

Abstract
Atrial fibrillation (AF) is a growing clinical problem, increasing in prevalence as the population of the United States and countries around the world ages. Intensive research aimed at improving prevention, diagnosis, and treatment of AF is ongoing. Although the use and efficacy of catheter ablation‐based approaches in AF treatment have increased significantly in the last decade, pharmacological agents remain the first‐line therapy for rhythm management of AF. Currently available anti‐AF agents are generally only moderately effective and associated with extracardiac toxicity and/or a risk for development of life‐threatening ventricular arrhythmias. Included among current investigational strategies for improving the effectiveness and safety of anti‐AF drugs is the development of (1) Agents that produce atrial‐specific or predominant inhibition of IKur, IK‐ACh, or INa; (2) “Upstream therapies” that effect nonion channel targets that reduce atrial structural remodeling, hypertrophy, dilatation, inflammation, oxidative injury, etc; (3) Derivatives of “old” anti‐AF drugs with an improved safety pharmacological profile; and (4) Gap junction therapy aimed at improving conduction without affecting sodium channels. This review focuses on new pharmacological approaches under investigation for the treatment of AF.
Keywords: antiarrhythmic drugs, pharmacology, cardiac arrhythmias, electrophysiology
INTRODUCTION
Atrial fibrillation (AF) is a major clinical problem with increasing prevalence due to the progressive increase in longevity. The two principal options for the management of AF are rhythm and rate control. The first option aims to maintain sinus rhythm; with its restoration when required (pharmacologically, surgically, or with direct current or catheter oblation). The second option leaves the atria fibrillating and focuses on reducing the detrimental effects of fibrillating atria on the ventricles (such as the development of cardiomyopathy) by prolonging the effective refractory period of impulse transmission through the atrioventricular (AV) node or by completely interrupting conduction through AV node. This may be accomplished either pharmacologically or with catheter ablation techniques.
Rate control and in some cases rhythm control approaches require anticoagulation therapy to reduce the risk of stroke. It is generally accepted that rhythm control is not superior to rate control in terms of survival and that rhythm control involving drugs may be complicated by adverse reactions and a greater rate of hospitalization. 1 , 2 The general consensus however is that rhythm control would be preferable for most AF patients if safer and more effective anti‐AF drugs were available. 3 , 4 , 5 This has prompted the search for such agents.
Although the effectiveness and use of catheter ablation techniques for the management of AF has increased importantly over the past decade, pharmacological agents remain first‐line therapy for rhythm control AF. 6 Currently available anti‐AF agents are in general only moderately effective and associated with a risk for induction of serious ventricular arrhythmias and/or organ toxicity. Agents that inhibit the early sodium current (INa) such as flecainide and propafenone have proven to be effective in terminating paroxysmal episodes of AF, but far less effective in dealing with persistent AF. 7 Because of a proclivity for arrhythmogenesis, these agents are contraindicated in patients with acute coronary syndrome and structural heart disease, which account for the majority of AF patients. 6 Agents that as a primary action inhibit the rapidly activating delayed rectified potassium current (IKr), such as dofetilide, also effectively terminate paroxysmal AF and less effectively persistent AF, but these drugs also cause acquired long QT syndrome (LQTS) and may be associated with the development of torsade de pointes (TdP) arrhythmias. The success rate for terminating persistent AF is greater for IKr blockers than for INa blockers. 6
Amiodarone, a mixed ion channel blocker, is widely used for the long‐term maintenance of sinus rhythm rather than for acute AF conversion. 8 The drug takes weeks to achieve its full effects on cardiac electrophysiological parameters. Advantages of amiodarone include the fact that it can be safely used in patients with structural heart disease and very rarely is associated with ventricular proarrhythmia. A major disadvantage of long‐term use of amiodarone is the relatively high rate of multiple organ toxicity.
Accordingly, there is a need for safer and more effective anti‐AF agents than those currently available. Several pharmacological strategies aimed at improving the effectiveness and safety of drugs used for rate control of AF have been proposed and tested in clinical and/or experimental settings in recent years (Fig. 1). This brief review provides an update of the present‐day view of these pharmacological approaches for the management of AF.
Figure 1.
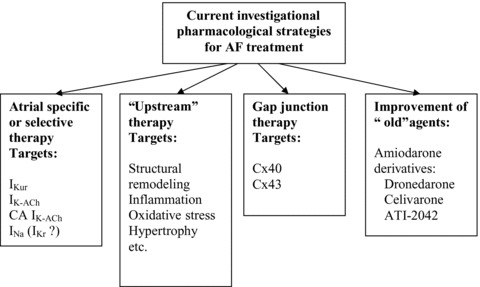
Current investigational strategies for rhythm control of atrial fibrillation.
Atrial‐Specific Ion Channel Block Approaches
A great deal of focus has been placed on the development of atrial‐specific ion channel blockers, in an effort to avoid the ventricular arrhythmogenic effects of currently available drugs. Atrial‐specific targets for AF treatment include the ultrarapid delayed rectified potassium current (IKur), the acetylcholine‐regulated inward rectifying potassium current (IK‐ACh), the constitutively active IK‐ACh (i.e., which does not require acetylcholine or muscarinic receptors for activation), and connexin 40 (Cx40). 9 , 10 The channels responsible for IKur and IK‐ACh are exclusively or nearly exclusively present in atria and largely absent in the ventricles and these channels are commonly referred to as atrial‐specific.
“IKur block for AF” is the most investigated strategy among the atrial‐specific approaches. Agents capable of blocking IKur (such as AVE0118, AVE1231, S9947, S20951, ISQ‐1, DPO‐1, vernakalant; AZD7009; NIP141, NIP‐142, acacetin) have been shown to selectively prolong atrial‐effective refractory period (ERP) and thus to effectively terminate AF and/or prevent its induction. 11 , 12 , 13 , 14 , 15 , 16 , 17 , 18 , 19 , 20 , 21 Most of these agents, however, at concentrations that effectively suppress AF, potently block other currents as well (e.g., INa is inhibited by vernakalant and AZD7009). 22 , 23 In fact, it is not clear if IKur or INa plays a greater role in the atrial selectivity and anti‐AF actions of these agents, since INa blockers may selectively prolong atrial ERP and effectively suppress AF. 24 An inhibition of transient outward current (Ito) and IK‐ACh by AVE0118 and AVE1231 also questions the relative role of IKur inhibition in AF termination by these agents. At concentrations that specifically inhibit IKur (≤50 μM), 4‐AP neither terminates sustained AF nor prevents its initiation in an acetylcholine‐mediated AF model. 25 Anti‐AF effects of 4‐aminopyridine (4‐AP) in this AF model appears only at concentrations that potently block Ito. It has been reported that IKur density is reduced at rapid activation rates, 26 , 27 which indicates that the relative contribution of IKur to atrial repolarization during AF may not be crucial and, thus, blockade of this current alone may not be sufficient for effective AF termination. The density of IKur has been shown to be reduced in cells isolated from chronic AF atria in some studies, but not all (for review see 28 ).
Important issues regarding the safety of IKur blockers have been raised recently with the finding that loss‐of‐function mutations in KCNA5 are associated with familial AF. 29 Because KCNA5 encodes the α subunit of the IKur channel, these results suggest that a reduction in IKur may predispose to the development of AF. Indeed, recent experimental studies have demonstrated different effects of IKur block on the action potential of “remodeled” versus “healthy” atria. 25 , 30 Block of IKur in “healthy” atria (displaying a plateau‐shaped AP morphology) abbreviates the atrial action potential duration measured at 70–90% repolarization (APD70–90) (Fig. 2) 25 , 30 , 31 In contrast, in remodeled atria (typically displaying a triangular‐shaped AP morphology) a reduction of IKur prolongs APD70–90. 25 , 30 Abbreviation of atrial repolarization is well known to be associated with an increase in AF vulnerability. Consistent with this observation, block of IKur with 10–50 μM of 4‐AP has been shown to promote the induction of nonsustained AF in “healthy” canine arterially perfused atrial preparations, apparently due to APD90/ERP abbreviation (Fig. 3). 25
Figure 2.
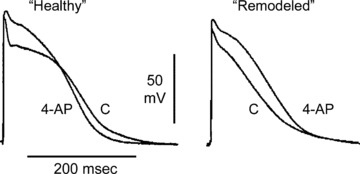
Block of IKur with 4‐aminopyridine (4‐AP, 50 μM) abbreviates APD90 in “healthy” (plateau‐shaped action potential), but prolongs it in “acutely remodeled” (triangular‐shaped action potential) canine coronary‐perfused atrial preparations (pectinate muscles). Low flow ischemia was used to generate the “acutely remodeled” atria. Modified from Burashnikov et al., 25 , 31 with permission.
Figure 3.
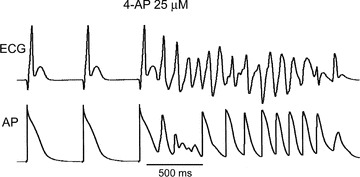
Nonsustained AF induced by a single premature beat (S1‐S2= 115 ms) in the presence of 25 μM 4‐AP in a “healthy” canine isolated coronary‐perfused atrial preparation. From Burashnikov and Antzelevitch, 25 with permission.
There is an apparent inconsistency between prolongation of ERP 11 , 12 , 13 , 14 , 15 , 16 , 17 , 18 , 19 and abbreviation of APD70–90 induced by IKur blockers in “healthy” atria. 25 , 30 , 31 Because inhibition of IKur alone abbreviates APD90, the prolongation of ERP measured in some studies is most readily explained by development of postrepolarization refractoriness (PRR), likely due to concurrent inhibition of sodium channels. Interestingly, atrial‐selective agents that block IKur such as vernakalant and AZD7009 also potently block INa. 22 , 23 AZD7009 has characteristics of an atrial‐selective sodium channel blocker, slowing conduction and increasing diastolic threshold of excitation in atria, but not in the canine ventricle in vivo. 16 isoquinolinone 3‐[(dimethylamino)‐methyl]‐6‐methoxy‐2‐methyl‐4‐phenylisoquinolin‐1(2H)‐one (ISQ1) also slows conduction velocity in atria in vivo, 32 indicating that it blocks INa. Camm and Savelieva 33 in their recent review noted that AVE1231 also blocks early INa. AVE0118 reduces maximal rate of rise of action potential upstroke (VMax) in canine coronary‐perfused atrial preparations, suggesting that AVE0118 also blocks INa.(Burashnikov et al., unpublished data). Atrial‐specific ERP prolongation can also be the result of atrial‐selective and ‐specific sodium channel blockade (Fig. 4). 24 , 34
Figure 4.
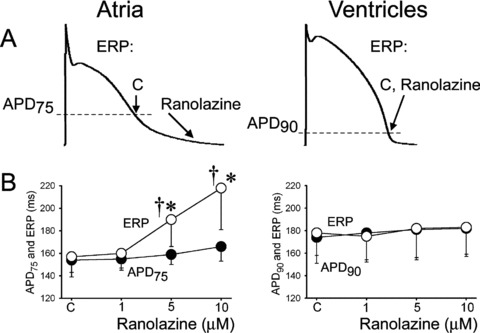
Ranolazine specifically induces prolongation of the effective refractory period (ERP) and development of postrepolarization refractoriness in atria (PRR, the difference between ERP and APD75 in atria and between ERP and APD90 in ventricles; ERP corresponds to APD75 in atria and APD90 in ventricles). CL = 500 ms. C = control. The arrows in panel A illustrate the position on the action potential corresponding to the end of the ERP in atria and ventricles and the effect of ranolazine to shift the end of the ERP in atria but not ventricles. *P < 0.05 versus control. †P < 0.05 versus APD75 values in atria and APD90 in ventricles; (n = 5–18). From Burashnikov et al., 24 with permission.
Thus, available experimental and clinical data suggest that “pure” IKur block may not suffice to effectively suppress AF and that inhibition of additional currents may be required (e.g., INa, Ito, and/or IKr). Moreover, recent data suggest that selective reduction of IKur may predispose to the development of AF in healthy atria.
It has been reported that a vagal component may importantly contribute to the initiation of some paroxysmal AF. 35 , 36 Under normal conditions, IK‐ACh is activated through the muscarinic receptors in response to release of the neurotransmitter acetylcholine (ACh) in vivo or addition of ACh into solution in vitro, with direct consequences being an abbreviation of atrial repolarization and promotion of AF. In contrast to atria, parasympathetic system stimulation or ACh produce little to no direct effects on ventricular electrophysiological parameters due to practical absence of the channels underlying IK‐ACh and respective receptors. Thus, block of IK‐ACh can specifically affect atria and may suppress vagally mediated AF. In atria isolated from humans with chronic AF, ACh‐activated IK‐ACh is reported to be either increased or decreased (for review see 28 ).
There is another form of IK‐ACh that does not require cholinergic agonist stimulation for activation, 37 , 38 which was recently termed constitutively active (CA) IK‐ACh (CA IK‐ACh) 9 , 39 This current is only marginally present in healthy nonfibrillating human or canine atria and is significantly increased in atria of chronic AF patients and canine tachycardia‐remodeled atria. 9 , 38 , 39 , 40 The augmentation of CA IK‐ACh in chronic AF patients has been related to abnormal protein kinase C function. 39 The CA IK‐ACh is likely to contribute to abbreviation of atrial APD and AF maintenance. 9 , 39 , 40 Block of IK‐ACh currents with tertiapin prolongs atrial APD and suppresses AF in experimental models. 40 , 41 Although CA IK‐ACh has been suggested recently as a new atrial‐ and pathology‐specific target for AF treatment, 39 , 42 there is no selective CA IK‐ACh blocker available at the present time and the feasibility of an atrial‐selective CA IK‐ACh approach is yet to be determined. The development of clinically safe IK‐ACh blockers must take into account the presence of the IK‐ACh channels and receptors in many organs other than the heart.
Connexins are the proteins that principally determine cardiac cell‐to‐cell communication. Cx40 is commonly included to the list of potential atrial‐specific targets for AF treatment, because Cx40 is found in atrial but not ventricular myocardium, with the exception of the conduction system in the ventricles. 10 , 43 Somatic mutations in Cx40 gene (GJA5) have recently been found in patients with idiopathic AF. 44 There are no specific Cx40 modulators available as yet and the there are no data demonstrating either safety or effectiveness of this approach in the management of AF.
Atria‐Selective or Predominant Antiarrhythmic Approaches to AF Management
In addition to atrial‐specific ionic channels, there are ionic channels that are present in both chambers of the heart but the inhibition of these channels can produce predominant electrophysiological changes in atria vs. ventricular. These atrial‐selective or predominant targets, include sodium channels responsible for fast INa 24 , 45 and, perhaps, channels underlying IKr. 46 , 47 , 48 , 49 , 50 , 51 , 52 Note that atrial‐predominant refers to a lesser degree of atrial selectivity.
We recently reported the results of experimental studies demonstrating that some early INa blockers affect sodium channel‐dependent parameters in an atrial selective manner (Fig. 4 and 5). 24 , 34 , 53 Ranolazine and chronic amiodarone reduced the maximum rate of rise of the action potential upstroke (Vmax) and conduction velocity (CV), increased diastolic threshold of excitation (DTE), and induced PRR predominantly in canine atrial vs. ventricular coronary‐perfused preparations. Ranolazine was more “atrial selective” than chronic amiodarone. 24 , 53 Propafenone showed no chamber selectivity in depression of sodium channel‐dependent parameters at a normal pacing rate (cycle length [CL]= 500 ms), but displayed some atrial predominance at rapid pacing rates (likely due to atrial‐specific APD prolongation, as discussed below). 54 Lidocaine turned out to also be an atrial‐predominant sodium channel blocker, but with a much lesser degree of atrial selectivity than either ranolazine or amiodarone. 24 , 53 Note that acute lidocaine is not effective in terminating AF in the clinics. 55 As mentioned above, AZD7009 also behaves as an atrial‐selective INa blocker, slowing conduction and increasing DTE only in atria. 16
Figure 5.
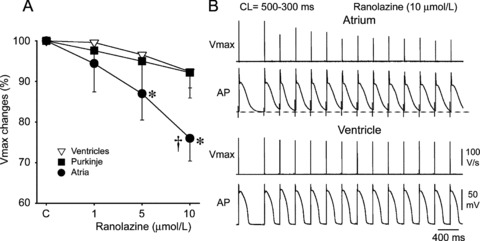
Ranolazine produces a much greater rate‐dependent inhibition of the maximal action potential upstroke velocity (Vmax) in atria than in ventricles. (A) Normalized changes in Vmax of atrial and ventricular cardiac preparations paced at a cycle length (CL) of 500 ms. (B) Ranolazine prolongs late repolarization in atria, but not ventricles and acceleration of rate leads to elimination of the diastolic interval (during which the recovery from sodium channel block largely occurs) in atria but not ventricles. *P < 0.05 versus control. †P < 0.05 versus respective values of M cell and Purkinje (n = 7–21). From Burashnikov et al., 24 with permission.
Interestingly, ranolazine, propafenone, and chronic amiodarone all block IKr, in addition to INa, and produce preferential APD90 prolongation in canine atria vs. ventricles at 300–500 ms pacing CLs studied. 24 , 34 , 54 At normal heart rates or pacing rates, selective IKr blockers (E‐4031, sotalol, d‐sotalol, dl‐sotalol, dofetilide, WAY‐123,398, ibutilide, MK499, and almokalant) preferentially prolong atrial vs. ventricular ERP and/or APD, but do not induce early afterdepolarizations (EADs) in atria. 46 , 47 , 48 , 49 , 50 , 51 , 52 In contrast, at slow pacing rates, ventricles, but not atria, display a significant APD prolongation, early after‐depolarization (EAD) and TdP when IKr is reduced. 56 , 57 Interestingly, recently published data showed no association of AF with the congenital LQT2 syndrome (IKr defect; in 0/174 patients) and only a marginal AF association in LQT3 syndrome (late INa defect, in 1/59 patients). 58 However, a higher prevalence of AF was found in the congenital IKs mutation‐related LQT1 syndrome (5/211 patients; 2.4% vs. 0.1% in <50 years age population). 58 The rate‐dependent atrioventricular differences in response to IKr inhibition are not well appreciated and underlying mechanisms of these differences are not unclearly defined.
The atrial‐selective action of ranolazine and chronic amiodarone is thought to be due to important distinctions in action potential characteristics and biophysical properties of sodium channels of atrial versus ventricular myocytes as well as to atrial‐predominant APD prolongation of these agents. 24 , 34 , 45 The half inactivation voltage (V0.5) of canine atrial sodium channels is 12–16 mV more negative than those of ventricular sodium channels; resting membrane potential (RMP) in atria is also less negative than in ventricles (approximately –83 vs –87 mV). 24 , 59 These factors indicate that there is a larger fraction of inactivated sodium channels at RMP in atria versus ventricles and a smaller fraction of resting sodium channels at RMP in atria versus ventricles. This is expected to slow the recovery of the sodium channel from block in atria compared to ventricles, since the recovery occurs principally during the resting state. 60 The inherently slow phase 3 and atrial‐selective APD prolongation contribute importantly to the atrial‐predominant suppression of INa by ranolazine and chronic amiodarone, and at rapid pacing rates by propafenone. Atrial‐selective APD prolongation leads to abbreviation or even elimination of diastolic intervals in atria, but not ventricles (Fig. 5). Since the recovery from the sodium channel block occurs largely during the diastolic interval, the effectiveness of sodium channel block is greater in atria versus ventricles.
Limited data are available regarding atrioventricular differences in the response to sodium channel blockers. Available data, summarized in Figure 6 (and discussed in details in our previous review 34 ), indicate that there are atrial‐selective, ventricular‐selective, as well as nonchamber‐selective sodium channel blockers. 16 , 23 , 24 , 53 , 61 , 62 , 63 , 64 , 65 It is noteworthy that a significant portion of these data was obtained using superfused preparations or isolated myocytes, where atrioventricular differences on the effects of INa block may be different from those recorded in arterially perfused preparation or in in vivo (for review see 34 ).
Figure 6.
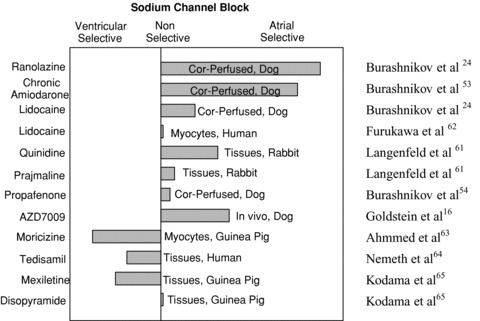
A semiquantitative assessment of atrial selectivity of INa blockers based on studies conducted in atrial and ventricular coronary‐perfused (cor‐perfused) and superfused (tissues) preparations, isolated myocytes, and in vivo (see text for details). Reproduced from Burashnikov and Antzelevitch, 34 with permission.
Ranolazine, propafenone, and chronic amiodarone effectively suppress ACh‐mediated arrhythmias in isolated canine coronary‐perfused right atrial preparations. 24 , 53 , 54 Lidocaine is far less effective in suppressing AF in these models. These antiarrhythmic effects of ranolazine, amiodarone, and propafenone were associated with both APD prolongation and the development a significant PRR. The concentration of ranolazine that effectively suppressed AF (10 μM) produced little to no effect in canine ventricular preparations, prompting us to suggest “atrial selective sodium channel block” as a novel strategy for suppression of AF. 24 The effectiveness of ranolazine to suppress AF in experimental models is consistent with the results of the recently reported MERLIN‐TIMI 36 study, where ranolazine treatment was associated with reduced incidence of the supraventricular arrhythmias and new onset AF in patients in non‐ST segment elevation acute coronary syndrome patients. 66 Ranolazine also reduced the incidence of ventricular arrhythmias, an effect attributed to the action of ranolazine to block late INa. 66 , 67
It seems obvious that atrial selectivity of pharmacological agents recorded in “healthy” heart may not be directly applied to pathophysiological conditions (such as ischemia, long QT syndrome, electrical remodeling, etc.), because responses of “healthy” and “diseased” hearts to IKr, INa, or IKur blockers can be very different (see Fig. 2). 30 , 34 Therefore, while ranolazine and AVE0118 selectively affect atrial electrophysiological parameters in “healthy” hearts, 11 , 24 these agents may significantly modify ventricular electrophysiology as well as suppress ventricular arrhythmias in the conditions of acute ischemia or long QT syndromes. 66 , 68 , 69 Ranolazine's potent action to suppress late INa contributes to the drug's antiarrhythmic efficacy under these conditions.
It is of interest that 4‐AP blocks Ito much more effectively in atria versus ventricles (with an IC50 in atrial myocytes one‐third that in ventricular myocytes). 70 , 71 If this is also the case with other Ito blockers, than Ito block should produce a greater changes in atrial versus ventricular repolarization. Block of Ito likely contributes to the atrial‐specific effects of IKur blockers on atrial repolarization since all agents that block IKur also inhibit Ito.
There are data indicating that adenosine triphosphate (ATP)‐sensitive potassium current (IK‐ATP) may be involved in the generation of some forms of AF. 28 , 72 Propafenone blocks IK‐ATP with four‐fold higher affinity in atrial than in ventricular rabbit myocytes. 73 Although it is not known whether IK‐ATP blockers such as glybenclamide are atrial‐selective, atrial‐selective block of IK‐ATP could conceivably be useful as a treatment for IK(ATP)‐mediated forms of AF.
“Upstream” Therapy for AF
In addition to further developing ion channel‐based AF therapy, there is rapid development of nonion‐channel approaches, aimed at reducing or reversing structural remodeling, inflammation, and oxidative injury associated with AF. These are generally referred to as “upstream therapies.” 74 , 75 Inflammation and oxidative injury promote structural remodeling, including interstitial fibrosis, fibroblast proliferation, accumulation/redistribution of collagen, dilatation, and hypertrophy. Proarrhythmic actions of atrial structural remodeling are generally related to the conduction disturbances, which promote reentrant arrhythmias.
A number of experimental and clinical studies have shown that interventions that affect structural remodeling, inflammation, and/or oxidative stress such as angiotensin‐converting enzyme (ACE) inhibitors, angiotensin II (Ang II) receptor blockers (ARBs), and statins may reduce the occurrence of AF, 74 , 75 , 76 although some studies question the anti‐AF efficacy of such therapies. 75 , 77 , 78 , 79 It seems that ACE, ARB, and statin therapies may be beneficial for AF patients with severe ventricular dysfunction and heart failure, and less so in moderately diseased or relatively normal hearts. These therapies may be more effective in paroxysmal versus persistent AF. 76 , 77 , 78 The anti‐AF mechanisms of ACE inhibitors, ARBs, and statins are not well established, and presumed to be largely due to their antihypertensive, antiinflammatory, and antioxidative stress actions.
Successful development of “upstream therapy” depends on our ability to identify factors and signaling pathways involved in the generation of atrial structural remodeling, inflammation, and oxidative stress. A number of mediating factors have been identified such as Ang II, Ang II receptors, transforming growth factor‐β1 (TGF‐β1), mitogen‐activated protein kinase (MAPK), platelet‐derived growth factor (PDGF), peroxisome proliferator‐activated receptor‐λ (PPAR‐ λ), Janus kinase (JAK), Rac1, nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, signal transducers and activators of transcription (STAT), and calcineurin 74 , 80 , 81 , 82 , 83 , 84 , 85 with Ang II and its angiotensin II type 1 (AT1) receptors are critically involved in the initiation of the signaling cascades. 74 , 82 The relative roles and contributions of these mediating factors in structural remodeling, inflammation, and oxidative stress are poorly understood. Moreover, the relative role of structural remodeling, inflammation, and oxidative stress in development of AF is still not fully understood. The contribution of structural remodeling, inflammation, and oxidative injury in the development of AF varies significantly among different AF pathologies. 82 , 86
Atria often develop structural remodeling to a greater degree than the ventricles. 80 , 83 , 85 , 87 , 88 , 89 , 90 , 91 Cardiac overexpression of a constitutively active form of TGF‐β1 (a profibrotic factor) promotes atrial but not ventricular fibrosis in mice. 80 , 89 The extent of atrial fibrosis in canine ventricular tachypacing‐induced congestive heart failure (CHF) was reported to be by far greater than that of ventricular fibrosis. 87 Mice with cardiac‐restricted ACE, producing overexpression of Ang II in the heart, display atrial but not ventricular structural and functional abnormalities. 88 Chronic cardiac‐specific overexpression of constitutively active Rac1 in mice significantly increases atrial size and the extent of fibrosis in atria to a greater extent compared to ventricles (at least in part due to increased NADPH oxidase activity). 90 A PDGF‐mediated signaling pathway causing atrial‐selective structural remodeling has been described recently. 83 Mechanisms underlying the greater preponderance of atria to develop structural remodeling are poorly understood. They may be related, in part, to a higher Ang II receptor density, 4 , 85 a higher basal STAT3 85 and PDGF 83 receptor expression in atria vs. ventricles. Fibroblast density is greater in atria versus ventricles in nonremodeled hearts. 83 A significant tyrosine phosphorylation of STAT3 in the atrium but not the ventricles has been reported to be induced by infusion of Ang II in rat in vivo. 85 These data point to potential atrial‐selective targets for “upstream” AF therapy.
Improved Derivatives of “Old” Drugs
Amiodarone is the most effective of the currently available anti‐AF agents for long‐term rhythm control of AF. A major drawback of long‐term use of amiodarone is its proclivity for multiple organ toxicity presumably related to the iodine moiety of the drug. In order to eliminate these adverse effects, several derivatives of amiodarone have been synthesized including dronedarone, celivarone, and ATI‐2042. 10 , 33 The most investigated of amiodarone's derivatives is dronedarone, which is a noniodinated benzofuran derivative of amiodarone with much faster pharmacodynamics. Like amiodarone, dronedarone blocks multiple ionic channels (such as IKr, IKs, INa, ICa(L), IK1) and is significantly more effective than placebo in reducing AF occurrence, but lacks the adverse effects of amiodarone. 92 The long‐term effectiveness of dronedarone to maintain sinus rhythm appears to be lower than that of amiodarone. A recently determined important limitation of dronedarone is that it increases early mortality in patients with severe heart failure and left ventricular systolic dysfunction. 93
“Gap Junction” Therapy for AF
Since conduction disturbances are associated with many cardiac arrhythmia syndromes including AF, it has long been appreciated that improved conduction may be antiarrhythmic. Improved conduction achieved by using the gap junction modulator rotigaptide has been shown to lead to antiarrhythmic effects. 94 , 95 The feasibility of this antiarrhythmic approach was demonstrated in canine ventricular ischemia model, 94 chronic mitral regurgitation AF model, 96 and in the canine acute ischemia AF model. 97 Rotigaptide, however, did not effect AF occurrence in AF models associated with heart failure. 96 , 97
Summary
Ongoing research aimed at development of new pharmacological strategies for the management of AF includes both ion channel and nonion channel‐mediated approaches to therapy. While success to date has been modest, the recent identification of atrial‐ and pathology‐selective agents and targets hold promise for the development of effective new treatments.
Financial support: Supported by grant HL47678 from NHLBI (CA) and NYS and Florida Grand Lodges F. & A.M.
REFERENCES
- 1. Wyse DG, Waldo AL, DiMarco JP, et al A comparison of rate control and rhythm control in patients with atrial fibrillation. N Engl J Med 2002;347:1825–1833. [DOI] [PubMed] [Google Scholar]
- 2. Roy D, Talajic M, Nattel S, et al Rhythm control versus rate control for atrial fibrillation and heart failure. N Engl J Med 2008;358:2667–2677. [DOI] [PubMed] [Google Scholar]
- 3. Morrow JP, Cannon CP, Reiffel JA. New antiarrhythmic drugs for establishing sinus rhythm in atrial fibrillation: What are our therapies likely to be by 2010 and beyond? Am Heart J 2007;154:824–829. [DOI] [PubMed] [Google Scholar]
- 4. Savelieva I, Camm J. Anti‐arrhythmic drug therapy for atrial fibrillation: Current anti‐arrhythmic drugs, investigational agents, and innovative approaches. Europace 2008;10:647–665. [DOI] [PubMed] [Google Scholar]
- 5. Mazzini MJ, Monahan KM. Pharmacotherapy for atrial arrhythmias: Present and future. Heart Rhythm 2008;5:S26–S31. [DOI] [PubMed] [Google Scholar]
- 6. Fuster V, Ryden LE, Cannom DS, et al ACC/AHA/ESC 2006 guidelines for the management of patients with atrial fibrillation—executive summary: A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the European Society of Cardiology Committee for Practice Guidelines (Writing Committee to Revise the 2001 Guidelines for the Management of Patients With Atrial Fibrillation). J Am Coll Cardiol 2006;48:854–906. [DOI] [PubMed] [Google Scholar]
- 7. Viskin S, Barron HV, Heller K, et al The treatment of atrial fibrillation: Pharmacologic and nonpharmacologic strategies. Curr Probl Cardiol 1997;22:37–108. [DOI] [PubMed] [Google Scholar]
- 8. Zimetbaum P. Amiodarone for atrial fibrillation. N Engl J Med 2007;356:935–941. [DOI] [PubMed] [Google Scholar]
- 9. Dobrev D, Friedrich A, Voigt N, et al The G protein‐gated potassium current I K,ACh is constitutively active in patients with chronic atrial fibrillation. Circulation 2005;112:3697–3706. [DOI] [PubMed] [Google Scholar]
- 10. Nattel S, Carlsson L. Innovative approaches to anti‐arrhythmic drug therapy. Nat Rev Drug Discov 2006;5:1034–1049. [DOI] [PubMed] [Google Scholar]
- 11. Blaauw Y, Gogelein H, Tieleman RG, et al “Early” class III drugs for the treatment of atrial fibrillation: Efficacy and atrial selectivity of AVE0118 in remodeled atria of the goat. Circulation 2004;110:1717–1724. [DOI] [PubMed] [Google Scholar]
- 12. Knobloch K, Brendel J, Rosenstein B, et al Atrial‐selective antiarrhythmic actions of novel Ikur vs. Ikr, Iks, and IKAch class Ic drugs and beta blockers in pigs. Med Sci Monit 2004;10:BR221–BR228. [PubMed] [Google Scholar]
- 13. Wirth KJ, Brendel J, Steinmeyer K, et al In vitro and in vivo effects of the atrial selective antiarrhythmic compound AVE1231. J Cardiovasc Pharmacol 2007;49:197–206. [DOI] [PubMed] [Google Scholar]
- 14. Regan CP, Stump GL, Wallace AA, et al In vivo cardiac electrophysiologic and antiarrhythmic effects of an isoquinoline IKur blocker, ISQ‐1, in rat, dog, and nonhuman primate. J Cardiovasc Pharmacol 2007;49:236–245. [DOI] [PubMed] [Google Scholar]
- 15. Dorian P, Pinter A, Mangat I, et al The effect of vernakalant (RSD1235), an investigational antiarrhythmic agent, on atrial electrophysiology in humans. J Cardiovasc Pharmacol 2007;50:35–40. [DOI] [PubMed] [Google Scholar]
- 16. Goldstein RN, Khrestian C, Carlsson L, et al Azd7009: A new antiarrhythmic drug with predominant effects on the atria effectively terminates and prevents reinduction of atrial fibrillation and flutter in the sterile pericarditis model. J Cardiovasc Electrophysiol 2004;15:1444–1450. [DOI] [PubMed] [Google Scholar]
- 17. Seki A, Hagiwara N, Kasanuki H. Effects of NIP‐141 on K currents in human atrial myocytes. J Cardiovasc Pharmacol 2002;39:29–38. [DOI] [PubMed] [Google Scholar]
- 18. Matsuda T, Takeda K, Ito M, et al Atria selective prolongation by NIP‐142, an antiarrhythmic agent, of refractory period and action potential duration in guinea pig myocardium. J Pharmacol Sci 2005;98:33–40. [DOI] [PubMed] [Google Scholar]
- 19. Li GR, Wang HB, Qin GW, et al Acacetin, a natural flavone, selectively inhibits human atrial repolarization potassium currents and prevents atrial fibrillation in dogs. Circulation 2008;117:2449–2457. [DOI] [PubMed] [Google Scholar]
- 20. Roy D, Pratt CM, Torp‐Pedersen C, et al Vernakalant hydrochloride for rapid conversion of atrial fibrillation. A phase 3, randomized, placebo‐controlled trial. Circulation 2008;117:1518–1525. [DOI] [PubMed] [Google Scholar]
- 21. Crijns HJ, Van GI, Walfridsson H, et al Safe and effective conversion of persistent atrial fibrillation to sinus rhythm by intravenous AZD7009. Heart Rhythm 2006;3:1321–1331. [DOI] [PubMed] [Google Scholar]
- 22. Fedida D. Vernakalant (RSD1235): A novel, atrial‐selective antifibrillatory agent. Expert Opin Investig Drugs 2007;16:519–532. [DOI] [PubMed] [Google Scholar]
- 23. Carlsson L, Chartier D, Nattel S. Characterization of the in vivo and in vitro electrophysiological effects of the novel antiarrhythmic agent AZD7009 in atrial and ventricular tissue of the dog. J Cardiovasc Pharmacol 2006;47:123–132. [DOI] [PubMed] [Google Scholar]
- 24. Burashnikov A, Di Diego JM, Zygmunt AC, et al Atrium‐selective sodium channel block as a strategy for suppression of atrial fibrillation: Differences in sodium channel inactivation between atria and ventricles and the role of ranolazine. Circulation 2007;116:1449–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Burashnikov A, Antzelevitch C. Can inhibition of IKur promote atrial fibrillation? Heart Rhythm 2008;5:1304–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Feng J, Xu D, Wang Z, et al Ultrarapid delayed rectifier current inactivation in human atrial myocytes: Properties and consequences. Am J Physiol 1998;275:H1717–H1725. [DOI] [PubMed] [Google Scholar]
- 27. Ehrlich JR, Ocholla H, Ziemek D, et al Characterization of human cardiac Kv1.5 inhibition by the novel atrial‐selective antiarrhythmic compound AVE1231. J Cardiovasc Pharmacol 2008;51:380–387. [DOI] [PubMed] [Google Scholar]
- 28. Workman AJ, Kane KA, Rankin AC. Cellular bases for human atrial fibrillation. Heart Rhythm 2008;5:S1–S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Olson TM, Alekseev AE, Liu XK, et al Kv1.5 channelopathy due to KCNA5 loss‐of‐function mutation causes human atrial fibrillation. Hum Mol Genet 2006;15:2185–2191. [DOI] [PubMed] [Google Scholar]
- 30. Wettwer E, Hala O, Christ T, et al Role of IKur in controlling action potential shape and contractility in the human atrium: Influence of chronic atrial fibrillation. Circulation 2004;110:2299–2306. [DOI] [PubMed] [Google Scholar]
- 31. Burashnikov A, Mannava S, Antzelevitch C. Transmembrane action potential heterogeneity in the canine isolated arterially‐perfused atrium: Effect of I Kr and I to /I Kur block. Am J Physiol 2004;286:H2393–H2400. [DOI] [PubMed] [Google Scholar]
- 32. Regan CP, Kiss L, Stump GL, et al Atrial antifibrillatory effects of structurally distinct IKur blockers 3‐[(dimethylamino)methyl]‐6‐methoxy‐2‐methyl‐4‐phenylisoquinolin‐1(2H)‐one and 2‐phenyl‐1,1‐dipyridin‐3‐yl‐2‐pyrrolidin‐1‐yl‐ethanol in dogs with underlying heart failure. J Pharmacol Exp Ther 2008;324:322–330. [DOI] [PubMed] [Google Scholar]
- 33. Camm AJ, Savelieva I. New antiarrhythmic drugs for atrial fibrillation: Focus on dronedarone and vernakalant. J Interv Card Electrophysiol 2008;23:7–14. [DOI] [PubMed] [Google Scholar]
- 34. Burashnikov A, Antzelevitch C. Atrial‐selective sodium channel blockers: Do they exist? J Cardiovasc Pharmacol 2008;52:121–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bettoni M, Zimmermann M. Autonomic tone variations before the onset of paroxysmal atrial fibrillation. Circulation 2002;105:2753–2759. [DOI] [PubMed] [Google Scholar]
- 36. Pappone C, Santinelli V, Manguso F, et al Pulmonary vein denervation enhances long‐term benefit after circumferential ablation for paroxysmal atrial fibrillation. Circulation 2004;109:327–334. [DOI] [PubMed] [Google Scholar]
- 37. Heidbuchel H, Callewaert G, Vereecke J, et al Membrane‐bound nucleoside diphosphate kinase activity in atrial cells of frog, guinea pig, and human. Circ Res 1992;71:808–820. [DOI] [PubMed] [Google Scholar]
- 38. Ehrlich JR, Cha TJ, Zhang L, et al Characterization of a hyperpolarization‐activated time‐dependent potassium current in canine cardiomyocytes from pulmonary vein myocardial sleeves and left atrium. J Physiol 2004;557:583–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Voigt N, Friedrich A, Bock M, et al Differential phosphorylation‐dependent regulation of constitutively active and muscarinic receptor‐activated I K,ACh channels in patients with chronic atrial fibrillation. Cardiovasc Res 2007;74:426–437. [DOI] [PubMed] [Google Scholar]
- 40. Cha TJ, Ehrlich JR, Chartier D, et al Kir3‐based inward rectifier potassium current: Potential role in atrial tachycardia remodeling effects on atrial repolarization and arrhythmias. Circulation 2006;113:1730–1737. [DOI] [PubMed] [Google Scholar]
- 41. Hashimoto N, Yamashita T, Tsuruzoe N. Tertiapin, a selective I K,ACh blocker, terminates atrial fibrillation with selective atrial effective refractory period prolongation. Pharmacol Res 2006;54:136–141. [DOI] [PubMed] [Google Scholar]
- 42. Ravens U. Potassium channels in atrial fibrillation: Targets for atrial and pathology‐specific therapy? Heart Rhythm 2008;5:758–759. [DOI] [PubMed] [Google Scholar]
- 43. Ehrlich JR, Biliczki P, Hohnloser SH, et al Atrial‐selective approaches for the treatment of atrial fibrillation. J Am Coll Cardiol 2008;51:787–792. [DOI] [PubMed] [Google Scholar]
- 44. Gollob MH, Jones DL, Krahn AD, et al Somatic mutations in the connexin 40 gene (GJA5) in atrial fibrillation. N Engl J Med 2006;354:2677–2688. [DOI] [PubMed] [Google Scholar]
- 45. Burashnikov A, Antzelevitch C. How do atrial‐selective drugs differ from antiarrhythmic drugs currently used in the treatment of atrial fibrillation? J Atrial Fibrillation 2008;1:98–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Spinelli W, Parsons RW, Colatsky TJ. Effects of WAY‐123,398, a new Class‐III antiarrhythmic agent, on cardiac refractoriness and ventricular fibrillation threshold in anesthetized dogs—a comparison with UK‐68798, e‐4031, and DL‐ Sotalol. J Cardiovasc Pharmacol 1992;20:913–922. [DOI] [PubMed] [Google Scholar]
- 47. Wiesfeld AC, De Langen CD, Crijns HJ, et al Rate‐dependent effects of the class III antiarrhythmic drug almokalant on refractoriness in the pig. J Cardiovasc Pharmacol 1996;27:594–600. [DOI] [PubMed] [Google Scholar]
- 48. Baskin EP, Lynch JJ., Jr . Differential atrial versus ventricular activities of class III potassium channel blockers. J Pharmacol Exp Ther 1998;285:135–142. [PubMed] [Google Scholar]
- 49. Stump GL, Wallace AA, Regan CP, et al In vivo antiarrhythmic and cardiac electrophysiologic effects of a novel diphenylphosphine oxide IKur blocker (2‐isopropyl‐5‐methylcyclohexyl) diphenylphosphine oxide. J Pharmacol Exp Ther 2005;315:1362–1367. [DOI] [PubMed] [Google Scholar]
- 50. Wang J, Feng J, Nattel S. Class III antiarrhythmic drug action in experimental atrial fibrillation. Differences in reverse use dependence and effectiveness between d‐sotalol and the new antiarrhythmic drug ambasilide. Circulation 1994;90:2032–2040. [DOI] [PubMed] [Google Scholar]
- 51. Echt DS, Berte LE, Clusin WT, et al Prolongation of the human monophasic action potential by sotalol. Am J Cardiol 1982;50:1082–1086. [DOI] [PubMed] [Google Scholar]
- 52. Buchanan LV, LeMay RJ, Walters RR, et al Antiarrhythmic and electrophysiologic effects of intravenous ibutilide and sotalol in the canine sterile pericarditis model. J Cardiovasc Electrophysiol 1996;7:113–119. [DOI] [PubMed] [Google Scholar]
- 53. Burashnikov A, Di Diego JM, Zygmunt AC, et al Atrial‐selective sodium channel block as a strategy for suppression of atrial fibrillation. Ann N Y Acad Sci 2008;1123:105–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Burashnikov A, Belardinelli L, Antzelevitch C. Ranolazine and propafenone both suppress atrial fibrillation but ranolazine unlike propafenone does it without prominent effects on ventricular myocardium. Heart Rhythm 2007;4:S163 (abstract). [Google Scholar]
- 55. Marrouche NF, Reddy RK, Wittkowsky AK, et al High‐dose bolus lidocaine for chemical cardioversion of atrial fibrillation: A prospective, randomized, double‐blind crossover trial. Am Heart J 2000;139:E8–11. [DOI] [PubMed] [Google Scholar]
- 56. Antzelevitch C, Shimizu W, Yan GX, et al The M cell: Its contribution to the ECG and to normal and abnormal electrical function of the heart. J Cardiovasc Electrophysiol 1999;10:1124–1152. [DOI] [PubMed] [Google Scholar]
- 57. Burashnikov A, Antzelevitch C. Late‐phase 3 EAD. A unique mechanism contributing to initiation of atrial fibrillation. Pacing Clin Electrophysiol 2006;29:290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Johnson JN, Tester DJ, Perry J, et al Prevalence of early‐onset atrial fibrillation in congenital long QT syndrome. Heart Rhythm 2008;5:704–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Li GR, Lau CP, Shrier A. Heterogeneity of sodium current in atrial vs epicardial ventricular myocytes of adult guinea pig hearts. J Mol Cell Cardiol 2002;34:1185–1194. [DOI] [PubMed] [Google Scholar]
- 60. Whalley DW, Wendt DJ, Grant AO. Basic concepts in cellular cardiac electrophysiology: Part II: Block of ion channels by antiarrhythmic drugs. Pacing Clin Electrophysiol 1995;18:1686–1704. [DOI] [PubMed] [Google Scholar]
- 61. Langenfeld H, Weirich J, Kohler C, et al Comparative analysis of the action of class I antiarrhythmic drugs (lidocaine, quinidine, and prajmaline) in rabbit atrial and ventricular myocardium. J Cardiovasc Pharmacol 1990;15:338–345. [DOI] [PubMed] [Google Scholar]
- 62. Furukawa T, Koumi S, Sakakibara Y, et al An analysis of lidocaine block of sodium current in isolated human atrial and ventricular myocytes. J Mol Cell Cardiol 1995;27:831–846. [DOI] [PubMed] [Google Scholar]
- 63. Ahmmed GU, Hisatome I, Kurata Y, et al Analysis of moricizine block of sodium current in isolated guinea‐pig atrial myocytes. Atrioventricular difference of moricizine block. Vascul Pharmacol 2002;38:131–141. [DOI] [PubMed] [Google Scholar]
- 64. Nemeth M, Virag L, Hala O, et al The cellular electrophysiological effects of tedisamil in human atrial and ventricular fibers. Cardiovasc Res 1996;31:246–248. [PubMed] [Google Scholar]
- 65. Kodama I, Toyama J, Takanaka C, et al Block of activated and inactivated sodium channels by class I antiarrhythmic drugs studied by using the maximum upstroke velocity (Vmax) of action potential in guinea‐pig cardiac muscles. J Mol Cell Cardiol 1987;19:367–377. [DOI] [PubMed] [Google Scholar]
- 66. Scirica BM, Morrow DA, Hod H, et al Effect of ranolazine, an antianginal agent with novel electrophysiological properties, on the incidence of arrhythmias in patients with non ST‐segment elevation acute coronary syndrome: Results from the Metabolic Efficiency With Ranolazine for Less Ischemia in Non ST‐Elevation Acute Coronary Syndrome Thrombolysis in Myocardial Infarction 36 (MERLIN‐TIMI 36) randomized controlled trial. Circulation 2007;116:1647–1652. [DOI] [PubMed] [Google Scholar]
- 67. Antzelevitch C. Ranolazine: A new antiarrhythmic agent for patients with non‐ST‐segment elevation acute coronary syndromes? Nat Clin Pract Cardiovasc Med 2008;5:248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Antzelevitch C, Belardinelli L, Wu L, et al Electrophysiologic properties and antiarrhythmic actions of a novel anti‐anginal agent. J Cardiovasc Pharmacol Therapeut 2004;9(Suppl 1):S65–S83. [DOI] [PubMed] [Google Scholar]
- 69. Billman GE, Kukielka M. Novel transient outward and ultra‐rapid delayed rectifier current antagonist, AVE0118, protects against ventricular fibrillation induced by myocardial ischemia. J Cardiovasc Pharmacol 2008;51:352–358. [DOI] [PubMed] [Google Scholar]
- 70. Amos GJ, Wettwer E, Metzger F, et al Differences between outward currents of human atrial and subepicardial ventricular myocytes. J Physiol 1996;491(Pt 1):31–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Nattel S, Matthews C, De Blasio E, et al Dose‐dependence of 4‐aminopyridine plasma concentrations and electrophysiological effects in dogs : Potential relevance to ionic mechanisms in vivo. Circulation 2000;101:1179–1184. [DOI] [PubMed] [Google Scholar]
- 72. Olson TM, Alekseev AE, Moreau C, et al KATP channel mutation confers risk for vein of Marshall adrenergic atrial fibrillation. Nat Clin Pract Cardiovasc Med 2007;4:110–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Christe G, Tebbakh H, Simurdova M, et al Propafenone blocks ATP‐sensitive K+ channels in rabbit atrial and ventricular cardiomyocytes. Eur J Pharmacol 1999;373:223–232. [DOI] [PubMed] [Google Scholar]
- 74. Goette A, Bukowska A, Lendeckel U. Non‐ion channel blockers as anti‐arrhythmic drugs (reversal of structural remodeling). Curr Opin Pharmacol 2007;7:219–224. [DOI] [PubMed] [Google Scholar]
- 75. Savelieva I, Camm J. Statins and polyunsaturated fatty acids for treatment of atrial fibrillation. Nat Clin Pract Cardiovasc Med 2008;5:30–41. [DOI] [PubMed] [Google Scholar]
- 76. Ducharme A, Swedberg K, Pfeffer MA, et al Prevention of atrial fibrillation in patients with symptomatic chronic heart failure by candesartan in the Candesartan in Heart failure: Assessment of Reduction in Mortality and morbidity (CHARM) program. Am Heart J 2006;152:86–92. [PubMed] [Google Scholar]
- 77. Salehian O, Healey J, Stambler B, et al Impact of ramipril on the incidence of atrial fibrillation: Results of the Heart Outcomes Prevention Evaluation study. Am Heart J 2007;154:448–453. [DOI] [PubMed] [Google Scholar]
- 78. Berkowitsch A, Neumann T, Kuniss M, et al Effects of angiotensin converting enzyme inhibitors and angiotensin receptor blockers in patients with hypertension and atrial fibrillation after pulmonary vein isolation. Heart Rhythm 2008;5:S265 (abstract). [Google Scholar]
- 79. Jang JK, Park JS, Kim YH, et al Effects of the therapy with statins, angiotensin‐converting enzyme inhibitors, and angiotensin II receptor blocker on the outcome after catheter ablation of atrial fibrillation. Heart Rhythm 2008;5:S324 (abstract). [Google Scholar]
- 80. Nakajima H, Nakajima HO, Salcher O, et al Atrial but not ventricular fibrosis in mice expressing a mutant transforming growth factor‐b1 transgene in the heart. Circ Res 2000;86:571–579. [DOI] [PubMed] [Google Scholar]
- 81. Dudley SC, Jr ., Hoch NE, McCann LA, et al Atrial fibrillation increases production of superoxide by the left atrium and left atrial appendage: Role of the NADPH and xanthine oxidases. Circulation 2005;112:1266–1273. [DOI] [PubMed] [Google Scholar]
- 82. Nattel S, Burstein B, Dobrev D. Atrial remodeling and atrial fibrillation: Mechanisms and implications. Circ Arrhythmia Electrophysiol 2008;1:62–73. [DOI] [PubMed] [Google Scholar]
- 83. Burstein B, Libby E, Calderone A, et al Differential behaviors of atrial versus ventricular fibroblasts: A potential role for platelet‐derived growth factor in atrial‐ventricular remodeling differences. Circulation 2008;117:1630–1641. [DOI] [PubMed] [Google Scholar]
- 84. Shimano M, Tsuji Y, Inden Y, et al Pioglitazone, a peroxisome proliferator‐activated receptor‐gamma activator, attenuates atrial fibrosis and atrial fibrillation promotion in rabbits with congestive heart failure. Heart Rhythm 2008;5:451–459. [DOI] [PubMed] [Google Scholar]
- 85. Tsai CT, Lai LP, Kuo KT, et al Angiotensin II activates signal transducer and activators of transcription 3 via Rac1 in atrial myocytes and fibroblasts: Implication for the therapeutic effect of statin in atrial structural remodeling. Circulation 2008;117:344–355. [DOI] [PubMed] [Google Scholar]
- 86. Lin CS, Pan CH. Regulatory mechanisms of atrial fibrotic remodeling in atrial fibrillation. Cell Mol Life Sci 2008;65:1489–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Hanna N, Cardin S, Leung TK, et al Differences in atrial versus ventricular remodeling in dogs with ventricular tachypacing‐induced congestive heart failure. Cardiovasc Res 2004;63:236–244. [DOI] [PubMed] [Google Scholar]
- 88. Xiao HD, Fuchs S, Campbell DJ, et al Mice with cardiac‐restricted angiotensin‐converting enzyme (ACE) have atrial enlargement, cardiac arrhythmia, and sudden death. Am J Pathol 2004;165:1019–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Verheule S, Sato T, Everett T, et al Increased vulnerability to atrial fibrillation in transgenic mice with selective atrial fibrosis caused by overexpression of TGF‐beta1. Circ Res 2004;94:1458–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Adam O, Frost G, Custodis F, et al Role of Rac1 GTPase activation in atrial fibrillation. J Am Coll Cardiol 2007;50:359–367. [DOI] [PubMed] [Google Scholar]
- 91. Burashnikov A. Are there atrial selective/predominant targets for “upstream” atrial fibrillation therapy? Heart Rhythm 2008;5:1294–1295. [DOI] [PubMed] [Google Scholar]
- 92. Singh BN, Connolly SJ, Crijns HJ, et al Dronedarone for maintenance of sinus rhythm in atrial fibrillation or flutter. N Engl J Med 2007;357:987–999. [DOI] [PubMed] [Google Scholar]
- 93. Kober L, Torp‐Pedersen C, McMurray JJ, et al Increased mortality after dronedarone therapy for severe heart failure. N Engl J Med 2008;358:2678–2687. [DOI] [PubMed] [Google Scholar]
- 94. Xing D, Kjolbye AL, Nielsen MS, et al ZP123 increases gap junctional conductance and prevents reentrant ventricular tachycardia during myocardial ischemia in open chest dogs. J Cardiovasc Electrophysiol 2003;14:510–520. [DOI] [PubMed] [Google Scholar]
- 95. Eloff BC, Gilat E, Wan X, et al Pharmacological modulation of cardiac gap junctions to enhance cardiac conduction: Evidence supporting a novel target for antiarrhythmic therapy. Circulation 2003;108:3157–3163. [DOI] [PubMed] [Google Scholar]
- 96. Guerra JM, Everett TH, Lee KW, et al Effects of the gap junction modifier rotigaptide (ZP123) on atrial conduction and vulnerability to atrial fibrillation. Circulation 2006;114:110–118. [DOI] [PubMed] [Google Scholar]
- 97. Shiroshita‐Takeshita A, Sakabe M, Haugan K, et al Model‐dependent effects of the gap junction conduction‐enhancing antiarrhythmic peptide rotigaptide (ZP123) on experimental atrial fibrillation in dogs. Circulation 2007;115:310–318. [DOI] [PubMed] [Google Scholar]