH2CH2C(CH3)3 | L | 1.7 | 0.71 | 0.74 | 0.94 | |
| 23 | -CH2CH2C(CH3)3 | D | 1 | 0.7 | 1.6 | 2.2 | |
| 24 | -CH2(CH2)4CH3 | D | 0.89 | 0.56 | 0.53 | 1.7 | |
| 25 | -CH2(CH2)5CH3 | L | 0.51 | 0.29 | 0.54 | 0.24 | |
| 26 | -CH2(CH2)5CH3 | D | 0.27 | 0.18 | 0.17 | 0.65 | |
| 27 | -CH2(CH2)14CH3 | L | 0.019 | 0.046 | 0.01 | 0.081 | |
| 28 |  |
D | 0.42 | 0.78 | 0.38 | 2.1 | |
| 29 | 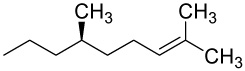 |
D | 6.4 | 14 | 4.9 | 18 | |
| 30 |  |
L | 0.073 | 0.075 | 0.056 | 0.14 | |
| 31 |  |
D | 1.5 | 0.77 | - | 1.9 | |
Data obtained following 6 day incubations in the presence of 5.0 mM DFMO, 0.5 µM spermidine and 1.0 mM aminoguanidine.
All EC50 values are the average of at least two independent determinations and were calculated from growth inhibition curves generated from the mean of three determinations per concentration (eight drug concentrations), with the variation between individual values being <15%.
These compounds’ abilities to inhibit the growth of a variety of cell line types were explored and the human melanoma A375 cell line was most consistently and potently affected. Additionally, oral hamster keratinocyte cells (HCPC cells) further demonstrated the polyamine depletion therapy’s potency against skin-derived cells (Fig. 4). These experiments demonstrated that increased growth inhibition correlated with increased lipophilicity of the PTI. The addition of a C8-acyl (8) or a C16-acyl group (11) to incrementally improve the potency of the original molecule (5) when used against this cell line (EC50 values 2.2 µM and 0.15 µM for 8 and 11, respectively compared to 20.5 µM for the original 5).
Figure 4.

HCPC Growth Inhibition with PTI and DFMO.a
aHCPC cells cultured for 6 days in combination with 5.0 mM DFMO, 0.5 µM spermidine and 1.0 mM aminoguanidine together with the amount of Lys-spm conjugates shown.
Preliminary cell-based results show that these agents are not able to cross the cell membrane and have limited impact on polyamine metabolizing enzymes in the cell. This suggests that the target for these compounds is on the extracellular surface of the cells. Compounds 5, 8 and 10 were incubated with human melanoma-derived SK-Mel cells at 10 µM for 24 hrs and various polyamine metabolic activities and polyamine levels were measured. When added alone, none of these agents influenced the level of cellular polyamines. A reduction in the activity of ODC was noted for 5 and 10 but not for 8 (40% and 46% vs. 110% activity compared to control, respectively). A slight reduction in the activity of polyamine metabolizing enzyme SSAT was noted for these three analogs (40% reduction in activity compared to control). These results demonstrate that these agents, when used on cells alone, do not influence cellular polyamine metabolism to any great degree. This is further demonstrated by the inability of any of these agents to rescue from the growth inhibitory effects of DFMO when it is given to cells in the absence of exogenous spermidine (data not shown). This result implies that these agents cannot replace the polyamine needs of the cell when their intracellular polyamines are depleted by a biosynthesis inhibitor. These results showed that these agents did not greatly impact cellular polyamine metabolism after 24 hours and we hypothesize that the target of these agents is the polyamine transport apparatus on the exterior of the cell.
Treatment of MDA-MB-231 cells with 5 or 11 in combination with DFMO for 5 days resulted in potent reduction in the polyamine levels of the cell (Fig. 5). Cytotoxicity of theseagents when used alone is defined as the IC50 value, which was 62 µM for 10 and 57 µM for 11 against MDA-MB-231 cells treated for 6 days. This translates into a 1033 or 750X toxic/therapeutic (IC50/EC50) dose ratio for compounds 10 & 11 in these cells, giving PDT a significant therapeutic window with which to further explore the in vivo efficacy. When the effects of decreasing intracellular polyamine levels by this combination treatment are examined one can see that the design goals are met: a relatively non-toxic agent can potently deplete the intracellular levels of polyamines of cells when used in conjugation with DFMO.
Figure 5.

Cellular polyamine depletion using the combination of PTI and DFMO.a
aConditions: MDA-MB-231 cells were treated for 5 days with 30 µM 5 (D-Lys-spm) or 0.1 µM 11 (Lipo-PA), 5.0 mM DFMO, or the combination of both Lys-spm conjugate and DFMO (all with 0.5 µM spermidine and 1.0 mM aminoguanidine). Polyamine levels determined by pre-column dansylation followed by fluorescent HPLC detection.
Combined DFMO and PTI 5 treatment leads to a dramatic response of SCCs
The combination therapy of polyamine biosynthesis/transport inhibition was then tested against the recently developed K6/ODC transgenic mouse murine squamous cell carcinoma (SCC) model.12 It was developed in an attempt to discover whether altered expression of ODC was a contributing cause or simply an effect of malignant transformation. Using a bovine keratin 6 (K6) promoter to drive high-level ODC expression specifically in proliferating cells of hair follicles (the presumed targets of carcinogens), we were able to demonstrate skin tumor development after only a single low dose of the carcinogen 7,12-dimethylbenz-(α)-anthracene (DMBA), as compared with non-transgenic mice of the same strain that did not show significant tumorigenesis in response to the same treatment. While most skin tumorigenesis models yielded benign squamous papillomas as the predominant tumor type, when the K6/ODC transgene was expressed on the FVB/N strain background, the majority of tumors that developed were aggressive squamous cell carcinomas. These SCCs appeared as early as 5 weeks after treatment and in high multiplicities (up to 4 tumors per mouse), making this a very efficient model for SCC induction. Using this model, we were thus able to conclude that over-expression of ODC is a sufficient condition for tumor promotion in mouse skin.13
We were extremely pleased to see that the promising in vitro results above were translated into positive results when initially tested using the combination of DFMO and our previously described therapeutic agent 5.14 Tumor-bearing animals were treated with DFMO alone and the combination of DFMO (either 0.25% or 0.5% in drinking water) with unsubstituted d-Lysine-spermine 5. Compound 5 was dissolved in phosphate-buffered saline at a concentration of 9.75 mM (5 mg/ml) and injected i.p at a dose of 50 mg/kg twice daily. While the DFMO alone group showed moderate tumor growth inhibitory effects, we found that there were many more complete responses to the combination therapy than to either drug alone (see ref. 14 for data using DFMO alone). In the combination group there were 13 complete responses (no visible tumor) at the end of the 4 week treatment period, out of 17 tumors treated, a complete response rate of 76%. In a second trial, the durability of this antitumor response was tested by following the mice for 6 weeks off-treatment after the 4 week treatment period: 100% of the completely regressed tumors did not recur. In contrast, the majority (11 of 12) of the DFMO alone treated tumors re-grew. Thus, the combination therapy can be considered curative, although the 6-week off-treatment period may be too short to draw definitive conclusions. As reported in our prior publication,14 a moderate increase in the number of apoptotic cells was observed compared to control SCC tumors from this model when extracts of these tumors were analyzed by a TUNEL assay. While not definitive, this observation may help determine the ultimate mechanism for tumor disappearance. We are hoping to extend these studies regarding the mechanism of action of this therapy and will report these results in due course.
Combined anti-tumor effects of DFMO and lipophilic PTI agents
Because of their substantially greater potency in vitro, we conducted an in vivo anti-tumor trial of DFMO combined with either the D-C16-acyl 11 Lys-spm conjugate (Fig. 6) or the L-C16-alkyl 27 analog (Fig. 7) in the K6/ODC SCC model. Also shown in Fig. 6 are the effects of treatment with compound 11 alone, demonstrating the improvements gained in using the combination with the biosynthesis inhibitor DFMO. The potency of these combinations of agents to inhibit tumor growth corresponded to their relative activities in tissue culture with slightly better results observed using 11 in the combination. Comparable efficacy of DFMO and 11 was achieved at a 100-fold lower dose compared to 5 (100 mg/kg/d vs. 1.4 mg/kg/d). With the combination of DFMO (at 0.5% in drinking water) and 0.5 mg/kg 11 (i.p. twice daily) most SCCs (71%) exhibited complete or near-complete responses (>95% volume reduction), in contrast to the weak effect of DFMO alone (data discussed in section above and reference 14). Furthermore, when the 9 out of 17 SCCs that exhibited complete responses were followed for an additional 6 weeks off-treatment, only one tumor recurrence was observed. Based on these results, obtained using a 100-fold lower dosage level of 11 compared with 5, and with no apparent toxicity at this dose level, clinical development of 11 instead of 5 holds much greater promise.
Figure 6.

Antitumor effects in SCC mouse model using either PTI analog 11 alone or combination of DFMO and agent 11.a
aResponse of SCC tumors to 4 weeks of treatment with Cmpd 11 alone (TOP; 0.5 mg/kg twice daily) or combined treatment with 0.5% DFMO orally and 11 (BOTTOM; 0.5 mg/kg i.p. twice daily). Numbers above each set represent the ratio of each tumor volume: tumor volume at the end of the trial period divided by the initial pretreatment volume. "–" represents complete disappearance of the tumor.
Figure 7.

Antitumor effects in SCC mouse model using combination of DFMO and agent 27.a
aResponse of 17 tumors to 4 weeks of a combined treatment with 0.5% DFMO and 0.5 mg/kg 27 (i.p. twice daily). Numbers above each set represent the ratio of each tumor volume: tumor volume at the end of the trial period divided by the initial pretreatment volume. "–" represents complete disappearance of the tumor.
As a further demonstration of the effectiveness of this combination therapy, a second, smaller trial was conducted to assess the efficacy of orally delivered 11 on SCC growth (Figure 8). The concentration of 11 delivered was 14 µg/ml, or an average daily dose of ~50–65 µg (equivalent to ~3 mg/kg/d). All SCCs responded over a 6-week treatment period, with two tumors exhibiting a complete response (>99% volume reduction). This preliminary result suggests an oral route of administration of polyamine transport inhibitors can be effective. Prior formulation work has shown that great improvements in oral bioavailability (from <1% to 60%) were made upon changing from the hydrochloride salt form of 5 to its free base in the presence of appropriate excipients. Therefore, it is expected that great improvements in the oral bioavailability of 11 would result from a similar drug formulation. Clearly, pharmacokinetic optimization of the oral delivery of 11 is in order.
Figure 8.

Antitumor activity following oral delivery of PTI analog 11 (~3 mg/kg) and DFMOa.
aResponse of 6 tumors to 6 weeks of a combined treatment with 0.5% DFMO and 11 both given in the drinking water. Numbers above each set represent the ratio of tumor volume at the end of the trial period divided by the initial pretreatment volume.
Discussion
A variety of naturally occurring lipophilic polycationic molecules have been discovered. Squalamine, which shows potent antibacterial activities, is a sterol-spermidine conjugate that was isolated from stomach extracts of the dogfish shark Squalus acanthias.15 Several acylated short peptides with antibacterial activity have been discovered in various microorganisms.16–19 The posttranslational modification of proteins through palmitoylation of lysine20 or cysteine21,22 residues has been shown to be a reversible mechanism to influence the protein’s interaction with membranes and their trafficking. Acylation with long chain aliphatic lipid groups has been an effective method for increasing the efficacy of antibacterial peptides.23–28 Naturally-occurring peptides known as protein transduction domains (PTDs) or cell-penetrating peptides (CPP) have been shown to contain sequences that provide a means to deliver molecules into mammalian cells. Included in this group are HIV-tat,29 Drosophila melanogaster antennapedia protein30 and the herpes simplex virus structural protein VP2231 all of which contain a group of basic Lys or Arg residues. The mechanisms underlying the uptake of these peptides have been reviewed recently.32,33
Synthetic chemists have followed Nature’s lead by attaching lipophilic features to molecules in order to increase their potency toward their targets. Wender’s group has demonstrated that conjugation of a polyarginine sequence allows cellular uptake of a variety of lipophilic drug molecules.34,35;36 A recent report showed the impact the anionic counterion has on this polyarginine uptake.37 Additionally, cationic lipids have shown promise as non-viral gene therapy transfection agents.38,39
Insights into the possible mechanism of the lipophilic PTI’s increased activity can be gathered by consideration of Schwyzer’s theory of “membrane-catalyzed” peptide-receptor interactions.40 Given that the cell membrane’s polyamine transporter apparatus can only occupy a small fraction of the cellular surface it might be considered more likely that the lipophilic PTI will interact with the lipid membrane of the cell before associating with the presumed polyanionic target of the transport apparatus. Furthermore, since now, the localized membrane concentration of inhibitor is increased, it might also be assumed more likely that the transporter will bind with these membrane-associated inhibitors. Additionally, the higher-binding efficiencies may have an entropic contribution from the reduction-of-dimensionality from a three-dimensional target search to a two-dimensional search. It has been estimated that this can result in an enhancement of 102-103 in binding.41 A recent review of these concepts has appeared.42 As demonstrated by the results reported here, and regardless of the exact order of binding events sequence, consideration of these effects for the design of drugs targeting membrane-associated biomolecules is well warranted.
The polyamine transport apparatus of the cell performs the inherently difficult biological task of transporting the poly-ammonium charged polyamines across the lipophilic cell membrane. Polyamine transport into mammalian cells is energy and temperature dependent, saturable, carrier mediated and operates against a substantial concentration gradient43;44 Ample experimental proof exists that polyamine concentration homeostasis is mediated via this transport system. Changes in the requirements for polyamines in response to growth stimulation are reflected by increases in the transport activity. Stimulation of human fibroblasts to cell proliferation by serum or epidermal growth factor was followed by an 18–100 fold increase in the uptake of putrescine.45 Tumors have been shown to have an increased rate of putrescine uptake46;47 Reduction of the systemic availability of polyamines by reduction of gastrointestinal bacterial polyamine production has been shown to increase the antitumor efficacy of DFMO.48–50 A promising recent report showed that the use of DFMO as a chemopreventative for the reoccurrence of colon cancer and demonstrates that the use of a polyamine-based chemotherapeutic is a viable approach in the fight against cancer.51
Several researchers have studied the ability of synthetic analogs of polyamines to inhibit the uptake of 3H-spermidine into cells.52–54 Notable examples of potent polyamine transport inhibitors include the work by Poulin55;56 and Cullis.57;58 Two groups have summarized much of this literature data by use of CoMFA and QSAR methods.59;60 Additionally, a group has exploited the higher polyamine transport activity of cancer cells as a drug-targeting strategy.61 In this work, it was demonstrated that triamine-anthracene conjugates were able to gain access to the interior of the cell whereas their tetraamine counterparts appeared to remain on the cell surface. These important observations contribute to the possibility that the tetraamino Lys-spm analogs described in this manuscript remain associated with the cell membrane. Several research groups have begun to explore the molecular mechanisms for transport of polycharged species. Regen’s work on the assisted delivery of bile-acid spermine conjugates might be considered as a synthetic model of a membrane transporter.62–64 A series of persulfated polyanionic molecular umbrellas based on spermine tetra-lysine conjugates have been evaluated as anti-HIV and anti-HSV agents.65 Peterson’s group has designed and characterized several series of cholesterol-linked of peptides as models for receptor-mediated endocytosis.66,67,68
Genes for the polyamine transport protein or complex have been cloned from Escherichia coli and yeast69 and a subunit of the transporter from E. coli has been crystallized and its X-ray structure has been determined.70;71 The genes for the mammalian transporter await identification. Despite this, several researchers have suggested that the polyamine transport system may involve binding first to a plasma membrane carrier followed by transport into pre-existing polyamine-sequestering vesicles72 which may be related to a complex S-nitrosylated glypican recycling system.73–75 In fact, the use of a HIV-Tat domain-derived peptide that potently inhibits polyamine uptake in combination with DFMO has been shown to dramatically increase its effectiveness in a mouse cancer model.76 Furthermore, a recent report demonstrated the effectiveness of polyamine depletion by dual use of DFMO and an antibody to the heparan sulfate.77
Conclusions
In this work, we extend our earlier studies characterizing amino acid-spermine conjugates as polyamine transport inhibitors by attaching various lipophilic groups to the epsilon nitrogen atom of the lysine portion of our Lys-spermine conjugate lead. Extensive biochemical characterization led to the selection of the C16-acyl derivative of the d-Lys-spermine conjugate 11 as our lead compound. An especially appealing aspect of this approach is the fact that the target resides on the extracellular membrane of the cell and the drug agent does not need to enter the cell. The use of this agent, in combination with the approved drug DFMO, represents a first-in-class therapy to deplete cellular polyamine levels in proliferative diseases such as cancer. We have demonstrated both in vitro and in vivo efficacy with this approach against skin derived tumors yet feel that its usefulness would apply equally to a variety of hyperproliferative diseases. As demonstrated by the positive results following oral delivery in the murine skin cancer model reported here, together with recent results obtained in spontaneous squamous cell carcinomas in housecats, this therapy holds promise for the treatment of cancer. We have initiated studies to explore the formulation, pharmacokinetic and toxicological profile of these agents in anticipation of future human clinical studies.
Materials and Methods
Compounds
All compounds were prepared as previously reported78 and were tested as their per-HCl salts. Drugs readily dissolved in H2O to at least 10 mM concentrations. Compounds were characterized by TLC (using 4:1:1:2 isopropanol/pyridine/glacial acetic acid/H2O), 1H and 13C NMR, LC/MS and elemental analysis and all data were consistent with structures assigned and supported >97% purities. Characterization details are given below for several representative compounds.
Representative acyl products
L-Lys-ε-(palmitoyl)-N1-spermine (10) - TLC analysis: Rf = 0.19. 1H NMR (D2O, δ): 3.93 (1H), 3.45 (1H), 3.03 (13H), 2.12 (2H), 2.02 (2H), 1.85 (4H), 1.75 (s, 4H), 1.43 (4H), 1.32 (2H), 1.11 (24H), 0.72 (t, 3H). 13C NMR (D2O, ppm): 175.7, 169.8, 53.4, 46.7 (m), 45.2, 44.6, 38.8, 36.5, 36.1, 31.9, 30.4, 30.0 (m), 29.9 (m), 29.6 (m), 29.4, 28.2, 25.7, 25.5, 23.6, 22.8, 22.7, 22.3, 21.7, 13.8. LC/MS (ret time, 7.2 min, 99% purity), calcd for C32H68N6O2 m/z 568, obsd 569 (MH+). Anal. Calcd for C32H72Cl4N6O2: C, 53.77; H, 10.15; N, 11.76. Found: C, 53.51; H, 10.09; N, 11.51.
D-Lys-ε-(palmitoyl)-N1-spermine (11) - TLC analysis: Rf = 0.19. 1H NMR (D2O, δ): 3.94 (1H), 3.47 (1H), 3.06 (13H), 2.13 (2H), 2.04 (2H), 1.87 (4H), 1.75 (4H), 1.47 (4H), 1.36 (2H), 1.16 (24H), 0.78 (3H). 13C NMR (D2O, ppm): 175.7, 169.8, 53.4, 47.2 (m), 45.6, 44.8, 39.0, 36.6 (m), 36.1, 31.9, 29.8 (m), 29.6, 29.3, 28.4, 25.9, 25.7, 23.8, 22.8, 23.1, 22.8, 22.1, 14.0. LC/MS (ret time, 7.2 min; 98% purity), calcd for C32H68N6O2 m/z 568, obsd 569 (MH+). Anal. Calcd for C32H72Cl4N6O2: C, 53.77; H, 10.15; N, 11.76. Found: C, 53.38; H, 10.20; N, 11.52.
Representative alkyl product
L-Lys-ε-(n-hexadecyl)-N1-spermine (27) - TLC analysis: Rf = 0.11. 1H NMR (D2O, δ): 3.97 (1H), 3.48 (1H), 3.04 (15H), 2.04 (2H), 1.91 (4H), 1.75 (8H), 1.48 (2H), 1.22 (26H), 0.91 (3H). 13C NMR (D2O, ppm): 168.8, 53.4, 48.0, 47.3, 47.1 (m), 45.4, 44.7, 36.7, 32.0, 30.6, 29.9 (m), 29.8, 29.5, 29.4, 29.1, 26.5, 25.9, 25.6, 25.5, 23.8, 22.9, 22.8, 21.7, 13.9. LC/MS (ret time, 7.2 min; 97% purity), calcd for C32H70N6O m/z 555, obsd 556 (MH+). Anal. Calcd for C32H75Cl5N6O·3/2H2O: C, 50.29; H, 10.29; N, 11.00. Found: C, 50.30; H, 10.05; N, 10.67.
Cell Growth Assay
All cell lines were obtained from ATCC (Manassas, VA) and cultured in the recommended media, serum, and CO2 concentration. Medias were obtained from Mediatech, Inc. (Herndon, VA) and serums from Gibco BRL (Gaithersburg, MD). 50 U/ml penicillin, 50 µg/ml streptomycin and 2 mM L-glutamine (all from BioWhittaker, Walkersville, MD) were included in all cultures. When cells were cultured with inhibitors, 1.0 mM aminoguanidine was included to inhibit serum amine oxidase activity. Cells were plated in 96-well plates such that they would be in log growth for the duration of the assay. The day after plating, inhibitors were added to the cells, and growth, if any, permitted to continue for six days in the presence 0.5 µM spermidine to insure that any growth inhibition was not the result of depletion of external polyamines in the media. At the end of the six days, cell growth was measured by MTS/PMS dye assay (Cell Titer 96 Aqueous Non-Radioactive Cell Proliferation Assay; Promega, Madison, WI). EC50 represents the concentration of inhibitor that resulted in 50% of maximum growth inhibition achievable in the presence of both DFMO (5.0 mM in all cell lines) and inhibitor (at different concentrations depending in part on the cell line used) compared to controls. IC50 represents the concentration of inhibitor that resulted in 50% maximum growth inhibition when used alone. EC50 values are shown in Table 1 and Table 2.
Polyamine Transport and Ki Assays
[2,9-3H]spermidine from DuPont NEN (Boston, MA) was added alone or simultaneously with inhibitors to 24-well plates containing MDA-MB-231 cells in log growth. The cells were incubated at 37°C for 15 min to determine initial rate polyamine uptake. The cells were then washed three times with cold PBS, lysed with 0.1% SDS, and the amount of polyamine incorporation into the cells was determined by scintillation counting of the cell lysates. To determine a Ki, four radioactive substrate concentrations (0.3–3 µM) and five inhibitor concentrations (0.01–1.0 µM) and a control were tested. The Ki values were determined using double reciprocal Lineweaver-Burke plot analyses. Ki values were determined from linear equations derived from graphing the slopes of Lineweaver-Burke plot vs. inhibitor concentration, with Ki = y-intercept / slope.
HPLC Analysis of Dansylated Derivatives
Polyamine levels and analogs were determined by prederivatization by dansyl chloride followed by reverse-phase HPLC by fluorescent detection.79
Mouse Squamous Cell Carcinoma Model
The K6/ODC mouse model on the FVB/N strain background instead of the original C57BL/6J background12 was used in all experiments. All experiments with mice were approved by the Institutional Animal Care and Use Committee of Lankenau Institute for Medical Research. To induce SCCs, newborn (1 day old) pups were treated once topically with 100 nmol of DMBA dissolved in 50 µl acetone. Both male and female mice were used. SCCs typically developed on the treated area (dorsal skin) beginning 5 weeks later. SCC-bearing mice were used in experiments between 8 and 18 weeks of age. Based on pilot data indicating a general lack of responsiveness of SCCs of the face and head, these tumors were excluded from analyses.
There was substantial variability in tumor growth rates, even among multiple SCCs in the same mouse. In an attempt to control for this variability, tumor-bearing mice were randomized based on tumor size distribution to one of four treatment groups: (1) control (phosphate-buffered saline (vehicle for PTI); (2) DFMO (either 0.25% or 0.5% in drinking water); (3) PTI analog and (4) DFMO plus PTI analog. Thus, each treatment group consisted of mice bearing SCCs with approximately equal tumor size distributions (small→large). DFMO solutions were changed every 4 days or less. Compound 11 was dissolved in phosphate-buffered saline at a concentration of 9.75 mM (5 mg/ml) and injected i.p at a dose of 0.7 mg/kg twice daily in early morning and late afternoon, except on weekends when mice were injected once. Animals were observed daily for signs of distress due to tumor burden and/or drug treatment. Tumor volume was calculated according to the equation V = l × w2/2 where l = length (longest dimension) and w = width. Responses of individual tumors to treatment were expressed as “ratio of tumor volume,” which is defined as the tumor volume after treatment divided by the initial (pretreatment) volume.
Acknowledgment
The authors wish to thank Debra Kramer at Roswell Park Cancer Institute, Buffalo, New York for providing measurements of enzyme activities, transport inhibitor accumulation and polyamine pool levels in SK-Mel cells. Part of this research was funded by NIH grant # 5R01CA094107-07 to T.O. and hereby gratefully acknowledged.
Abbreviations
- ATCC
American type culture collection
- CoMFA
comparative molecular field analysis
- DFMO
α-difluoromethylornithine
- DMBA
7,12-dimethylbenz-(α)-anthracene
- ODC
ornithine decarboxylase
- PTI
polyamine transport inhibitor
- QSAR
quantitative structure activity relationship
- SCC
squamous cell carcinoma
- SSAT
spermine/spermidine N1-acetyltransferase
Reference List
- 1.Leveque J, Foucher F, Bansard JY, Havouis R, Grall JY, Moulinoux JP. Polyamine profiles in tumor, normal tissue of the homologous breast, blood, and urine of breast cancer sufferers. Breast Cancer Res. Treat. 2000;60:99–105. doi: 10.1023/a:1006319818530. [DOI] [PubMed] [Google Scholar]
- 2.Cipolla B, Guille F, Moulinoux JP, Quemener V, Staerman F, Corbel L, Lobel B. Polyamines and prostatic carcinoma: clinical and therapeutic implications. Eur. Urol. 1993;24:124–131. doi: 10.1159/000474279. [DOI] [PubMed] [Google Scholar]
- 3.Janne J, Poso H, Raina A. Polyamines in rapid growth and cancer. Biochim. Biophys. Acta. 1978;473:241–293. doi: 10.1016/0304-419x(78)90015-x. [DOI] [PubMed] [Google Scholar]
- 4.Scalabrino G, Ferioli ME. Polyamines in mammalian tumors. Part II. Adv. Cancer Res. 1982;36:1–102. doi: 10.1016/s0065-230x(08)60422-4. [DOI] [PubMed] [Google Scholar]
- 5.Scalabrino G, Ferioli ME. Polyamines in mammalian tumors. Part I. Adv. Cancer Res. 1981;35:151–268. doi: 10.1016/s0065-230x(08)60911-2. [DOI] [PubMed] [Google Scholar]
- 6.Bogle RG, Mann GE, Pearson JD, Morgan DM. Endothelial polyamine uptake: selective stimulation by L-arginine deprivation or polyamine depletion. Am. J. Physiol. 1994;266:C776–C783. doi: 10.1152/ajpcell.1994.266.3.C776. [DOI] [PubMed] [Google Scholar]
- 7.Alhonen-Hongisto L, Seppanen P, Janne J. Intracellular putrescine and spermidine deprivation induces increased uptake of the natural polyamines and methylglyoxal bis(guanylhydrazone) Biochem. J. 1980;192:941–945. doi: 10.1042/bj1920941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Geisse JK. Comparison of treatment modalities for squamous cell carcinoma. Clin. Dermatol. 1995;13:621–626. doi: 10.1016/0738-081x(95)00098-z. [DOI] [PubMed] [Google Scholar]
- 9.Miller DL, Weinstock MA. Nonmelanoma skin cancer in the United States: incidence. J.Am. Acad. Dermatol. 1994;30:774–778. doi: 10.1016/s0190-9622(08)81509-5. [DOI] [PubMed] [Google Scholar]
- 10.Rowe DE, Carroll RJ, Day CL., Jr Long-term recurrence rates in previously untreated (primary) basal cell carcinoma: implications for patient follow-up. J. Dermatol. Surg. Oncol. 1989;15:315–328. doi: 10.1111/j.1524-4725.1989.tb03166.x. [DOI] [PubMed] [Google Scholar]
- 11.Burns MR, Carlson CL, Vanderwerf SM, Ziemer JR, Weeks RS, Cai F, Webb HK, Graminski GF. Amino acid/spermine conjugates: polyamine amides as potent spermidine uptake inhibitors. J. Med. Chem. 2001;44:3632–3644. doi: 10.1021/jm0101040. [DOI] [PubMed] [Google Scholar]
- 12.Megosh L, Gilmour SK, Rosson D, Soler AP, Blessing M, Sawicki JA, O'Brien TG. Increased frequency of spontaneous skin tumors in transgenic mice which overexpress ornithine decarboxylase. Cancer Res. 1995;55:4205–4209. [PubMed] [Google Scholar]
- 13.O'Brien TG, Megosh LC, Gilliard G, Soler AP. Ornithine decarboxylase overexpression is a sufficient condition for tumor promotion in mouse skin. Cancer Res. 1997;57:2630–2637. [PubMed] [Google Scholar]
- 14.Chen Y, Weeks RS, Burns MR, Boorman DW, Klein-Szanto A, O'Brien TG. Combination therapy with 2-difluoromethylornithine and a polyamine transport inhibitor against murine squamous cell carcinoma. Int. J. Cancer. 2006;118:2344–2349. doi: 10.1002/ijc.21621. [DOI] [PubMed] [Google Scholar]
- 15.Moore KS, Wehrli S, Roder H, Rogers M, Forrest JN, Jr, McCrimmon D, Zasloff M. Squalamine: an aminosterol antibiotic from the shark. Proc. Natl. Acad. Sci. U. S. A. 1993;90:1354–1358. doi: 10.1073/pnas.90.4.1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bassarello C, Lazzaroni S, Bifulco G, Lo CP, Iacobellis NS, Riccio R, Gomez-Paloma L, Evidente A. Tolaasins A--E, five new lipodepsipeptides produced by Pseudomonas tolaasii. J. Nat. Prod. 2004;67:811–816. doi: 10.1021/np0303557. [DOI] [PubMed] [Google Scholar]
- 17.Peggion C, Formaggio F, Crisma M, Epand RF, Epand RM, Toniolo C. Trichogin: a paradigm for lipopeptaibols. J. Pept. Sci. 2003;9:679–689. doi: 10.1002/psc.500. [DOI] [PubMed] [Google Scholar]
- 18.Swanson PE, Paddy MR, Dahlquist FW, Storm DR. Characterization of octapeptin-membrane interactions using spin-labeled octapeptin. Biochemistry. 1980;19:3307–3314. doi: 10.1021/bi00555a032. [DOI] [PubMed] [Google Scholar]
- 19.Tsubery H, Ofek I, Cohen S, Fridkin M. N-terminal modifications of Polymyxin B nonapeptide and their effect on antibacterial activity. Peptides. 2001;22:1675–1681. doi: 10.1016/s0196-9781(01)00503-4. [DOI] [PubMed] [Google Scholar]
- 20.Hackett M, Guo L, Shabanowitz J, Hunt DF, Hewlett EL. Internal lysine palmitoylation in adenylate cyclase toxin from Bordetella pertussis. Science. 1994;266:433–435. doi: 10.1126/science.7939682. [DOI] [PubMed] [Google Scholar]
- 21.Linder ME, Deschenes RJ. New insights into the mechanisms of protein palmitoylation. Biochemistry. 2003;42:4311–4320. doi: 10.1021/bi034159a. [DOI] [PubMed] [Google Scholar]
- 22.Smotrys JE, Linder ME. Palmitoylation of intracellular signaling proteins: regulation and function. Annu. Rev. Biochem. 2004;73:559–587. doi: 10.1146/annurev.biochem.73.011303.073954. [DOI] [PubMed] [Google Scholar]
- 23.Avrahami D, Shai Y. Conjugation of a magainin analogue with lipophilic acids controls hydrophobicity, solution assembly, and cell selectivity. Biochemistry. 2002;41:2254–2263. doi: 10.1021/bi011549t. [DOI] [PubMed] [Google Scholar]
- 24.Chicharro C, Granata C, Lozano R, Andreu D, Rivas L. N-terminal fatty acid substitution increases the leishmanicidal activity of CA(1–7)M(2–9), a cecropin-melittin hybrid peptide. Antimicrob. Agents Chemother. 2001;45:2441–2449. doi: 10.1128/AAC.45.9.2441-2449.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lockwood NA, Haseman JR, Tirrell MV, Mayo KH. Acylation of SC4 dodecapeptide increases bactericidal potency against Gram-positive bacteria, including drug-resistant strains. Biochem. J. 2004;378:93–103. doi: 10.1042/BJ20031393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Majerle A, Kidric J, Jerala R. Enhancement of antibacterial and lipopolysaccharide binding activities of a human lactoferrin peptide fragment by the addition of acyl chain. J. Antimicrob. Chemother. 2003;51:1159–1165. doi: 10.1093/jac/dkg219. [DOI] [PubMed] [Google Scholar]
- 27.Mak P, Pohl J, Dubin A, Reed MS, Bowers SE, Fallon MT, Shafer WM. The increased bactericidal activity of a fatty acid-modified synthetic antimicrobial peptide of human cathepsin G correlates with its enhanced capacity to interact with model membranes. Int. J. Antimicrob. Agents. 2003;21:13–19. doi: 10.1016/s0924-8579(02)00245-5. [DOI] [PubMed] [Google Scholar]
- 28.Wakabayashi H, Matsumoto H, Hashimoto K, Teraguchi S, Takase M, Hayasawa H. N-Acylated and D enantiomer derivatives of a nonamer core peptide of lactoferricin B showing improved antimicrobial activity. Antimicrob. Agents Chemother. 1999;43:1267–1269. doi: 10.1128/aac.43.5.1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Frankel AD, Pabo CO. Cellular uptake of the tat protein from human immunodeficiency virus. Cell. 1988;55:1189–1193. doi: 10.1016/0092-8674(88)90263-2. [DOI] [PubMed] [Google Scholar]
- 30.Derossi D, Chassaing G, Prochiantz A. Trojan peptides: the penetratin system for intracellular delivery. Trends Cell Biol. 1998;8:84–87. [PubMed] [Google Scholar]
- 31.Elliott G, O'Hare P. Intercellular trafficking and protein delivery by a herpesvirus structural protein. Cell. 1997;88:223–233. doi: 10.1016/s0092-8674(00)81843-7. [DOI] [PubMed] [Google Scholar]
- 32.Fischer R, Fotin-Mleczek M, Hufnagel H, Brock R. Break on through to the other side-biophysics and cell biology shed light on cell-penetrating peptides. Chembiochem. 2005;6:2126–2142. doi: 10.1002/cbic.200500044. [DOI] [PubMed] [Google Scholar]
- 33.Patel LN, Zaro JL, Shen WC. Cell penetrating peptides: intracellular pathways and pharmaceutical perspectives. Pharm. Res. 2007;24:1977–1992. doi: 10.1007/s11095-007-9303-7. [DOI] [PubMed] [Google Scholar]
- 34.Rothbard JB, Kreider E, Vandeusen CL, Wright L, Wylie BL, Wender PA. Arginine-rich molecular transporters for drug delivery: role of backbone spacing in cellular uptake. J. Med. Chem. 2002;45:3612–3618. doi: 10.1021/jm0105676. [DOI] [PubMed] [Google Scholar]
- 35.Rothbard JB, Jessop TC, Lewis RS, Murray BA, Wender PA. Role of membrane potential and hydrogen bonding in the mechanism of translocation of guanidinium-rich peptides into cells. J. Am. Chem. Soc. 2004;126:9506–9507. doi: 10.1021/ja0482536. [DOI] [PubMed] [Google Scholar]
- 36.Wright LR, Rothbard JB, Wender PA. Guanidinium rich peptide transporters and drug delivery. Curr. Protein Pept. Sci. 2003;4:105–124. doi: 10.2174/1389203033487252. [DOI] [PubMed] [Google Scholar]
- 37.Sakai N, Matile S. Anion-mediated transfer of polyarginine across liquid and bilayer membranes. J. Am. Chem. Soc. 2003;125:14348–14356. doi: 10.1021/ja037601l. [DOI] [PubMed] [Google Scholar]
- 38.Miller AD. Cationic liposome systems in gene therapy. IDrugs. 1998;1:574–583. [PubMed] [Google Scholar]
- 39.Karmali PP, Chaudhuri A. Cationic liposomes as non-viral carriers of gene medicines: resolved issues, open questions, and future promises. Med. Res. Rev. 2007;27:696–722. doi: 10.1002/med.20090. [DOI] [PubMed] [Google Scholar]
- 40.Sargent DF, Schwyzer R. Membrane lipid phase as catalyst for peptide-receptor interactions. Proc. Natl. Acad. Sci. U. S. A. 1986;83:5774–5778. doi: 10.1073/pnas.83.16.5774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Haugh JM, Lauffenburger DA. Physical modulation of intracellular signaling processes by locational regulation. Biophys. J. 1997;72:2014–2031. doi: 10.1016/S0006-3495(97)78846-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Castanho MA, Fernandes MX. Lipid membrane-induced optimization for ligand-receptor docking: recent tools and insights for the "membrane catalysis" model. Eur. Biophys. J. 2006;35:92–103. doi: 10.1007/s00249-005-0007-9. [DOI] [PubMed] [Google Scholar]
- 43.Seiler N, Dezeure F. Polyamine transport in mammalian cells. Int. J. Biochem. 1990;22:211–218. doi: 10.1016/0020-711x(90)90332-w. [DOI] [PubMed] [Google Scholar]
- 44.Seiler N, Delcros JG, Moulinoux JP. Polyamine transport in mammalian cells. An update. Int. J. Biochem. Cell Biol. 1996;28:843–861. doi: 10.1016/1357-2725(96)00021-0. [DOI] [PubMed] [Google Scholar]
- 45.Pohjanpelto P. Putrescine transport is greatly increased in human fibroblasts initiated to proliferate. J. Cell Biol. 1976;68:512–520. doi: 10.1083/jcb.68.3.512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Volkow N, Goldman SS, Flamm ES, Cravioto H, Wolf AP, Brodie JD. Labeled putrescine as a probe in brain tumors. Science. 1983;221:673–675. doi: 10.1126/science.6603020. [DOI] [PubMed] [Google Scholar]
- 47.Moulinoux JP, Quemener V, Khan NA. Biological significance of circulating polyamines in oncology. Cell Mol. Biol. 1991;37:773–783. [PubMed] [Google Scholar]
- 48.Leveque J, Foucher F, Havouis R, Desury D, Grall JY, Moulinoux JP. Benefits of complete polyamine deprivation in hormone responsive and hormone resistant MCF-7 human breast adenocarcinoma in vivo. Anticancer Res. 2000;20:97–101. [PubMed] [Google Scholar]
- 49.Seiler N, Sarhan S, Grauffel C, Jones R, Knodgen B, Moulinoux JP. Endogenous and exogenous polyamines in support of tumor growth. Cancer Res. 1990;50:5077–5083. [PubMed] [Google Scholar]
- 50.Quemener V, Blanchard Y, Chamaillard L, Havouis R, Cipolla B, Moulinoux JP. Polyamine deprivation: a new tool in cancer treatment. Anticancer Res. 1994;14:443–448. [PubMed] [Google Scholar]
- 51.Meyskens FL, McLaren CE, Pelot D, Fujikawa-Brooks S, Carpenter PM, Hawk E, Kelloff G, Lawson MJ, Kidao J, McCracken J, Albers CG, Ahnen DJ, Turgeon DK, Goldschmid S, Lance P, Hagedorn CH, Gillen DL, Gerner EW. Difluoromethylornithine Plus Sulindac for the Prevention of Sporadic Colorectal Adenomas: A Randomized Placebo-Controlled, Double-Blind Trial. Cancer Prev. Res. Phila Pa. 2008;1:9–11. doi: 10.1158/1940-6207.CAPR-08-0042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bergeron RJ, McManis JS, Liu CZ, Feng Y, Weimar WR, Luchetta GR, Wu Q, Ortiz-Ocasio J, Vinson JR, Kramer D. Antiproliferative properties of polyamine analogues: a structure-activity study. J. Med. Chem. 1994;37:3464–3476. doi: 10.1021/jm00047a004. [DOI] [PubMed] [Google Scholar]
- 53.Bergeron RJ, Feng Y, Weimar WR, McManis JS, Dimova H, Porter C, Raisler B, Phanstiel O. A comparison of structure-activity relationships between spermidine and spermine analogue antineoplastics. J. Med. Chem. 1997;40:1475–1494. doi: 10.1021/jm960849j. [DOI] [PubMed] [Google Scholar]
- 54.Porter CW, Bergeron RJ. Spermidine requirement for cell proliferation in eukaryotic cells: structural specificity and quantitation. Science. 1983;219:1083–1085. doi: 10.1126/science.6823570. [DOI] [PubMed] [Google Scholar]
- 55.Huber M, Pelletier JG, Torossian K, Dionne P, Gamache I, Charest-Gaudreault R, Audette M, Poulin R. 2,2'-Dithiobis(N-ethyl-spermine-5-carboxamide) is a high affinity, membrane-impermeant antagonist of the mammalian polyamine transport system. J. Biol. Chem. 1996;271:27556–27563. doi: 10.1074/jbc.271.44.27556. [DOI] [PubMed] [Google Scholar]
- 56.Covassin L, Desjardins M, Soulet D, Charest-Gaudreault R, Audette M, Poulin R. Xylylated dimers of putrescine and polyamines: influence of the polyamine backbone on spermidine transport inhibition. Bioorg. Med. Chem. Lett. 2003;13:3267–3271. doi: 10.1016/s0960-894x(03)00668-1. [DOI] [PubMed] [Google Scholar]
- 57.Cullis PM, Green RE, Merson-Davies L, Travis N. Probing the mechanism of transport and compartmentalisation of polyamines in mammalian cells. Chem. Biol. 1999;6:717–729. doi: 10.1016/s1074-5521(00)80019-8. [DOI] [PubMed] [Google Scholar]
- 58.Cullis PM, Green RE, Merson-Davies L, Travis NG. Chemical highlights of polyamine transport. Biochem. Soc. Trans. 1998;26:595–601. doi: 10.1042/bst0260595. [DOI] [PubMed] [Google Scholar]
- 59.Li Y, MacKerell AD, Jr, Egorin MJ, Ballesteros MF, Rosen DM, Wu YY, Blamble DA, Callery PS. Comparative molecular field analysis-based predictive model of structure-function relationships of polyamine transport inhibitors in L1210 cells. Cancer Res. 1997;57:234–239. [PubMed] [Google Scholar]
- 60.Xia CQ, Yang JJ, Ren S, Lien EJ. QSAR analysis of polyamine transport inhibitors in L1210 cells. J. Drug Target. 1998;6:65–77. doi: 10.3109/10611869808997882. [DOI] [PubMed] [Google Scholar]
- 61.Wang C, Delcros JG, Biggerstaff J, Phanstiel O. Molecular requirements for targeting the polyamine transport system. Synthesis and biological evaluation of polyamine-anthracene conjugates. J. Med. Chem. 2003;46:2672–2682. doi: 10.1021/jm020598g. [DOI] [PubMed] [Google Scholar]
- 62.Zhang J, Jing B, Regen SL. Kinetic evidence for the existence and mechanism of formation of a barrel stave structure from pore-forming dendrimers. J. Am. Chem. Soc. 2003;125:13984–13987. doi: 10.1021/ja036390h. [DOI] [PubMed] [Google Scholar]
- 63.Bandyopadhyay P, Janout V, Zhang LH, Regen SL. Ion conductors derived from cholic acid and spermine: importance of facial hydrophilicity on NA(+) transport and membrane selectivity. J. Am. Chem. Soc. 2001;123:7691–7696. doi: 10.1021/ja010926m. [DOI] [PubMed] [Google Scholar]
- 64.Janout V, Lanier M, Deng G, Regen SL. Evidence for highly cooperative binding between molecular umbrella-spermine conjugates and DNA. Bioconjug. Chem. 1997;8:891–895. doi: 10.1021/bc970142r. [DOI] [PubMed] [Google Scholar]
- 65.Jing B, Janout V, Herold BC, Klotman ME, Heald T, Regen SL. Persulfated molecular umbrellas as anti-HIV and anti-HSV agents. J. Am. Chem. Soc. 2004;126:15930–15931. doi: 10.1021/ja044400o. [DOI] [PubMed] [Google Scholar]
- 66.Boonyarattanakalin S, Hu J, Dykstra-Rummel SA, August A, Peterson BR. Endocytic delivery of vancomycin mediated by a synthetic cell surface receptor: rescue of bacterially infected Mammalian cells and tissue targeting in vivo. J. Am. Chem. Soc. 2007;129:268–269. doi: 10.1021/ja067674f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Boonyarattanakalin S, Martin SE, Sun Q, Peterson BR. A synthetic mimic of human Fc receptors: defined chemical modification of cell surfaces enables efficient endocytic uptake of human immunoglobulin-G. J. Am. Chem. Soc. 2006;128:11463–11470. doi: 10.1021/ja062377w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sun Q, Cai S, Peterson BR. Selective disruption of early/recycling endosomes: release of disulfide-linked cargo mediated by a N-alkyl-3beta-cholesterylamine-capped peptide. J. Am. Chem. Soc. 2008;130:10064–10065. doi: 10.1021/ja803380a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tomitori H, Kashiwagi K, Sakata K, Kakinuma Y, Igarashi K. Identification of a gene for a polyamine transport protein in yeast. J. Biol. Chem. 1999;274:3265–3267. doi: 10.1074/jbc.274.6.3265. [DOI] [PubMed] [Google Scholar]
- 70.Sugiyama S, Matsuo Y, Maenaka K, Vassylyev DG, Matsushima M, Kashiwagi K, Igarashi K, Morikawa K. The 1.8-A X-ray structure of the Escherichia coli PotD protein complexed with spermidine and the mechanism of polyamine binding. Protein Sci. 1996;5:1984–1990. doi: 10.1002/pro.5560051004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sugiyama S, Vassylyev DG, Matsushima M, Kashiwagi K, Igarashi K, Morikawa K. Crystal structure of PotD, the primary receptor of the polyamine transport system in Escherichia coli. J. Biol. Chem. 1996;271:9519–9525. doi: 10.1074/jbc.271.16.9519. [DOI] [PubMed] [Google Scholar]
- 72.Soulet D, Gagnon B, Rivest S, Audette M, Poulin R. A fluorescent probe of polyamine transport accumulates into intracellular acidic vesicles via a two-step mechanism. J. Biol. Chem. 2004;279:49355–49366. doi: 10.1074/jbc.M401287200. [DOI] [PubMed] [Google Scholar]
- 73.Belting M, Mani K, Jonsson M, Cheng F, Sandgren S, Jonsson S, Ding K, Delcros JG, Fransson LA. Glypican-1 is a vehicle for polyamine uptake in mammalian cells: a pivital role for nitrosothiol-derived nitric oxide. J. Biol. Chem. 2003;278:47181–47189. doi: 10.1074/jbc.M308325200. [DOI] [PubMed] [Google Scholar]
- 74.Belting M, Persson S, Fransson LA. Proteoglycan involvement in polyamine uptake. Biochem. J. 1999;338(Pt 2):317–323. [PMC free article] [PubMed] [Google Scholar]
- 75.Belting M, Havsmark B, Jonsson M, Persson S, Fransson LA. Heparan sulphate/heparin glycosaminoglycans with strong affinity for the growth-promoter spermine have high antiproliferative activity. Glycobiology. 1996;6:121–129. doi: 10.1093/glycob/6.2.121. [DOI] [PubMed] [Google Scholar]
- 76.Mani K, Sandgren S, Lilja J, Cheng F, Svensson K, Persson L, Belting M. HIV-Tat protein transduction domain specifically attenuates growth of polyamine deprived tumor cells. Mol. Cancer Ther. 2007;6:782–788. doi: 10.1158/1535-7163.MCT-06-0370. [DOI] [PubMed] [Google Scholar]
- 77.Welch JE, Bengtson P, Svensson K, Wittrup A, Jenniskens GJ, Ten Dam GB, Van Kuppevelt TH, Belting M. Single chain fragment anti-heparan sulfate antibody targets the polyamine transport system and attenuates polyamine-dependent cell proliferation. Int. J. Oncol. 2008;32:749–756. [PubMed] [Google Scholar]
- 78.Burns MR, Wood SJ, Miller KA, Nguyen T, Cromer JR, David SA. Lysine-spermine conjugates: hydrophobic polyamine amides as potent lipopolysaccharide sequestrants. Bioorg. Med. Chem. 2005;13:2523–2536. doi: 10.1016/j.bmc.2005.01.038. [DOI] [PubMed] [Google Scholar]
- 79.Kabra PM, Lee HK, Lubich WP, Marton LJ. Solid-phase extraction and determination of dansyl derivatives of unconjugated and acetylated polyamines by reversed-phase liquid chromatography: improved separation systems for polyamines in cerebrospinal fluid, urine and tissue. J. Chromatogr. 1986;380:19–32. doi: 10.1016/s0378-4347(00)83621-x. [DOI] [PubMed] [Google Scholar]