

Summary
We investigated the molecular determinants of aquaporin-4 (AQP4) assembly in orthogonal arrays of particles (OAPs) by visualizing fluorescently labeled AQP4 mutants in cell membranes using quantum-dot single-particle tracking and total internal reflection fluorescence microscopy. The full-length `long' (M1) form of AQP4 diffused freely in membranes and did not form OAPs, whereas the `short' (M23) form of AQP4 formed OAPs and was nearly immobile. Analysis of AQP4 deletion mutants revealed progressive disruption of OAPs by the addition of three to seven residues at the AQP4-M23 N-terminus, with polyalanines as effective as native AQP4 fragments. OAPs disappeared upon downstream deletions of AQP4-M23, which, from analysis of point mutants, involves N-terminus interactions of residues Val24, Ala25 and Phe26. OAP formation was also prevented by introducing proline residues at sites just downstream from the hydrophobic N-terminus of AQP4-M23. AQP1, an AQP4 homolog that does not form OAPs, was induced to form OAPs upon replacement of its N-terminal domain with that of AQP4-M23. Our results indicate that OAP formation by AQP4-M23 is stabilized by hydrophobic intermolecular interactions involving N-terminus residues, and that absence of OAPs in AQP4-M1 results from non-selective blocking of this interaction by seven residues just upstream from Met23.
Keywords: AQP4, OAP, Water channel, Water permeability, Single particle tracking
Introduction
Orthogonal arrays of particles (OAPs) are regular, square arrays of intramembrane particles that have been visualized by freeze-fracture electron microscopy (FFEM) in cell membranes in brain (Landis and Reese, 1974; Rash et al., 1974), kidney (Orci et al., 1981; Verbavatz et al., 1994), skeletal muscle (Ellisman et al., 1976; Hatton et al., 1987) and other tissues (reviewed by Wolburg, 1995). The biological significance of these unique structures has long been the subject of speculation, because their appearance has been correlated with various neuromuscular diseases (Hatton and Ellisman, 1982; Schotland et al., 1981). We first speculated that the water channel aquaporin-4 (AQP4) is the major constituent of OAPs based on its expression in several cell types known to exhibit array structures (Frigeri et al., 1995), which was confirmed by demonstrating OAP formation in AQP4-transfected cells (Yang et al., 1996) and absence of OAPs in tissues from knockout mice lacking AQP4 (Verbavatz et al., 1997). Label-fracture electron microscopy confirmed that OAPs contain AQP4 (Rash et al., 1998). Since AQP4 functions as a water-transporting protein in glial cell foot processes, it has been suggested that OAPs might enhance water movement through AQP4 pores (Silberstein et al., 2004; Van Hoek et al., 2000; Yang et al., 1997) and maintain AQP4 polarization in foot processes (Amiry-Moghaddam et al., 2004).
The AQP4 transcript contains alternative translation initiation sites (Hasegawa et al., 1994), yielding a full-length `long' (M1) isoform (AQP4-M1) of ∼34 kDa, and a `short' (M23) isoform (AQP4-M23) of ∼31 kDa (Jung et al., 1994; Lu et al., 1996). Both AQP4 isoforms function as water channels, although their relative abundances are tissue specific, with brain expressing both isoforms, and kidney, skeletal muscle, stomach and lung expressing predominantly AQP4-M23 (Frigeri et al., 1995; Hatton et al., 1987; Neely et al., 1999). A recent study of the rat brain genome suggested four additional AQP4 isoforms, one of which is a 39 kDa functional water channel containing an additional 41 residues at the AQP4-M1 N-terminus, which was detected at very low levels by western blot in rat brain (Moe et al., 2008).
FFEM in transfected cells has shown that AQP4-M23 assembles into large OAPs of >100 particles, whereas AQP4-M1 is predominantly dispersed, forming few, if any, small arrays. In primary astrocytes, and in cells cotransfected with AQP1-M1 and AQP4-M23, OAPs are considerably smaller on average than those in cells transfected with only AQP4-M23 (Furman et al., 2003; Silberstein et al., 2004), indicating an interaction between these predominant AQP4 isoforms that probably limits the size to which arrays assemble in vivo. The specific nature and sites of intermolecular interactions responsible for OAP formation and disruption is currently the subject of intense speculation and study.
We recently developed fluorescence methods to visualize OAPs in live cells (Crane et al., 2008). Diffusion of individual AQP4 molecules, labeled with quantum dots at an engineered external epitope, was measured by single-particle tracking. AQP4-M1 diffused freely and rapidly on cell plasma membranes, whereas AQP4-M23 diffusion was highly restricted because of OAP formation. Quantitative analysis of single-molecule trajectories allowed determination of the fraction of AQP4 molecules in OAPs and their diffusional characteristics. Direct visualization of OAPs in cell membranes was accomplished using total internal reflection fluorescence microscopy in which AQP4 was labeled heavily with a small fluorophore and cluster intensity taken as a measure of OAP size.
The purpose of this study was to establish, using live-cell imaging, the molecular determinants of OAP assembly in cell membranes. To isolate the role of specific domains within AQP4 isoforms, we studied the diffusive behavior of a large number of AQP4 mutants individually transfected into COS-7 cells. We found that OAP formation in AQP4-M23 requires a hydrophobic interaction involving specific residues just downstream of Met23, whereas residues in AQP4-M1 upstream of Met23 prevent OAP formation by a nonspecific blocking interaction.
Results
Visualizing AQP4 OAPs in cell membranes
We used single-particle tracking (SPT) of quantum-dot (Qdot)-labeled AQP4 to distinguish between AQP4 molecules in rapidly diffusing individual tetramers and those that are immobilized by accumulation in OAPs (Fig. 1A). Qdots were attached via antibody binding to an engineered Myc epitope in the second extracellular loop of AQP4 (Fig. 1B). This insertion does not inhibit AQP4 trafficking or water-channel function, nor does it prevent AQP4 OAP formation (Crane et al., 2008). Trajectories from AQP4-M1 and AQP4-M23 acquired at 91 Hz over 6 seconds show very different behavior (Fig. 1A, right). AQP4-M1 diffusion is largely Brownian, with an average diffusion coefficient D1-3 ∼5×10–10 cm2/second, whereas the diffusion of AQP4-M23 is highly restricted. The MSD versus time plot computed from many Qdot trajectories for AQP4-M23 shows the characteristic negative curvature expected for confined or anomalous diffusion (Fig. 1C), with the average diffusion coefficient and range at 1 second being an order of magnitude lower than that of AQP4-M1 (Fig. 1C,D). We also used TIRFM to directly visualize the distribution of epitope-labeled AQP4 in cell membranes (Fig. 1E). For TIRFM imaging, 20-fold higher primary antibody and 100-fold higher secondary antibody concentrations compared with those used for SPT were used to heavily stain AQP4 with Alexa Fluor 555. Cells were fixed prior to labeling to prevent AQP4 clustering due to crosslinking that occurs with high concentrations of antibodies. Transfected AQP4-M1 and AQP4-M23 was seen in all regions of the cell plasma membrane, with AQP4-M23 clustered in distinct puncta of diffraction-limited size and AQP4-M1 relatively uniformly distributed over the cell surface (Fig. 1E).
Fig. 1.

Membrane assembly and diffusional mobility of M1 and M23 isoforms of AQP4. (A) Schematic showing the organization of AQP4 tetramers (left) and representative single particle trajectories (right) of Qdot-labeled AQP4 molecules in the plasma membranes of COS-7 cells expressing AQP4-M1 (top) or AQP4-M23 (bottom). Each grey cylinder represents one AQP4 tetramer. A subset of AQP4 molecules are labeled with quantum dots (red) for single particle tracking. (B) AQP4 sequence and topology. Black indicates Met1 and Met23 translation initiation sites; orange, engineered Myc site; blue, residues where single mutations had no effect on OAP formation or disruption; red, residues where single mutations significantly disrupted OAPs; pink, residues where single mutations mildly disrupted OAPs; yellow, residues where mutation produced loss of plasma membrane expression; and green, C-terminal PDZ-binding domains. Horizontal lines indicate sites of C-terminal truncations. (C) Combined MSD vs time plots and averaged diffusion coefficients for AQP4-M1 (grey) and AQP4-M23 (black) in COS-7 cells. (D) Cumulative probability distribution of ranges at 1 second [P(range)] for AQP4-M1 (grey) and AQP4-M23 (black), with dashed lines indicating median range. (E) TIRF micrographs of Alexa Flour 555-labeled AQP4-M1 and AQP4-M23 in COS-7 cells. Inset shows expanded 4×4 μm area. Scale bar: 10 μm.
AQP4 deletion mutants show remarkably altered ability to form OAPs
We made serial deletion mutants to investigate whether specific residues in AQP4-M1 between Met1 and Met23 are responsible for preventing OAP formation. The deletion mutants were named for the position of the N-terminal methionine relative to Met1 in full-length AQP4-M1. Deletion of up to 16 residues from the N-terminus (M1-M16) had little effect on AQP4 diffusion (Fig. 2A), indicating the presence of few, if any, OAPs. Deletion of Cys17, however, resulted in a large fraction of AQP4 with restricted diffusion (e.g. supplementary material Movie 1). The estimated percentage of AQP4 mutants in OAPs, based on the 95th percentile of ranges for AQP4-M23 at 1 second (dashed lines in Fig. 2A,B,C) (see Materials and Methods) is summarized in Fig. 2D. Using this criterion, 42-48% of M17 were present in OAPs. Further deletions increased the percentage of AQP4 in OAPs, with 84-90% for M20 and 92-96% for M21 (Fig. 2A,D). These data are in general agreement with a recent FFEM study of similar deletion mutants (Suzuki et al., 2008), except that the authors found an unexplained decrease in OAP formation for mutants M18 and M19, whereas here we found a continuously increasing fraction of AQP4 in OAPs between M17 and AQP4-M23 (Fig. 2D). Truncations downstream of Met23 produced progressive loss of OAPs from M25 to M27 (Fig. 2B,D), although further truncations resulted in a loss of membrane expression.
Fig. 2.

N-terminal deletion mutants of AQP4 show altered ability to form OAPs. (A) P(range) for indicated AQP4 truncation mutants upstream of Met23: M16 (red), M17 (green), M18 (dark blue), M19 (light blue), M20 (orange), M21 (purple). (B) P(range) for AQP4 truncations downstream from Met23: M24 (red), M25 (green), M26 (blue), M27 (orange). (C) P(range) for alanine mutant M23-R108A (blue) and C-terminal deletion mutants: M23Δ6 (red), M23Δ71 (green), M1Δ6 (purple), M1Δ71 (orange). P(range) for AQP4-M23 (black) and AQP4-M1 (grey) are shown in A-C for reference. Dashed line indicates 95th percentile of the range of AQP4-M23. (D) Estimated percentage of indicated AQP4 mutants in OAPs based on the 95th percentile of range for AQP4-M23. (E) TIRF micrographs of COS-7 cells transfected with AQP4-M1, AQP4-M23, and N-terminal deletion mutants M16, M17 and M20. Each image is a 5×5 μm square. Scale bar: 2 μm. (F) BN-PAGE (top) and SDS-PAGE (bottom) immunoblots of cell lysates from COS-7 cells transfected with AQP4-M23, AQP4-M1 and N-terminal deletion mutants M16, M17 and M20. Molecular size markers are indicated to the right.
AQP4 also contains a 73 residue cytoplasmic C-terminal domain (Fig. 1B). Deletion of up to 71 residues from the C-terminus did not affect the diffusion of AQP4-M1 or of AQP4-M23 (Fig. 2C,D). Deletion or mutation of residue 72 from the C-terminus (Phe252) led to a loss of plasma membrane expression. From crystal contacts in the electron crystallographic structure of AQP4, Hiroaki and colleagues hypothesized that a salt bridge between Arg108 and Tyr250 is primarily responsible for OAP formation (Hiroaki et al., 2006). To test this, we generated alanine mutants at these residues. We found that the diffusion of M23-R108A was identical to that of AQP4-M23 (Fig. 2C,D), whereas M23-Y250A did not express at the plasma membrane. Therefore, the subunit-subunit contacts observed in 2D AQP4 crystals are not the same interactions responsible for OAP formation in live-cell membranes.
As independent confirmation that the observed changes in diffusion of deletion mutants between M16 and AQP4-M23 were due to OAP formation, we performed TIRFM imaging on heavily labeled fixed COS-7 cells, and Blue-Native polyacrylamide gel electrophoresis (BN-PAGE) on lysates from COS-7 cells. Fig. 2E shows 5×5 μm TIRF micrographs. Nearly all of the fluorescence from AQP4-M23 came from distinct, diffraction-limited puncta, corresponding to dense OAPs, whereas the fluorescence from AQP4-M1 was fairly uniform over the cell surface. For BN-PAGE, we used non-denaturing conditions as recently reported (Sorbo et al., 2008) to separate AQP4 tetramers from higher-order AQP4 complexes. An immunoblot with anti-Myc primary antibody in Fig. 2F showed a diffuse band migrating at >1200 kDa for AQP4-M23, indicating large AQP4 complexes, along with a smaller band at ∼300 kDa, corresponding to AQP4 tetramers. AQP4-M1 showed a dense tetramer band and a faint band at ∼600 kDa, but no very high molecular mass band. Like AQP4-M1, TIRFM of M16 in the plasma membrane lacked distinct diffraction-limited spots, and BN-PAGE showed primarily a dense tetramer band. For M17 and M20, where diffusion began to resemble that of AQP4-M23, BN-PAGE yielded several prominent bands above that correspond to AQP4 tetramers, indicating higher-order AQP4 structures (Fig. 2F). Likewise, by TIRFM, diffraction-limited puncta became visible in M17-transfected cells, and were more prominent with M20 (Fig. 2E). No bands migrating faster than the AQP4 tetramer were visible for any species following BN-PAGE. SDS-PAGE confirmed the molecular identity of proteins visualized in the native western blot (Fig. 2F).
Molecular determinants of prevention of OAP formation in AQP4-M1
To explore the mechanism by which the seven residues upstream of Met23 appear to disrupt OAP formation, we measured the diffusion of a series of AQP4-M1 point mutants. Alanine substitutions at Cys17, Ser18, Arg19, Glu20, individually, or at all four positions together, did not affect AQP4-M1 diffusion (Fig. 3A,B). The recent study by Suzuki and co-workers (Suzuki et al., 2008) mentioned above reported that palmitoylation of Cys13 or Cys17 is required for disruption of OAPs. We tested this conclusion by measuring diffusion of the double mutant in which both cysteines were changed to alanines (M1-C13A/C17A), which cannot undergo palmitoylation. In contrast to the conclusion of these authors, we found no reduction in diffusion, indicating that palmitoylation is not required for OAP disruption (Fig. 3A,B). To further delineate the requirements for OAP disruption, we constructed mutants in which polyalanine or polyglutamine appendages of various lengths were added to the N-terminus of AQP4-M23 (Fig. 3C). Unexpectedly, the polyalanine appendages were effective at disrupting OAPs, with MA6M23 having identical diffusion to M16 and AQP4-M1 (Fig. 3D,E). However, a polyglutamine appendage (MQ6M23) only reduced OAP content by ∼10% compared with that of AQP4-M23. We also constructed a mutant (MVAFKGVM23), in which the first seven residues of AQP4-M23 were repeated at the N-terminus (Fig. 3C). This `repeat' mutant, which had the same length as M16 and MA6M23, also showed complete disruption of OAP formation (Fig. 3D,E). OAP disruption thus occurs independently of palmitoylation, and does not depend on specific AQP4 sequence elements of residues upstream of Met23, with nonpolar residues more effective at disruption than polar residues.
Fig. 3.

Disruption of OAPs by residues upstream of Met23 is not sequence specific. (A) P(range) for alanine mutations in AQP4-M1: M1-C13A/C17A (red), M1-C17A (green), M1-S18A (dark blue), M1-R19A (light blue), M1-E20A (orange), AQP4-M23 (black) and AQP4-M1 (grey). (B) Estimated percentage of AQP4-M1 alanine mutants in OAPs. (C) N-terminal sequences of selected AQP4-M23 mutants containing polypeptide additions upstream of Met23. (D) P(range) resulting from polypeptide additions upstream of Met23: MA2M23 (red), MA4M23 (green), MA6M23 (dark blue), MQ6M23 (purple), MVAFKGVM23 (orange), M16 (light blue), AQP4-M23 (black) and AQP4-M1 (grey). (E) Estimated percentage of indicated polypeptide addition mutants of AQP4-M23 in OAPs.
Molecular determinants of OAP formation in AQP4-M23
From the results above, we postulated that the AQP4 residues just upstream of Met23 disrupt OAPs in AQP4-M1 by interfering with interactions involving residues just downstream of Met23. Fig. 4A shows the first 25 residues of AQP4-M23 plotted on a Kyte-Doolittle hydropathy scale (Kyte and Doolittle, 1982), which is a useful estimate of the propensity for domains to form hydrophobic peptide-peptide interactions or to partition into the membrane. As is commonly done to compensate for effects of nearby residues on local hydropathy, the value plotted at each position in Fig. 4A is a running sum of five individual values from the Kyte-Doolittle scale, centered at the named residue. The plot shows that the N-terminus of AQP4-M23 is highly hydrophobic, with progressively decreasing hydropathy moving inward toward the beginning of the first transmembrane helix at Gln32 (Hiroaki et al., 2006).
Fig. 4.

N-terminus residues just downstream of Met23 are responsible for AQP4-M23 OAP formation. (A) Kyte-Doolittle hydropathy plot of the first 25 residues of AQP4-M23 using a 5-residue running sum. The dashed line indicates the start of the first transmembrane helix according to the published crystal structure (Hiroaki et al., 2006). * indicates aromatic residues. (B) P(range) for alanine mutations in AQP4-M23: M23-V24A (red), M23-F26A (green), M23-V24A/F26A (orange), M23-K27A (dark blue), M23-G28A (pink), M23-V29A (light blue), M23-W30A (purple), M23-T31A (yellow), M23-Q32A (dark green). (C) P(range) for glutamine point mutations of hydrophobic and aromatic residues: M23-V24Q (red), M23-A25Q (green), M23-F26Q (dark blue), M23-V24Q/A25Q/F26Q (orange), M23-W30Q (purple) and M23-A33Q (light blue). (D) Estimated percentage of indicated AQP4-M23 point mutants in OAPs. (E) P(range) following mutations of AQP4-M23 at Ala25: M23-A25L (red), M23-A25S (blue), M23-A25Q (green). (F) P(range) following mutations of AQP4-M23 at Phe26: M23-F26L (red), M23-F26A (green), M23-F26Y (purple), M23-F26Q (dark blue), M23-F26E (light blue), M23-F26K (orange). (G) P(range) following proline mutations in AQP4-M23: M23-K27P (green), M23-K27A (blue), M23-G28P (red), M23-G28A (pink), with estimated percentage of proline mutants in OAPs given below. P(range) for AQP4-M23 (black) and AQP4-M1 (grey) are shown in B, C and E-G for reference.
We first constructed a series of single alanine mutations between Val24 and Phe34 (Fig. 4B,D). Of these single mutations, alanine substitution at Phe26 had the greatest effect on diffusion, with only 66-73% of M23-F26A incorporating in OAPs. Although a single alanine mutation at Val24 (M23-V24A) had no effect, a double mutant (M23-V24A/F26A) was less efficient at OAP formation (47-57% in OAPs) than M23-F26A. Alanine substitution for the charged polar residue Lys27 had no disruptive effect. OAP formation was only slightly reduced in M23-V29A and M23-W30A (76-90% in OAPs). M23-G28A, M23-T31A and M23-Q32A behaved similarly to AQP4-M23, whereas M23-F34A was not expressed at the plasma membrane. Results from this initial set of mutations suggested that hydrophobic and aromatic residues downstream of Met23 are involved in OAP formation.
We next generated a series of more severe glutamine mutations on all hydrophobic and aromatic residues in this region (Fig. 4C,D). Glutamine substitutions of Ala25 or Phe26 resulted in a very low OAP content (14-25% in OAPs), whereas glutamine substitution of Val24 was less disruptive (39-45% in OAPs). A triple mutant M23-V24Q/A25Q/F26Q behaved similarly to the single mutants M23-A25Q or M23-F26Q. These data suggest that a hydrophobic interaction involving N-terminal residues is important for OAP formation, and that this interaction might occur very near to the cytoplasmic surface of the plasma membrane. Because mutations at Ala25 and Phe26 had the largest effect on OAP formation of the residues tested, we studied additional mutants of these residues (Fig. 4E,F). Surprisingly, mutation of Ala25 to a hydrophobic leucine (M23-A25L), or conservatively reducing hydrophobicity by addition of a hydroxyl group (M23-A25S), both led to a reduction in OAP content (64-77% in OAPs). Leucine substitution of Phe26 (M23-F26L) had no effect, whereas addition of a hydroxyl group at this position (M23-F26Y) resulted in even more restricted diffusion than AQP4-M23, suggesting a more rigid OAP structure (supplementary material Movie 2). Charged residues glutamate (M23-F26E) and lysine (M23-F26K) were effective at reducing OAP content, but neither was more disruptive than the neutral polar residue glutamine at the same position (Fig. 4F).
We hypothesized that the efficient intermolecular interactions producing OAPs might require the correct positioning of the hydrophobic N-terminus of AQP4-M23 relative to a hydrophobic binding pocket in adjacent AQP4 tetramers. A proline substitution just downstream from the N-terminus of AQP4-M23 might thus prevent OAP forming interactions by disrupting the secondary structure of this domain or by introducing a rigid kink that prevents required hydrophobic interactions. We targeted residues Lys27 and Gly28 for proline substitution because these residues are removed from the direct hydrophobic interactions responsible for OAP formation owing to their low hydropathy, and because alanine substitution at these positions did not prevent OAP formation (Fig. 4B). Fig. 4G shows that proline substitution at Lys27 or Gly28 produced complete disruption of OAP formation, with diffusion identical to that of AQP4-M1. Therefore, mutations altering N-terminus structure are even more effective at OAP prevention than mutations that greatly reduce hydropathy.
To test the prediction that interactions in the N-terminal domain of AQP4-M23 are wholly responsible for OAP formation, we modified AQP1, a homolog that does not form OAPs, to contain putative OAP-forming elements from the AQP4 N-terminus. Sequence alignment of AQP4 and AQP1 indicated no similarity in the short N-terminal cytoplasmic domains of the proteins, whereas the transmembrane regions have high sequence identity (de Groot et al., 2001; Hiroaki et al., 2006) (Fig. 5A). Therefore, in the first AQP4-AQP1 chimera (AQP1ch1), we replaced Met1-Lys8 in AQP1 with Met23-Thr31 of AQP4-M23, completely substituting the cytoplasmic domain but leaving the first transmembrane helix of AQP1 intact (Fig. 5B). As a template, we used an AQP1 construct that has been previously described (Crane and Verkman, 2008; Lu et al., 2000), which contains a Myc epitope in the same extracellular location as all of the AQP4 constructs. AQP1 diffusion in COS-7 cells is mostly Brownian and even faster than AQP4-M1, with D1-3 ∼8×10–10 cm2/second (Crane and Verkman, 2008). Replacement of the N-terminal domain of AQP1 with that of AQP4-M23 produced a dramatic reduction in diffusion (Fig. 5C; supplementary material Movie 3), with 76-89% of AQP1ch1 exhibiting OAP-like behavior (Fig. 5D). Plasma membrane expression of AQP1ch1 was lower than that of most AQP4 constructs, precluding OAP visualization by TIRFM or FFEM. To learn more about the minimum requirements for OAP formation, three additional AQP4-AQP1 chimeras were constructed. AQP1ch2 has the same overall length as AQP1ch1, but instead of replacing the entire N-terminal domain of AQP1, only the first three residues (Met-Ala-Ser) were replaced with the first four residues (Met-Val-Ala-Phe) of AQP4-M23. These four residues compose the hydrophobic domain where mutations or deletions in AQP4-M23 led to significant increases in diffusion (Fig. 4) (P<0.01). Diffusion of AQP1ch2 was comparable to that of unmodified AQP1 (Fig. 5C,D), with D1-3 ∼6×10–10 cm2/second, indicating that the four N-terminal residues of AQP4-M23 are not sufficient for OAP formation. In AQP1ch3, the whole nine-residue N-terminal domain of AQP4-M23 was again added, but shifted further toward the N-terminus of AQP1, replacing only Met1 through Glu4 instead of all eight N-terminal residues of AQP1. In AQP1ch4, the same nine AQP4 residues were again shifted all the way to the N-terminal end of AQP1 (Fig. 5B). Both AQP1ch3 and AQP1ch4 showed slower diffusion than native AQP1 and AQP1ch2, but did not exhibit the restricted diffusion seen in AQP1ch1 (Fig. 5C). Therefore, the position of the nine N-terminal residues of AQP4-M23 relative to transmembrane helix 1 is critical to OAP formation. Finally, we constructed a revertant mutant of AQP1ch1 (ch1-F26Q), analogous to the OAP-disrupting mutant M23-F26Q (Fig. 5B). As in AQP4-M23, mutation of Phe26 to glutamine in AQP1ch1 resulted in significantly increased diffusion (P<0.001), corresponding to an estimated OAP fraction of 26-35% (Fig. 5C,D).
Fig. 5.

AQP1 forms OAPs after replacement of its N-terminal domain with that of AQP4-M23. (A) Sequence alignment of the first 20 residues of AQP4-M23 and the first 18 residues of AQP1. Red and yellow highlight identical and conserved residues, respectively. Blue and yellow rectangles represent the beginning of transmembrane domains of AQP4 and AQP1, respectively, according to published structures. (B) Crystal structure of AQP1 (de Groot et al., 2001) (yellow) shown with the addition of nine N-terminal residues from AQP4-M23 (blue), forming AQP1ch1. Below is a schematic of various AQP4-AQP1 chimeras, showing the relative contributions from AQP4 (blue) and AQP1 (yellow), with numbers indicating the initial and final residues taken from each protein, and chimera names listed to the right. (C) P(range) of AQP4-AQP1 chimeras: AQP1ch1 (red), AQP1ch2 (green), AQP1ch3 (dark blue), AQP1ch4 (light blue), ch1-F26Q (orange), AQP4-M23 (black) and AQP1 (grey). (D) Estimated percentage of indicated AQPs and chimeras in OAPs.
Discussion
The identity and determinants of OAPs have been of continued interest since their discovery in freeze-fracture electron micrographs from brain tissue in the 1970s. Numerous studies have revealed correlations between OAPs and various diseases in brain and skeletal muscle, with the biological role(s) of OAPs remaining the subject of intense speculation. Here, we have exploited changes in the plasma membrane diffusion of AQP4 mutants to establish the molecular determinants of OAP formation. We used a combination of N-terminal deletions and additions, site-directed mutagenesis, and construction of AQP4-AQP1 chimeras to define a key domain necessary for OAP stability. Our data indicate that a single residue is not responsible for tetramer-tetramer interactions leading of OAP formation, but indicate the involvement of several hydrophobic residues in the N-terminus of AQP4-M23. The inability of AQP4-M1 to form OAPs results from non-selective blocking of N-terminal interactions by residues just upstream from Met23.
The diffusive behaviors of the various AQP4 mutants and chimeras were quantified in live cells by single-particle tracking of Qdots bound to an extracellular-facing Myc epitope. This technique was recently established as a reliable readout of OAP formation, association and dynamics (Crane et al., 2008). Comparison of cumulative probability distributions for diffusion of an AQP4 variant to known distributions of natural AQP4 isoforms provided an estimate of AQP4 accumulation in OAPs. Until recently, the only method available to identify OAPs in cell membranes was FFEM. When coupled with immunogold labeling, FFEM has been used to estimate the fraction of natural AQP4 isoforms (Furman et al., 2003) and AQP4 mutants (Suzuki et al., 2008) that associate in OAPs. However, in addition to technical challenges, limitations of FFEM include fixation artifacts, difficulty in obtaining statistically rigorous information about numbers of AQP4 tetramers in OAPs, and difficulty in identifying OAPs in cells expressing low levels of AQPs. FFEM often requires significant enrichment of cell populations or creation of stable AQP-overexpressing cell lines. Qdot labeling of epitope-tagged AQPs and SPT measurement obviates these limitations. Recently, BN-PAGE was demonstrated as an alternative to FFEM for OAP identification (Sorbo et al., 2008), which was verified here and used as an independent method to confirm the interpretation of diffusion data in terms of AQP4 supermolecular complexes. However, although BN-PAGE is technically simple, the use of detergents precludes meaningful quantitative determination of the fraction of AQP4 that accumulates in OAPs. As seen in Fig. 2F, even in cells transfected with AQP4-M23, where it is known that nearly all AQP4 is locked in OAPs, a significant tetramer band is visible in the immunoblot. Although relatively non-denaturing, the detergent docecyl maltoside apparently solubilized a fraction of the OAPs, leading to the appearance of a tetramer band when in fact very few individual tetramers are present in intact cell membranes. SPT provides quantitative information on OAP content in real time in live cells, avoiding artifacts of fixation or detergents. It allowed us to quantitatively examine many AQP4 mutant and chimeras more efficiently than is possible with alternative methods, because experiments are rapid, taking only 30-45 minutes from labeling to data acquisition. TIRFM on heavily labeled cells is a relatively low-resolution method when compared with SPT, but its advantage is the ability to visualize intact cells to determine the spatial distribution of labeled proteins throughout the membrane. In combination, these fluorescence techniques provide a powerful tool to elucidate the nature of AQP4 OAPs. It should be noted that the use of external epitopes was crucial in these studies, because additions at either the N- or C-terminus of AQP4 significantly alter its behavior. GFP-AQP1 and GFP-AQP2 chimeras have been used successfully by our lab in the past (Umenishi et al., 2000) and GFP-AQP4 chimeras have been reported in other studies (Zelenina et al., 2002). We attempted to use an N-terminal GFP-AQP4-M23 chimera, but found that it diffused freely in the membrane (our unpublished observations), indicating disruption of OAPs by the GFP moiety. In addition to the N-terminus, as discussed extensively below, a C-terminal PDZ-binding domain is probably involved in targeting or anchoring of OAPs to cytoplasmic binding partners (Amiry-Moghaddam et al., 2004; Crane et al., 2008).
Hydrophobic interactions involving residues Ala25 and Phe26 have great influence on OAP formation. Successively reducing hydropathy by mutating Phe26 to alanine and then glutamine, or mutating Ala25 to serine and glutamine, produced successive reductions in OAP content (Fig. 4). Of course, the effective hydropathy of a residue depends on its environment. It might therefore be more useful to consider the overall hydropathy of the N-terminus. Our data suggest the existence of low and high hydropathy `thresholds' in this region, beyond which further changes have no effect on OAP formation. For example, Ala25 is flanked by Val24 and Phe26, so although it alone is not overly hydrophobic, it resides in a highly hydrophobic region (Fig. 4A). Mutation to a bulky leucine at this position increases the local hydropathy above the effective threshold, and reduces OAP formation probably because of packing defects related to increased steric hindrance. At the other end of the spectrum, the triple glutamine mutant M23-V24Q/A25Q/F26Q was no more disruptive to OAP formation than glutamine mutations of Phe26 or Ala25 individually, indicating that a single mutation at either site lowers the hydropathy of the domain below the effective threshold. Further mutations at Phe26 indicate that hydropathy is not the only determinant of OAP stability. For example, lysine substitution was less effective at OAP disruption than glutamine or glutamate, even thought its hydropathy is similarly low by most scales (Kyte and Doolittle, 1982; White and Wimley, 1999). Additionally, conservative substitution of Phe26 with a more polar tyrosine residue led to an even more rigid, possibly hyperstable OAP (Fig. 4F), suggesting that the bulky aromatic structure at this position is more crucial to OAP stability than hydropathy.
At least seven residues upstream of Met23 are required to completely disrupt OAP formation (Figs 2 and 3). No single mutation in the N-terminal domain of AQP4-M1 could prevent OAP disruption (Fig. 3A,B). We found that a palmitoylation-null mutant (M1-C13A/C17A) did not diffuse differently from wild-type AQP4-M1, indicating that OAP prevention in AQP4-M1 does not require palmitoylation (Fig. 3A). These results do not agree with those recently reported, in which >90% of M1-C13A/C17A was found to be associated with OAPs (Suzuki et al., 2008). We do not know the reasons for the difference in our results and those of Suzuki and co-workers, although we note that other cysteine-free polypeptide additions at the end of AQP4-M23 were also capable of completely preventing OAP formation. Of the numerous polypeptide sequences we tested, only polyglutamine was ineffective at OAP disruption (Fig. 3E). Therefore, the exact sequence of the disrupting domain in AQP4-M1 is not important, provided that it is partially nonpolar. This could explain why sequence conservation in the first 22 amino acids of AQP4-M1 is relatively low (72% identity between rat and human) when compared with the entire AQP4-M23 molecule (96% identity between rat and human). Our evidence suggests that competitive auto-inhibition of hydrophobic interactions disrupts intermolecular contacts near Met23, and is responsible for the different behavior of the M1 and M23 isoforms of AQP4.
Truncation mutations and site-directed mutagenesis of AQP4-M23 suggest that the four most N-terminal residues of AQP4-M23 (Met-Val-Ala-Phe) form a key hydrophobic domain responsible for OAP formation. However, our results indicate that other residues near the N-terminus of AQP4-M23 are also involved. First, proline substitution of hydrophilic residues Lys27 and Gly28 completely prevented OAP formation in AQP4-M23, whereas alanine substitution at the same positions had no effect (Fig. 4). Second, simply adding the four N-terminal residues of AQP4-M23 onto the end of AQP1, a non-OAP-forming homolog of AQP4, was not sufficient to induce AQP1 to form OAPs. We found that replacement of the entire N-terminal cytoplasmic domain of AQP1 (Met1 to Lys8) with the entire N-terminal domain of AQP4-M23 (Met23 to Thr31) was required to produce AQP1 OAPs (Fig. 5). Furthermore, the position of these nine residues relative to the initial residue of the first transmembrane helix was crucial. There are several possible explanations of these observations. An intriguing possibility is that the nine residues in the N-terminal domain of AQP4-M23 form a secondary structural element that is crucial for OAP formation. The four-residue spacing between Trp30 and Phe26 suggests the possibility of a one-turn amphipathic helix anchored in the cytoplasmic face of the membrane by these aromatic residues that favor the lipid-water interface (White and Wimley, 1999). The introduction of a proline residue between Trp30 and Phe26 would certainly prevent the formation of a short helix, and might account for the complete loss of OAP formation. In the published structure of AQP4-M23, the N-terminal domain between Met23 and Gln32 was found to be unstructured (Hiroaki et al., 2006), but the authors used an N-terminal 6×His affinity purification tag in their sample preparation, which might disrupt any secondary structure in this region. In AQP4-M23, the linking domain between Phe26 and the first transmembrane helix (Lys-Gly-Val-Trp-Thr) has a net hydropathy of about zero (Fig. 4A) and contains residues that partition favorably into the membrane interface, likely maintaining the N-terminus in a membrane-associated state where tetramer-tetramer contacts may be optimal. The five residues adjacent to the first transmembrane helix in AQP1 (Glu-Phe-Lys-Lys-Lys) constitute a very polar stretch of peptide, which, in the case of AQP1ch2, might drive the N-terminus out of the membrane interface, away from its ideal position for hydrophobic interaction. Studies with reconstituted model peptides have suggested that `hydrophobic pockets' created by soluble N-terminal domains near the membrane interface may stabilize oligomerization of transmembrane helices (Arluison et al., 2004).
Coalescence of AQP4 tetramers in the plasma membrane to OAPs nearly abolishes their lateral mobility on a time scale of seconds. We have utilized the vastly differing diffusion of AQP4 mutants in OAPs compared diffusion outside OAPs to define, in live cells, the domains involved in OAP formation and disruption. Our data indicate that a hydrophobic intermolecular interaction at the N-terminus of AQP4-M23 occurs near the cytoplasmic membrane interface, leading to OAP formation. This interaction is disrupted completely by residues Cys17 to Ser21 of AQP4-M1; however, the specific sequence of this disrupting domain is not important. Although the most likely explanation from these and previous experiments is that AQP4 tetramers directly interact to form OAPs, we cannot formally exclude the possibility that other proteins are involved in OAP formation and/or disruption. It also remains to be determined whether OAP prevention in AQP4-M1 is caused by a direct block of the hydrophobic binding pocket near Met23, displacement of this pocket from its optimal position, or a change in the required secondary structure at this site.
Materials and Methods
DNA constructs
Plasmids AQP1.T120.myc, AQP4M1.myc and AQP4M23.myc, which encode human AQP1 and rat AQP4 isoforms, respectively and a ten-residue Myc epitope (NH2-EQKLISEEDL-COOH) in their second extracellular loop, were generated previously (Crane et al., 2008; Crane and Verkman, 2008). All AQP4 truncations, point mutants, and AQP4-AQP1 chimeras were generated by standard PCR methods using the above plasmids as templates. All constructs were fully sequenced.
Cell culture and transfections
COS-7 (ATCC CRL-1651) cell cultures were maintained at 37°C in 5% CO2, 95% air in DME-H21 medium containing 10% fetal bovine serum, 100 U/ml penicillin and 100 μg/ml streptomycin. Six hours before transfection, cells were transferred to 12-well plates containing 18-mm-diameter glass coverslips using antibiotic-free medium. Cells were transfected using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to the manufacturer's protocol 24-48 hours before experiments.
Labeling and cell treatment
Prior to labeling of AQPs with quantum dots (Qdots), cells were washed with 3 ml PBS containing 6 mM glucose and 1 mM pyruvate (GP buffer) and incubated for 5 minutes in blocking buffer (GP buffer containing 1% bovine serum albumin), followed by 5 minutes with 70 ng/ml mouse anti-Myc antibody (Covance, Emeryville, CA) in blocking buffer. Cells were then rinsed with GP buffer, incubated for 5 minutes with 0.1 nM goat F(ab′)2 anti-mouse IgG-conjugated Qdot 655 (Invitrogen) in blocking buffer, then rinsed again with GP buffer. Coverslips were transferred to a custom-built perfusion chamber and maintained in GP buffer throughout the experiment. Under these labeling conditions, cells expressing AQP4 or AQP1 showed multiple bright Qdots, whereas non-expressing cells were completely dark with no background.
For the heavy fluorescence labeling used in total internal reflection fluorescence microscopy (TIRFM), cells were fixed for 10 minutes with 4% paraformaldehyde, then rinsed with PBS. Fixed cells were incubated for 30 minutes in blocking buffer, then for 1 hour with 1.4 μg/ml anti-Myc antibody in blocking buffer. Cells were again rinsed with PBS and incubated for 1 hour with 10 μg/ml goat F(ab′)2 anti-mouse IgG-conjugated Alexa Fluor 555 (Invitrogen) in blocking buffer. Cells were then rinsed extensively in PBS, and coverslips were mounted with VectaMount hard-set medium for microscopy (Vector Laboratories, Burlingame, CA).
Microscopy instrumentation and measurements
SPT was performed on a Nikon Eclipse TE2000S inverted epifluorescence microscope (Nikon, Melville, NY) equipped with a Nikon ×100 TIRF oil-immersion objective (numerical aperture 1.45) and a deep-cooled CCD camera (Hamamatsu EM-CCD, Bridgewater, NJ). Qdot fluorescence was excited using a E460SPUV excitation filter and 475DCXRU dichroic mirror, and detected through a D655/40m emission filter (Chroma, Rockingham, VT). Data were acquired continuously at 11 milliseconds per frame (91 Hz) for 6 seconds, at 37°C, within 30 minutes of the final wash step. The spatial resolution of the system, determined from the s.d. of x,y coordinates of immobilized Qdots on a coverslip (Fujiwara et al., 2002), was 18 nm at 91 Hz. We previously demonstrated, using cluster pixel intensity analysis of heavily labeled AQP4-transfected cells, that approximately one primary and secondary antibody sandwich labels each AQP4 tetramer in our system (Crane et al., 2008). This is probably due to steric hindrance preventing multiple antibodies from accessing more than one Myc epitope in a single tetramer. Intermittent blinking to background levels confirmed that only single Qdots were tracked.
TIRFM was done using a Nikon Eclipse TE2000E microscope with a through-objective TIRF attachment and a ×100 TIRF oil-immersion objective (numerical aperture 1.49) mounted on a perfect focus module (Nikon). An argon-ion laser (Spectra Physics, Mountain View, CA) on a custom-built launch was coupled through a fiber-optic to the TIRF module. Alexa Fluor 555-labeled AQP4 was excited using a Z514/10x excitation filter and Z514RDC dichroic mirror, and detected through a D605/40m emission filter (Chroma). Images were acquired using a QuantEM 512SC deep-cooled CCD camera (Photometrics, Tucson, AZ).
SPT analysis
Image sequences were analyzed and trajectories constructed using IDL software (Research Systems, Boulder, CO) with algorithms available as shareware at www.physics.emory.edu/faculty/weeks/idl/. Blinking of individual Qdots was accounted for during trajectory constructions. Trajectories were considered to be continuous if a blinking Qdot was rediscovered within a 4-pixel radius and 20-frame window. The mean squared displacement (MSD) as a function of time 〈r2(t)〉 was constructed for each trajectory, and the diffusion coefficient D1-3 and offset (due to noise) were determined by a linear fit of the first three time steps of the MSD:
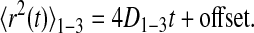 |
(1) |
The offset was subtracted from each point, and the first 25% of the curve (Saxton, 1997) was fitted using a weighted Levenberg-Marquardt nonlinear least-squares fitting algorithm to a combined quadratic, polynomial and exponential function (Haggie et al., 2006) with fitting parameters a1, a2, a3≥0, such that 〈r2(t)〉fit=a1t2+a2[1–exp(–a3t)]. The fit was weighted by the variance in the MSD at each time step.
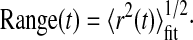 |
(2) |
Diffusion data are primarily reported in the form of the cumulative distribution of ranges at 1 second (Fig. 1D), where P(range) is defined as the probability that a particle's range is less than or equal to a given distance at t=1 second. Only trajectories longer than 200 steps were analyzed, with data sets composed of at least 200 trajectories from at least 15 cells. Statistical significance of differences in the mean values of range was determined using the Student's t-test. Based on the finding that ∼95% of AQP4 is present in OAPs in AQP4-M23-expressing cells (Furman et al., 2003; Suzuki et al., 2008), and a spatial resolution of 18 nm for our SPT system we estimated the percentage of AQP4 in OAPs for the various mutants as P(79±18 nm), the range at which P(range)=0.95 for AQP4-M23. These estimates are presented as bar graphs in Figs 2, 3, 4, 5, with the low and high extremes of the error bars equal to P(61 nm) and P(97 nm), respectively.
Electrophoresis and immunoblot analysis
Transfected cells were lysed with NativePAGE sample buffer containing 1% dodecyl-β-D-maltoside (DDM) for 10 minutes on ice. All materials were purchased from Invitrogen unless specified otherwise. The lysate was centrifuged at 20,000 g for 30 minutes at 4°C and the pellet discarded. Total protein content was determined by Bradford assay. For Blue-Native PAGE, 10 μg protein was mixed with Coomassie G-250 at a DDM:Coomassie ratio of 4:1, loaded onto a NativePAGE 3%-12% Bis-Tris gel along with NativeMARK molecular mass markers, and run with NativePAGE running buffers according to the manufacturer's protocol. For SDS-PAGE, the same cell lysates were denatured with NuPAGE LDS sample buffer and reducing agent, loaded onto a NuPAGE 12% Bis-Tris gel along with SeeBlue Plus2 molecular mass markers, and run with NuPAGE MES SDS running buffer at 100 V. Proteins were blotted onto PVDF membranes. Following transfer, native proteins were fixed by soaking membranes for 15 minutes in 8% acetic acid and destained with methanol. Membranes were blocked with 5% nonfat milk and incubated overnight with 0.7 μg/ml anti-Myc primary antibody at 4°C. Membranes were then rinsed, incubated for 1 hour with horseradish-peroxidase-conjugated goat anti-mouse IgG (Bio-Rad Laboratories, Hercules, CA), rinsed extensively, and labeled proteins were detected with ECL Plus enzymatic chemiluminescence kit (Amersham).
Supplementary Material
This work was supported by NIH grants DK35124, EB00415, HL73856, HL59198, EY13574 and DK72517, and Research Development Program and Drug Discovery grants from the Cystic Fibrosis Foundation. J.M.C. was supported in part by NIH NRSA award GM808512. Deposited in PMC for release after 12 months.
Supplementary material available online at http://jcs.biologists.org/cgi/content/full/122/6/813/DC1
References
- Amiry-Moghaddam, M., Frydenlund, D. S. and Ottersen, O. P. (2004). Anchoring of aquaporin-4 in brain: molecular mechanisms and implications for the physiology and pathophysiology of water transport. Neuroscience 129, 999-1010. [DOI] [PubMed] [Google Scholar]
- Arluison, V., Seguin, J., Le Caer, J. P., Sturgis, J. N. and Robert, B. (2004). Hydrophobic pockets at the membrane interface: an original mechanism for membrane protein interactions. Biochemistry 43, 1276-1282. [DOI] [PubMed] [Google Scholar]
- Crane, J. M. and Verkman, A. S. (2008). Long-range nonanomalous diffusion of quantum dot-labeled aquaporin-1 water channels in the cell plasma membrane. Biophys. J. 94, 702-713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crane, J. M., Van Hoek, A. N., Skach, W. R. and Verkman, A. S. (2008). Aquaporin-4 dynamics in orthogonal arrays in live cells visualized by quantum dot single particle tracking. Mol. Biol. Cell 19, 3369-3378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Groot, B. L., Engel, A. and Grubmuller, H. (2001). A refined structure of human aquaporin-1. FEBS Lett. 504, 206-211. [DOI] [PubMed] [Google Scholar]
- Ellisman, M. H., Rash, J. E., Staehelin, L. A. and Porter, K. R. (1976). Studies of excitable membranes. II. A comparison of specializations at neuromuscular junctions and nonjunctional sarcolemmas of mammalian fast and slow twitch muscle fibers. J. Cell Biol. 68, 752-774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frigeri, A., Gropper, M. A., Umenishi, F., Kawashima, M., Brown, D. and Verkman, A. S. (1995). Localization of MIWC and GLIP water channel homologs in neuromuscular, epithelial and glandular tissues. J. Cell Sci. 108, 2993-3002. [DOI] [PubMed] [Google Scholar]
- Fujiwara, T., Ritchie, K., Murakoshi, H., Jacobson, K. and Kusumi, A. (2002). Phospholipids undergo hop diffusion in compartmentalized cell membrane. J. Cell Biol. 157, 1071-1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furman, C. S., Gorelick-Feldman, D. A., Davidson, K. G., Yasumura, T., Neely, J. D., Agre, P. and Rash, J. E. (2003). Aquaporin-4 square array assembly: opposing actions of M1 and M23 isoforms. Proc. Natl. Acad. Sci. USA 100, 13609-13614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haggie, P., Kim, J., Lukacs, G. and Verkman, A. (2006). Tracking of quantum dot-labeled CFTR shows near immobilization by C-terminal PDZ interactions. Mol. Biol. Cell 17, 4937-4945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa, H., Ma, T., Skach, W., Matthay, M. A. and Verkman, A. S. (1994). Molecular cloning of a mercurial-insensitive water channel expressed in selected water-transporting tissues. J. Biol. Chem. 269, 5497-5500. [PubMed] [Google Scholar]
- Hatton, J. D. and Ellisman, M. H. (1982). The distribution of orthogonal arrays in the freeze-fractured rat median eminence. J. Neurocytol. 11, 335-349. [DOI] [PubMed] [Google Scholar]
- Hatton, J. D., Cox, G. F., Miller, A. L., Nichol, J. A. and Ellisman, M. H. (1987). Identification of polypeptides associated with sarcolemmal vesicles enriched in orthogonal arrays. Biochim. Biophys. Acta 904, 373-380. [DOI] [PubMed] [Google Scholar]
- Hiroaki, Y., Tani, K., Kamegawa, A., Gyobu, N., Nishikawa, K., Suzuki, H., Walz, T., Sasaki, S., Mitsuoka, K., Kimura, K. et al. (2006). Implications of the aquaporin-4 structure on array formation and cell adhesion. J. Mol. Biol. 355, 628-639. [DOI] [PubMed] [Google Scholar]
- Jung, J. S., Bhat, R. V., Preston, G. M., Guggino, W. B., Baraban, J. M. and Agre, P. (1994). Molecular characterization of an aquaporin cDNA from brain: candidate osmoreceptor and regulator of water balance. Proc. Natl. Acad. Sci. USA 91, 13052-13056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyte, J. and Doolittle, R. F. (1982). A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 157, 105-132. [DOI] [PubMed] [Google Scholar]
- Landis, D. M. and Reese, T. S. (1974). Arrays of particles in freeze-fractured astrocytic membranes. J. Cell Biol. 60, 316-320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, M., Lee, M. D., Smith, B. L., Jung, J. S., Agre, P., Verdijk, M. A., Merkx, G., Rijss, J. P. and Deen, P. M. (1996). The human AQP4 gene: definition of the locus encoding two water channel polypeptides in brain. Proc. Natl. Acad. Sci. USA 93, 10908-10912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, Y., Turnbull, I. R., Bragin, A., Carveth, K., Verkman, A. S. and Skach, W. R. (2000). Reorientation of aquaporin-1 topology during maturation in the endoplasmic reticulum. Mol. Biol. Cell 11, 2973-2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moe, S. E., Sorbo, J. G., Sogaard, R., Zeuthen, T., Petter Ottersen, O. and Holen, T. (2008). New isoforms of rat Aquaporin-4. Genomics 91, 367-377. [DOI] [PubMed] [Google Scholar]
- Neely, J. D., Christensen, B. M., Nielsen, S. and Agre, P. (1999). Heterotetrameric composition of aquaporin-4 water channels. Biochemistry 38, 11156-11163. [DOI] [PubMed] [Google Scholar]
- Orci, L., Humbert, F., Brown, D. and Perrelet, A. (1981). Membrane ultrastructure in urinary tubules. Int. Rev. Cytol. 73, 183-242. [DOI] [PubMed] [Google Scholar]
- Rash, J. E., Staehelin, L. A. and Ellisman, M. H. (1974). Rectangular arrays of particles on freeze-cleaved plasma membranes are not gap junctions. Exp. Cell Res. 86, 187-190. [DOI] [PubMed] [Google Scholar]
- Rash, J. E., Yasumura, T., Hudson, C. S., Agre, P. and Nielsen, S. (1998). Direct immunogold labeling of aquaporin-4 in square arrays of astrocyte and ependymocyte plasma membranes in rat brain and spinal cord. Proc. Natl. Acad. Sci. USA 95, 11981-11986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxton, M. (1997). Single-particle tracking: the distribution of diffusion coefficients. Biophys. J. 72, 1744-1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schotland, D. L., Bonilla, E. and Wakayama, Y. (1981). Freeze fracture studies of muscle plasma membrane in human muscular dystrophy. Acta Neuropathol 54, 189-197. [DOI] [PubMed] [Google Scholar]
- Silberstein, C., Bouley, R., Huang, Y., Fang, P., Pastor-Soler, N., Brown, D. and Van Hoek, A. N. (2004). Membrane organization and function of M1 and M23 isoforms of aquaporin-4 in epithelial cells. Am. J. Physiol. Renal Physiol. 287, F501-F511. [DOI] [PubMed] [Google Scholar]
- Sorbo, J. G., Moe, S. E., Ottersen, O. P. and Holen, T. (2008). The molecular composition of square arrays. Biochemistry 47, 2631-2637. [DOI] [PubMed] [Google Scholar]
- Suzuki, H., Nishikawa, K., Hiroaki, Y. and Fujiyoshi, Y. (2008). Formation of aquaporin-4 arrays is inhibited by palmitoylation of N-terminal cysteine residues. Biochim. Biophys. Acta 1778, 1181-1189. [DOI] [PubMed] [Google Scholar]
- Umenishi, F., Verbavatz, J. M. and Verkman, A. S. (2000). cAMP regulated membrane diffusion of a green fluorescent protein-aquaporin 2 chimera. Biophys. J. 78, 1024-1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Hoek, A. N., Ma, T., Yang, B., Verkman, A. S. and Brown, D. (2000). Aquaporin-4 is expressed in basolateral membranes of proximal tubule S3 segments in mouse kidney. Am. J. Physiol. Renal Physiol. 278, F310-F316. [DOI] [PubMed] [Google Scholar]
- Verbavatz, J. M., Van Hoek, A. N., Ma, T., Sabolic, I., Valenti, G., Ellisman, M. H., Ausiello, D. A., Verkman, A. S. and Brown, D. (1994). A 28 kDa sarcolemmal antigen in kidney principal cell basolateral membranes: relationship to orthogonal arrays and MIP26. J. Cell Sci. 107, 1083-1094. [DOI] [PubMed] [Google Scholar]
- Verbavatz, J. M., Ma, T., Gobin, R. and Verkman, A. S. (1997). Absence of orthogonal arrays in kidney, brain and muscle from transgenic knockout mice lacking water channel aquaporin-4. J. Cell Sci. 110, 2855-2860. [DOI] [PubMed] [Google Scholar]
- White, S. H. and Wimley, W. C. (1999). Membrane protein folding and stability: physical principles. Annu. Rev. Biophys. Biomol. Struct. 28, 319-365. [DOI] [PubMed] [Google Scholar]
- Wolburg, H. (1995). Orthogonal arrays of intramembranous particles: a review with special reference to astrocytes. J. Hirnforsch. 36, 239-258. [PubMed] [Google Scholar]
- Yang, B., Brown, D. and Verkman, A. S. (1996). The mercurial insensitive water channel (AQP-4) forms orthogonal arrays in stably transfected Chinese hamster ovary cells. J. Biol. Chem. 271, 4577-4580. [PubMed] [Google Scholar]
- Yang, B., van Hoek, A. N. and Verkman, A. S. (1997). Very high single channel water permeability of aquaporin-4 in baculovirus-infected insect cells and liposomes reconstituted with purified aquaporin-4. Biochemistry 36, 7625-7632. [DOI] [PubMed] [Google Scholar]
- Zelenina, M., Zelenin, S., Bondar, A. A., Brismar, H. and Aperia, A. (2002). Water permeability of aquaporin-4 is decreased by protein kinase C and dopamine. Am. J. Physiol. Renal Physiol. 283, F309-F318. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.