

Abstract
To detect and repair damaged DNA, DNA damage response proteins need to overcome the barrier of condensed chromatin to gain access to DNA lesions1. ATP-dependent chromatin remodeling is one of the fundamental mechanisms used by cells to relax chromatin in DNA repair2–3. However, the mechanism mediating their recruitment to DNA lesions remains largely unknown. BRIT1 (also known as MCPH1) is an early DNA damage response protein that is mutated in human primary microcephaly4–8. We report here a previously unknown function of BRIT1 as a regulator of ATP-dependent chromatin remodeling complex SWI/SNF in DNA repair. Upon DNA damage, BRIT1 increases its interaction with SWI/SNF through the ATM/ATR-dependent phosphorylation on the BAF170 subunit. This increase of binding affinity provides a means by which SWI/SNF can be specifically recruited to and maintained at DNA lesions. Loss of BRIT1 causes impaired chromatin relaxation owing to reduced association of SWI/SNF with chromatin. This explains the decreased recruitment of repair proteins to DNA lesions and reduced efficiency of repair in BRIT1-deficient cells, resulting in impaired survival from DNA damage. Our findings, therefore, identify BRIT1 as a key molecule that links chromatin remodeling with DNA damage response in the control of DNA repair, and its dysfunction contributes to human disease.
BRIT1 (BRCT-repeat inhibitor of hTERT expression) was initially identified as a transcriptional repressor of human telomerase reverse transcriptase (hTERT)4. Its sequence was later matched to that of a disease gene called microcephalin (MCPH1)7. In human, loss-of-function mutations in BRIT1 cause primary microcephaly (MCPH), which is inherited in an autosomal recessive pattern and characterized by a reduction in brain size to one third of normal size7,8. BRIT1 contains three BRCT domains and functions as an early DNA damage response protein5,6. In addition, dysfunction of BRIT1 impairs the recruitment of DNA damage signaling proteins to DNA lesions5. However, how dysfunction of BRIT1 in DNA damage response leads to MCPH remains unknown.
To answer this question, we systematically identified the binding partners of BRIT1, among which we found five core subunits of the human SWI/SNF complex: BRG1/BRM, BAF170, BAF155 and SNF5 (ref 9) (Fig. 1a). SWI/SNF is an ATP-dependent chromatin remodeling complex that utilizes ATP hydrolysis to alter chromatin structure10. The validation of our mass spectrometry result was shown in Fig.1b and Supplementary Fig. 1a.
Figure 1. BRIT1 interacts with the SWI/SNF complex.

(a) Silver staining of the BRIT1 complex separated by SDS-PAGE. The whole cell extracts were prepared from U2OS cells transiently transfected with empty vector or Flag-BRIT1. Subunits of SWI/SNF are indicated. (b) Co-IP of SWI/SNF with BRIT1 analyzed by Western blotting from cells transfected with empty vector (V) or Flag-BRIT1 (B). (c) BRIT1-SWI/SNF interaction dependent on BAF170 (Left) and BAF155 (Right). (d) N-terminal BRIT1 is required for BRIT1-SWI/SNF interaction. (Top) Schematic diagram of BRIT1 deletions. (Bottom) Co-immunoprecipitation of BAF170 with Flag-BRIT1 containing indicated deletions. (e) SWI/SNF interacts specifically with the N-terminal BRIT1 in vitro. Lysates of 293T cells were treated with or without lambda phosphatase, and incubated with purified GST or GST-N-terminal BRIT1. MCM-2 phosphorylation (S40/41) was used as a positive control for effective phosphatase treatment.
To further characterize the BRIT1-SWI/SNF interaction, we first sought to identify the subunit(s) of the SWI/SNF complex that mediated this interaction with BRIT1. Depletion of BAF170 entirely abolished this interaction. Depletion of BAF155 also resulted in loss of interaction between BRG1/BRM and BRIT1, and significantly reduced the interaction between BAF170/SNF5 and BRIT1 (Fig. 1c). In contrast, the two catalytic subunits, BRG1 and BRM, as well as SNF5, were not necessary for BRIT1-SWI/SNF interaction (Supplementary Fig. 1b–e). In addition, Endogenous SNF5 can pulldown other subunits of SWI/SNF in BAF155- or BAF170-deficient cells, excluding the possibility of an unstable SWI/SNF complex due to BAF155- or BAF177 deficiency (Supplementary Fig. 2f). Our data, therefore, showed that the core subunits BAF170 and BAF155 mediate BRIT1-SWI/SNF interaction. Next, we analyzed the critical regions that mediated these interactions. An N-terminal region of BRIT1 was required for its interaction with SWI/SNF (Fig. 1d). We also confirmed the direct binding of this region with SWI/SNF using GST pull-down assay, which was not affected by λ-phosphatase treatment (Fig. 1e), indicating that BRIT1-SWI/SNF interaction is not phosphorylation-dependent in the absence of DNA damage. When analyzing a series of deletion mutants of BAF15511 and BAF170, A conserved SANT domain (595–839aa) of BAF155 and a region (571–645aa) of BAF170 were required for their binding to BRIT1 (Supplementary Fig. 1g, h). Taken together, our data clearly establish an interaction between BRIT1 and the SWI/SNF complex, likely mediated through the N-terminal region of BRIT1 and the specific domains of BAF170 and BAF155 subunits of SWI/SNF.
As BRIT1 is an early DNA damage response protein5,6, we next examined whether the BRIT1-SWI/SNF interaction is responsive to DNA damage. The interaction between BRIT1 and SWI/SNF was indeed enhanced 15 mins after DNA damage with ionizing radiation (IR) (Fig. 2a). To gain mechanistic insights into this DNA damage-enhanced BRIT1-SWI/SNF interaction, we first determined whether this interaction is dependent on ATM and/or ATR, two central kinases in the DNA-damage response network. No apparent change was observed when either ATM or ATR was depleted (Supplementary Fig. 2a, b). However, deficiency of both ATM and ATR abolished the damage-enhanced interaction without affecting the basal binding affinity (Fig. 2b). These results suggest that ATM/ATR kinases are required for the DNA-damage enhanced BRIT1-SWI/SNF interaction. ATM/ATR substrates share a common motif S/TQ. Interestingly, we identified BAF170 (not BAF155) as a potential ATM/ATR substrate, which could be pulled down by the phospho-S/TQ (p-S/TQ) antibody in an ATM/ATR-dependent manner (Fig. 2c). We then generated a series of mutations (serines/threonines to alanines) on BAF170 S/TQ sites and found that when S969 was mutated, there was a significantly decreased the p-S/TQ antibody-binding affinity (Fig. 2d). The sequences around this site are very similar to the sequences around BRCA1 (S1432), a known ATM/ATR target site12 (Supplementary Fig. 2c), which allowed us to detect p-BAF170 (S969) by the antibody against p-BRCA1 (S1432) but not the S969A mutant (Supplementary Fig. 2c). The phosphorylation of this site was further confirmed by the in vitro kinase assay (Fig. 2e).
Figure 2. BRIT1-SWI/SNF interaction is responsive to DNA damage.
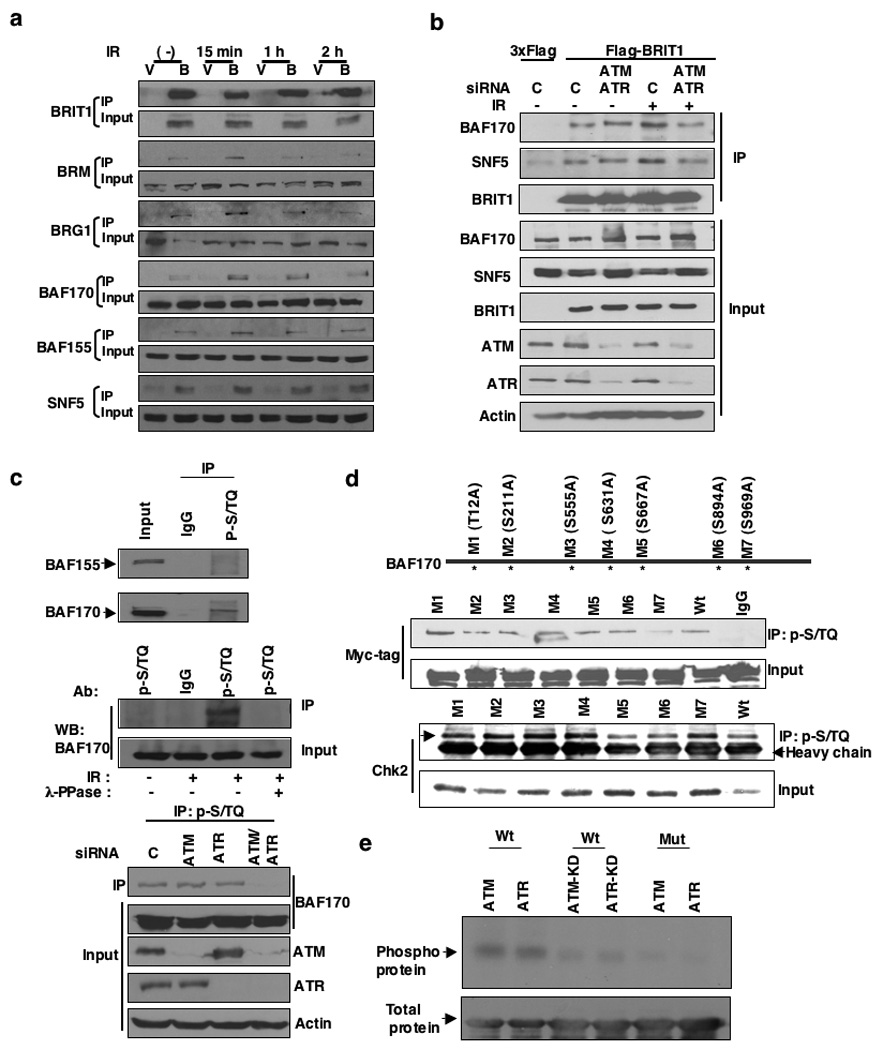
(a) BRIT1-SWI/SNF interaction is enhanced in the presence of DNA damage signaling. Lysates were prepared from cells transfected with empty vector (V) or Flag-BRIT (B) at the indicated time points after exposure to IR (10 Gy) and immunoprecipitated. (b) DNA-damage induced BRIT1-SWI/SNF interaction is dependent on ATM/ATR signaling. Cells were harvested 15 minutes after exposure to IR (10 Gy). (c) BAF170 is a substrate candidate for ATM/ATR (Top and Middle) and its phosphorylation is dependent on the presence of ATM/ATR (Bottom). (d) The mutation on BAF170 (S969) suppressed the recognition of BAF170 by the p-S/TQ antibody after IR. A series of mutations on BAF170 were generated to replace potential ATM/ATR target sites S/TQ to AQ. Chk2-pulldown by p-S/TQ was used as a positive control to show that the general binding activity of p-S/TQ to other ATM/ATR substrates was not affected in cells transfected with M7 mutant. (e) BAF170 (S969) is phosphorylated in vitro by ATM/ATR kinase assay. The sequence around BAF170 (S969) was cloned into GST vector and the phospho-mutant (S969A) was made in this vector.
To test the hypothesis that the phosphorylation of S969 by ATM/ATR upon DNA damage may increase the BRIT1-SWI/SNF binding, we compared the binding affinity of BRIT1 to BAF170 and S969A mutant and found that the mutation of BAF170 on S969 blocked its DNA-damage enhanced but not basal binding affinity to BRIT1 (Supplementary Fig. 2d). We then reconstituted BAF170 expression with either the wild-type or the S969A mutant constructs in BAF170-depleted cells. Although the mutant BAF170 could restore the BRIT1-SWI/SNF interaction in the absence of DNA damage, it failed to recover their DNA-damage enhanced interaction. In contrast, wild-type BAF170 readily rescued both (Supplementary Fig. 2e), suggesting that the enhanced BRIT1-SWI/SNF interaction in response to DNA damage is mediated through an ATM/ATR-dependent BAF170 phosphorylation.
Recent studies have demonstrated a role of ATP-dependent chromatin remodeling complexes in repairing DNA double-strand breaks (DSBs)1–3 and in the maintenance of cell survival after DNA damage13,14. We have previously shown that BRIT1 deficiency leads to prolonged H2AX phosphorylation upon DNA damage, indicating potentially impaired DNA repair5. Therefore, we tested whether the BRIT1-SWI/SNF interaction may function in repairing DSB.
Firstly, we used neutral pH comet assays to demonstrate BRIT1 deficiency resulted in a significantly defect in DSB repair (Supplementary Fig. 3a, b). In mammalian cells, two conserved pathways are involved in DSB repair, homologous recombination (HR) and nonhomologous end joining (NHEJ)15,16. To confirm BRIT1’s role in DSB repair and determine which repair pathway it acted in, we analyzed BRIT1-deficient cells using an HR repair analysis system17 (supplementary Fig. 3c, d) and found BRIT1 knockdown cells showed a significant decrease (40–60%) in HR repair induced-GFP+ cells, indicating defective HR repair (Fig. 3a). Decreased GFP signal was not due to effects on cell cycle distribution, transfection efficiency or cutting efficiency of I-SceI (Supplementary Fig. 3e–g). An indirect reduction in HR repair, through reduced expression of BRCA1 by BRIT1 knockdown, another component of HR repair 18–20 was also excluded (Supplementary Fig. 3h, i). During the course of our studies, another study also confirmed this new function of BRIT121 in HR repair.
Figure 3. BRIT1 depletion impairs DNA DSB repair.
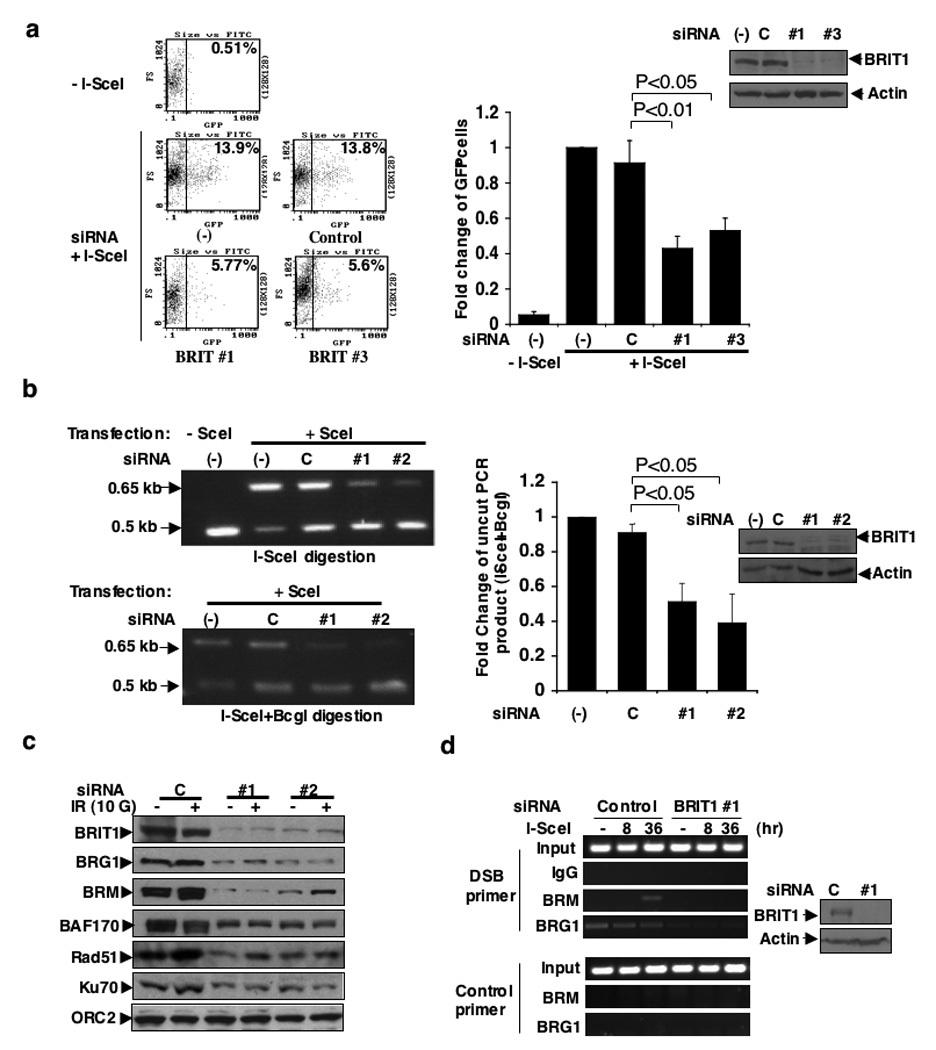
(a) Defective HR repair in BRIT1-depleted-cells transfected with BRIT1 siRNA #1 (#1) and siRNA #2 (#2) upon DSB induced by I-SceI. (Left) Representative flow cytometry profile. (Right) Quantitative summary of at least three independent experiments. Each value is relative to the percentage of GFP+ cells in I-SceI-transfected cells without siRNA transfection, which was set to 1 and represents the mean ± SD of three independent experiments; Student’s t-test. (b) Defective NHEJ repair in BRIT1-depleted-cells transfected with BRIT1 siRNA #1 (#1) and siRNA #3 (#3) upon DSB induced by I-SceI. (Left) Representative image of PCR products digested by I-SceI or I-SceI+BcgI. (Right) Quantification of NHEJ repair product (uncut by I-SceI+BcgI). Percentage of uncut products in cells without siRNA transfection was set as 1. Each value represents the mean ± SD of three independent experiments; Student’s t-test. Western blot analyses to demonstrate the effective BRIT1 knockdown were shown next to the graph. (c) Impaired recruitment of SWI/SNF and DNA repair proteins to chromatin in BRIT1-depleted cells (Time course study and densitometry analyses were shown in supplementary Fig. 4a, b). (d) Impaired recruitment of SWI/SNF to the DNA damage site. ChIP analyses were performed at the indicated time points after induction of DSB by I-SceI transfection.
In parallel, we used the method as described in Supplementary Fig. 3j to analyze NHEJ repair22 and found that NHEJ repair efficiency was also decreased in BRIT1 knockdown cells (50–60%) (Fig. 3b), which was not reversed by ectopic expression of BRCA1, but by the ectopic expression of siRNA-resistant BRIT1 (Supplementary Fig. 3k). Altogether, our data reveal a critical function of BRIT1 in both HR and NHEJ repair.
HR and NHEJ are two distinct mechanisms for DSB repair. However, both mechanisms are confronted with DNA wrapped into highly condensed chromatin structure. Therefore, BRIT1’s involvement in both HR and NHEJ could be explained by both pathways requiring chromatin relaxation to allow access of repair proteins to DNA lesions. Such access could be provided by BRIT1 facilitating association of SWI/SNF complex with chromatin and so promoting chromatin relaxation. In the first experiment to examine this possibility, we found BRIT1 depletion significantly reduced the amount of chromatin-associated BRG1, BRM, BAF170 and two key DNA repair proteins Rad51 and Ku7015,16, while their total expression remained constant (Fig. 3c and Supplementary Fig. 4a–c).
To address whether SWI/SNF recruitment was altered specifically at sites of induced DSBs, chromatin immunoprecipitation assays were performed utilizing the I-SceI GFP system described above. BRM and BRG1 are two catalytic subunits of SWI/SNF complex. The recruitment of BRM after I-SceI induced DSB was abolished in BRIT1 knockdown cells (Fig. 3d). Both basal and damage-induced DNA localization of BRG1 was also undetectable in BRIT1 knockdown cells (Fig. 3d). In contrast, depletion of individual SWI/SNF subunit affected neither the association of BRIT1 to chromatin nor its recruitment to the DNA damage loci (Supplementary Fig. 4d), placing SWI/SNF functions downstream of BRIT1.
As SWI/SNF relaxes chromatin and hence facilitates protein access to chromatin, we reasoned that impaired recruitment of SWI/SNF to chromatin in BRIT1-deficient cells might affect the state of chromatin relaxation and consequently the recruitment of the downstream DNA repair proteins to DNA lesions. To test this hypothesis, we assessed the extent of chromatin condensation using a micrococcal nuclease (MNase) sensitivity assay, which provides a measure of chromatin compaction1,23. BRIT1 knockdown cells were less sensitive to MNase digestion in both the absence and presence of DNA damage, indicating that chromatin structure is more compact in BRIT1-deficient cells (Fig. 4a and Supplementary Fig. 7h). Consistently, the impaired chromatin relaxation and the defective HR repair were also observed in SWI/SNF knockdown cells (Supplementary Fig. 5d–f).
Figure 4. BRIT promotes the function of SWI/SNF.
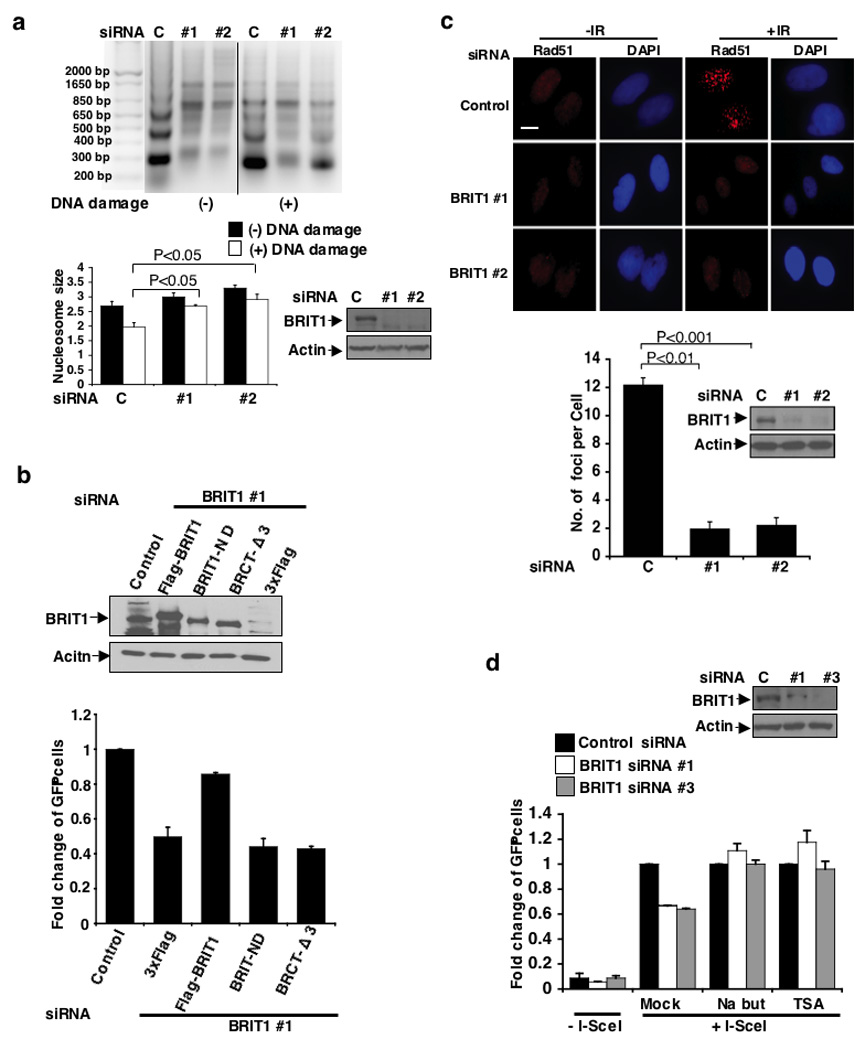
(a) Impaired chromatin relaxation analyzed by MNase sensitivity assay. Cells were exposed to 4 Gy IR. (Top) Representative image. (Bottom) Quantification of average nucleosome size. Each value represents the mean ± SEM of of three independent experiments; Student’s t-test. (b) The function of BRIT1 in DNA repair is dependent on its interaction with SWI/SNF. BRIT1-deficient cells were reconstituted with indicated plasmids. BRIT1 with N-terminal deletion (BRIT1-ND), which lacks its SWI/SNF interaction domain, was unable to rescue the defect of DNA repair to a similar extent to the BRIT1 BRCT-Δ3 mutant that lost its activity to be recruited to DNA damage lesions. Each value is relative to the percentage of GFP+ cells in I-SceI-transfected cells with control siRNA transfection, which was set to 1 and represents the mean ± SD of three independent experiments. (c) Requirement of BRIT1 for Rad51 foci formation. (Top) Representative immunostaining images. Scale bar is 10 µM. (Bottom) The bar graph represents the mean ± SD of three independent experiments; Student’s t-test. At least 50 cells were scored in each sample. (d) Restored HR repair in BRIT1 deficient cells in the presence of chromatin relaxation agents. Each value is relative to the percentage of GFP+ cells without siRNA transfection, which was set as 1, and represents the mean ± SD of three independent experiments. Western blot analyses to demonstrate the effective BRIT1 knockdown were shown next to the graph.
To demonstrate that the function of BRIT1 in chromatin relaxation and DNA repair is dependent on SWI/SNF, we made a small deletion (1-48aa) on N-terminal of BRIT1 (BRIT1-ND), which abolished its interaction with SWI/SNF but preserved its ability to form DNA-damage-induced foci (Supplementary Fig. 5a, b). By reconstitution of wild-type BRIT1 or BRIT1-ND to BRIT1-deficient cells, we observed that in contrast to wild-type construct, BRIT1-ND was unable to restore the defects in chromatin relaxation and DNA repair in BRIT1 knockdown cells, a phenomenon similar to our observations in BRCT1-Δ3 reconstituted cells (Fig. 4b, Supplementary Fig. 5a). As a consequence, the BRIT1-ND reconstituted cells still exhibited increased sensitivity to IR (Supplementary Fig. 5c). It is worthwhile to mention that since BRIT1 BRCT-Δ3 mutant could not form DNA-damage induced foci, it is not surprising that this mutant also failed to restore chromatin relaxation and DNA repair activity.
We also tested whether the mutants of BAF155 or BAF170 which lacked BRIT1-binding activity could exert dominant-negative effects to block proper DNA damage response such as DNA damage repair (Supplementary Fig. 5g–i). By sequence analysis, we found that BAF155 contained a highly hydrophobic sequence on its BRIT1-interacting domain (SANT), which has been reported to be essential for the function of SANT domain24. Interestingly, this sequence is also conserved in the BRIT1-interacting domain of BAF170. Therefore we replaced 4 consecutive leucines on BAF155 (629–632aa) and BAF170 (607–610aa) to arginines. These subtle mutations abolished their binding activity to BRIT1 without affecting their incorporations into the endogenous SWI/SNF complex. Notably, overexpression of these mutants reduced the binding of BRIT1 to other SWI/SNF subunits and thus exerted dominant-negative effects that impaired HR repair in the cells. Collectively, these data further support that dysfunction of SWI/SNF is the underlying mechanism responsible for impaired chromatin relaxation, HR repair and cell survival in BRIT1-deficient cells.
We next tested whether impaired chromatin relaxation would lead to defects in the recruitment of DNA repair proteins to DNA damage sites. The foci formation of Rad51 and phospho-replication protein A (p-RPA), key players in DSB repair15,25 was significantly reduced in BRIT1-depleted cells (Fig. 4c, Supplementary Fig. 6a). Chromatin binding of p-RPA34 was also impaired. However, treatment of chromatin relaxation agents significantly reversed the effects of BRIT1 depletion on RPA foci formation, phosphorylation and binding to chromatin (Supplementary Fig. 6a–d). Consistent with this notion, reduced HR repair efficiency in BRIT1 knockdown cells was reversed in the presence of chromatin relaxation agents (Fig. 4d), indicating that the impaired recruitment of DNA repair proteins is a direct consequence of impaired access to chromatin in BRIT1-deficient cells.
To confirm the physiological relevance of our findings, we examine MCPH patient lymphoblastoid cell lines (LCLs) with homozygous loss-of-function mutations in BRIT1 (Supplementary Fig. 7a).
Comet assays demonstrated a significantly reduced DSB repair efficiency in BRIT1 LCLs (Fig. 5a, Supplementary Fig. 7b). Consistent with this, BRIT1 LCL also exhibited increased sensitivity to the topoisomerase inhibitors camptothecin and etoposide, which generate DSBs during S phase, a cell cycle phase in which lesions are predominantly repaired by HR26. This increased sensitivity was consistent with DSB generation during S-phase as the effects were abrogated when cells were treated with the DNA replication inhibitor aphidicolin (Fig. 5b). In addition, increased sensitivity to IR-induced DNA damage was observed in BRIT1 LCLs arrested in G1 phase, a cell cycle exclusively utilizing NHEJ to repair DSBs (Supplementary Fig. 7c). Together, our data suggested that BRIT1 LCL might have impaired cell survival as a result of generated DSBs being un-repaired because of both the defective HR and NHEJ repair. Furthermore, repair foci formation was also impaired in these cells with significantly reduced recruitment of RPA and Rad51 (Fig. 5c). These results were further confirmed by our detection of a decreased association of DNA repair proteins to chromatin in patients’ cells, while total protein levels were unaffected (Supplementary Fig. 7d–f).
Figure 5. BRIT1 promotes DNA repair and chromatin relaxation in two human MCPH lymphoblastoid cell lines (MCPH #1 and MCPH #2).
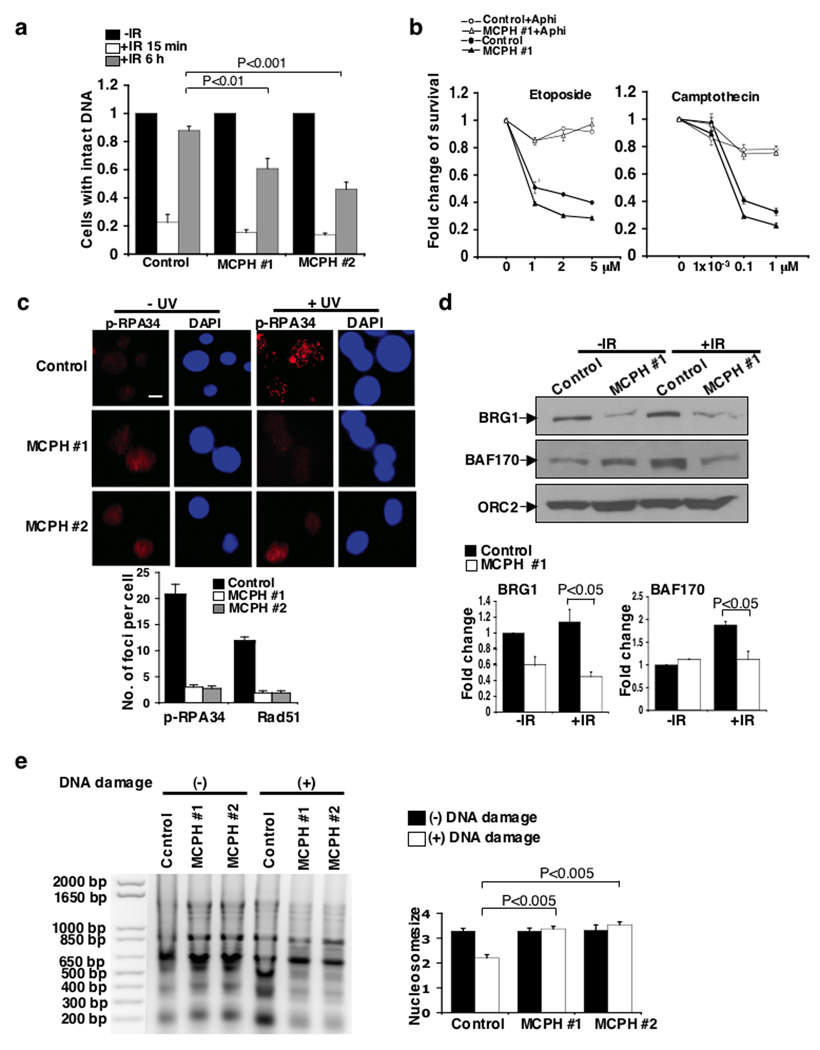
(a) Impaired DNA repair analyzed by comet assay after exposure of cells to IR. Percentage of cells with intact DNA (tail moment less than 2) in cells without IR exposure was set as 1. At least 100 cells were scored in each sample and each value represents the mean ± SEM of three independent experiments; Student’s t-test. Representative images are shown in Supplementary Fig. 7b. (b) Impaired cell survival in MCPH #1 cells exposed to DSB-inducing agents. Cells were pre-treated with the DNA replication inhibitor aphidicolin (Aphi) where indicated, and then treated with etoposide (Left) or camptothecin (Right). The graphs represent the mean ± SD of three independent experiments (c) Requirement of BRIT1 for foci formation of DNA repair proteins. (Top) Immunostaining of p-RPA foci. Scale bar is 10 µM. (Bottom) Quantification of p-RPA and Rad51 foci formation from three independent experiments (mean ± SD). At least 50 cells were scored in each sample for each experiment. (d) Reduced association of SWI/SNF to chromatin. (Top) Representative Western blots. (Bottom) Densitometry analyses of indicated protein values normalized against ORC2. Each value represents the mean ± SD of three independent experiments; Student’s t-test. (e) Impaired chromatin relaxation analyzed by MNase sensitivity assay. Cells were exposed to the DSB inducing agent neocarzinostatin (400 ng/mL). (Left) Representative image. (Right) Quantification of average nucleosome size. Each value represents the mean ± SEM of three independent experiments; Student’s t-test.
SWI/SNF binding to chromatin was also severely impaired in BRIT1 LCL cells (Fig. 5d). Again, reduced binding of SWI/SNF to chromatin was not due to decreased total cellular protein levels (Supplementary Fig. 7e). Importantly, in keeping with our RNAi studies, BRIT1 LCLs did not undergo chromatin relaxation after DNA damage, unlike control LCL. Control LCL chromatin exhibited increased sensitivity to MNase after neocarzinostatin induced DNA damage, while BRIT1 LCLs chromatin remained more resistant to MNase digestion (Fig. 5e, and time course, Supplementary Fig. 7h). Induction of chromatin relaxation also restored damaged-induced phosphorylation of RPA in BRIT1 LCLs (Supplementary Fig. 7g). Notably, the defects of cell survival, and chromatin relaxation could be rescued by the introduction of wild-type Flag-BRIT1 into BRIT1 deficient-LCLs, but not by the introduction of BRCT-Δ1 mutant, which abrogated its SWI/SNF-binding activity (Supplementary Fig. 7i–l). We also found a partial rescuing effect from BRCT-Δ2 mutant which might have been due to the requirement of C-terminal BRCT domain in other cellular functions6. Therefore our findings in BRIT1 LCLs are again consistent with a requirement for BRIT1 to mediate chromatin relaxation and the recruitment of DNA repair proteins to DNA lesions after DNA damage.
In summary, our results suggested a model for BRIT1 function. BRIT1 interacts with SWI/SNF via the core subunits BAF170 and BAF155. These interactions are enhanced in response to DNA damage through an ATM/ATR-dependent phosphorylation of BAF170. We suspect that BRIT1 is required for the recruitment and maintenance of SWI/SNF at DNA lesions and through which BRIT1 promotes chromatin relaxation and in turn facilitates the recruitment of DNA repair proteins to DNA lesions for efficient repair. Therefore, loss of BRIT1 would lead to impaired chromatin relaxation and DNA DSB repair, which may contribute to the development of MCPH and cancer.
Also, besides its recognition of histone modifications2,3, our findings reveal a mechanism by which the SWI/SNF complex is recruited to DNA lesions without containing intrinsic specificity for particular nuclear process10,27–28. Indeed, multiple mechanisms may be involved regulating chromatin structure in order to cope with different stages of damage response and/or response to different types of DNA lesions and/or repair DNA lesions located in different regions of chromatin (euchromatin or heterochromation)1,23,29. In addition, our studies reveal that post-translational modifications such as phosphorylation may serve as critical mechanisms to regulate the functions of SWI/SNF. Therefore it will be of future interests to illustrate the additional roles of phosphorylation on other SWI/SNF subunits in DNA damage response12 and impaired its function in the pathogenesis of human diseases30,31.
METHODS
Cell culture
U2OS and 293T cells were purchased from the American Type Culture Collection. The U2OS cells were maintained in McCoy’s 5A medium supplemented with 10% fetal bovine serum (FBS). 293T wre grown in Dulbecco’s modified Eagle’s medium (DMEM) with 10% FBS. Lymphoblastoid control cell line and two MCPH cell lines [MCPH#1 (C74G)7; MCPH#2 (G321C; Personal communication, A.P. Jackson & E. Griffth)] were grown as a suspension culture in RPMI 1640 medium supplemented with 20% FBS.
Plasmids, small interfering RNAs (siRNAs), and transfection
siRNA #1, #2, #3 sequences, control siRNAs, Flag-BRIT1, Flag-BRIT1 mutant resistant to siRNA#1, BRCA1 plasmids and the procedures for BRIT1 knockdown and ectopic expression of BRCA1-HA, Flag-BRIT1, or deletion mutants of BRIT1 in BRIT1 knockdown cells were all previously described5,18. On-target smart pool siRNAs against BAF170, BAF155, SNF5, ATM and ATR were purchased from Dharmacon Research (Lafayette, CO). Short hairpin RNA (shRNA) vectors targeting BRG1 or BRM were purchased from Sigma. The deletions of BRIT1 were generated from Flag-BRIT1 plasmid via polymerase chain reaction (PCR) using primers with restriction sites and subcloned into N-terminal p3xFlag-CMV plasmid in frame. A series of deletion mutants of BAF155 are kindly provided by Dr. Archer, T.K. (NIH, North Carolina)12. Flag-tagged ATM, ATR, ATM-KD (catalytic dead) and ATR-KD plasmids were generously provided by Dr. Kastan, M. (St. Jude Children’s Research Hospital, Memphis, TN.), Dr. Cimprich, K. (Stanford University) and Dr. Zou, L. (Harvard University). BAF170 was cloned from cDNA of HMEC cells (normal breast epithelial cells). The deletions of BAF170 were generated via PCR using primers with restriction sites and subcloned into pCMV/myc/nuc (Invitrogen). All the mutations described in the paper were generated by QuickChange II Site-Directed Mutagenesis Kit (Stratagene). Fragments containing BAF170 (S969) and BAF170 (S969A) were sub-cloned into pGEX vectors (GE Healthcare). Detailed cloning information is available upon request. Plasmids were verified by DNA sequencing. Oligofectamine (Invitrogen) was used for all siRNA transfections and FuGENE 6 (Roche) was used for all plasmids transfection following the manufacturers’ protocols. Transfection in LCLs was done as previously described8.
Affinity purification of BRIT1 protein complex
U2OS cells were transiently transfected with empty Flag plasmid or Flag-BRIT1 plasmid. Forty-eight hours later, whole cellular extracts were prepared with RIPA buffer (50 mM Tris Hcl pH7.4, 1% NP-40, 150 mM NaCl, 1 mM EDTA, 10% Na-deoxycholate, freshly added with 1 mM PMSF, 1 mM Na3VO4, and 1 mM NaF) and immunoprecipitated with anti-Flag M2 affinity gel (Sigma) overnight. Bead-bound immunocomplexes were eluted with 3xFlag peptide (Sigma) and subjected to SDS-PAGE. The silver staining was performed with SilverSNA kit for Mass spectrometry (Pierce). Specific bands were excised, digested and the peptides were analyzed by a mass spectrometry analysis at the M. D. Anderson Cancer Center Proteomic Facility.
Purification of GST-fusion proteins and GST pull down assay
Purification and GST pull down methods were adapted from previous publication32. BL21 bacteria containing indicated plasmids were allowed to grow 6 hrs after addition of IPTG. Cell pellets were resuspend in lysis buffer and sonicated. The supernatant was incubated with glutathione-agarose beads at 4°C for overnight. After washing, GST fusion proteins were eluted with glutathione. Then cell lysates (1 mg) were incubated with 2–5 µg GST fusion protein and 40 µl Gluthatione-agarose beads in a total 1 ml RIPA buffer at 4°C on a rotator for 2–4 hrs. After washing the beads with RIPA buffer for 3 times, elute the protein for SDS-PAGE gel analysis.
In vitro ATM and ATR kinase assay
Immunoprecipitations of ATM and ATR and in vitro kinase assay were performed as described earlier33. Briefly, 293T cells were transfected with 8 µg Flag-tagged ATM, ATR, ATM-KD and ATR-KD plasmids. Cell extracts were prepared in lysis buffer containing (50 mM Tris, pH 7.5, 150 mM NaCl, 1% Tween 20, 0.3% Nonidet P-40, 1 mM sodium fluoride, 1 mM Na3VO4, 1 mM phenylmethylsulfonyl fluoride, 50 mM glyycerophosphate, 1 mM DTT, 1mM EGTA, 10% glycerol and 1 _ protease inhibitor mixture from Roche Molecular Biochemicals). Cleared supernatants were immunoprecipitated with anti-Flag M2 antibody (sigma). After washing with lysis buffer and kinase buffer (20 mM HEPES, pH 7.5, 50 mM NaCl, 10 mM MgCl2, 1 mM dithiothreitol, and 10 mM MnCl2) 5 times, the immunoprecipitant was resuspended in 50 µl of kinase buffer containing 10 µCi of [-32P]ATP, 10 µM ATP, 1 mM sodium fluoride, 1 mM Na3VO4, 20 mM glyycerophosphate and 1 µg of GST fusion substrate. The kinase reaction was performed at 30 °C for 20 min and stopped by the addition of SDS sample buffer. Proteins were separated on SDS PAGE gel and transferred to PVDF membrane. Radiolabeled proteins were visualized by autoradiography.
Antibodies, immunoprecipitation, chromatin fractionation and Western blot analysis
BRIT1, BRCA1, RPA34, and p-RPA34 antibodies were previously described5,22. Rabbit anti-SWI/SNF subunits antibodies (BRG1, BRM, BAF170, BAF155 and SNF5), MCM2 and p-MCM2 (Ser40/Ser41) were purchase from Bethyl Laboratory. Rabbit anti-Rad51 (Ab-1) antibody was purchased from Calbiochem. Mouse anti-Ku70 antibody was purchased from Novus. Control rabbit Immunoglobulin G (IgG) and anti-ATR antibody were purchased from Santa Cruz. Anti-Mcy tag antibody, Chk1, p-Chk1 (Ser345), Chk2, p-Chk2 (Ser68), p-S/TQ antibodies, and anti-ATM antibody was ordered from Cell signaling. The immunoprecipitation with anti-Flag affinity beads was done as described above. For reciprocal immunoprecipitation, whole cellular extracts were prepared in RIPA buffer as indicated above and precleaned with protein A/G plus-agarose beads (Santa Cruz). Then cellular extracts were subjected to incubation with antibodies against BRG1 or BRGM (2 µg) for 2 h followed by incubation with protein A/G agarose beads overnight at 4 °C. The immunocomplex was eluted in loading buffer by boiling at 95 °C for 5 min. The preparation of chromatin fractions and Western blot analysis including the conditions for RPA analysis were done as previously described5,22. For the chromatin fractions to analyze SWI/SNF and DNA repair proteins, cells were exposed to IR (10 Gy), then harvested 2 h or 5 h later respectively. Densitometry analysis was done using NIH IMAGE software.
Chromatin immunoprecipitation (ChIP) assay
DSBs were induced in cells transfected with control siRNA or BRIT1 siRNA#1 by I-SceI expression. At indicated time points, cells were crosslinked with formaldehyde and ChIPs were performed using an EZ ChIP kit (Upstate) following the manufacturer’s instructions. Cellular lysates were subjected to five sets of sonication on wet ice with a 60 Sonic Dismembrator (Fisher Scientific). Each set consists of 8 seconds of sonication with 1 min interval on ice. BRG1, BRM (10 µl) antibodies were used for immunoprecipitation. The ChIP primers used to analyze proteins binding at the site of DSBs were: sense 5′-TACGGCAAGCTGACCCTGAA-3′; antisense, 5′-GCCCATATATGGAGTTCCGC-3′.
The primers used as a negative control on chromosome 12 were: sense, 5′-ATGGTTGCCACTGGGGATCT-3′; antisense 5′-TGCCAAAGCCTAGGGGAAGA-3′.
Methods for the following assays are available in Supplementary Information
DNA repair assays (Comet assay, HR repair analysis, NHEJ repair analysis); Cell cycle analysis; Micrococcal nuclease (MNase) sensitivity assay; Colony-forming assay; Cell viability assay; Immunofluorescent staining for foci formation;
Statistical analysis
All statistical analysis was done by one-tailed Student’s t- test.
Supplementary Material
ACKNOWLEDGEMENTS
We thank S. Rosenberg, X. Shen, and F. Meric-Bernstam for critical reading of the manuscript; S. Deming for proofreading the manuscript; M. Jasin for reagents; and M. D. Anderson Cancer Center core facilities for mass-spec and FACS. We also thank Dr. E. Griffith for identification of the mutation in the MCPH#2 cell line. This work was supported by grants from the NCI (R01CA112291) and American Cancer Society Research Scholar Award to S. Y. L.
Footnotes
COMPETING FINANCIAL INTERESTS The authors declare that we have no competing financial interests.
REFERENCE
- 1.Tsukuda T, Fleming AB, Nickoloff MA. Chromatin remodeling at a DNA double-strand break site in Saccharomyces cerevisiae. Nature. 2005;438:379–383. doi: 10.1038/nature04148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Morrison AJ, et al. IN80 and gamma-H2AX interaction links ATP-dependent chromatin remodeling to DNA damage repair. Cell. 2004;119:767–775. doi: 10.1016/j.cell.2004.11.037. [DOI] [PubMed] [Google Scholar]
- 3.van Attikum H, Fritsch O, Hohn B, Gasser SM. Recruitment of the INO80 complex by H2A phosphorylation links ATP-dependent chromatin remodeling with DNA double-strand break repair. Cell. 2004;119:777–788. doi: 10.1016/j.cell.2004.11.033. [DOI] [PubMed] [Google Scholar]
- 4.Lin SY, Elledge SJ. Multiple tumor suppressor pathways negatively regulate telomerase. Cell. 2003;113:881–889. doi: 10.1016/s0092-8674(03)00430-6. [DOI] [PubMed] [Google Scholar]
- 5.Rai R, et al. BRIT1 regulates early DNA damage response, chromosomal integrity, and cancer. Cancer Cell. 2006;10:145–157. doi: 10.1016/j.ccr.2006.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wood JL, Singh N, Mer G, Chen J. MCPH1 functions in an H2AX-dependent but MDC1-independent pathway in response to DNA damage. J. Biol. Chem. 2007;282:35416–35423. doi: 10.1074/jbc.M705245200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jackson AP, et al. Identification of microcephalin, a protein implicated in determining the size of the human brain. Am. J. Hum. Genet. 2002;71:136–142. doi: 10.1086/341283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alderton GK, et al. Regulation of mitotic entry by microcephalin and its overlap with ATR signaling. Nat. Cell. Biol. 2006;8:725–733. doi: 10.1038/ncb1431. [DOI] [PubMed] [Google Scholar]
- 9.Phelan ML, Sif S, Narlikar GJ, Kingston RE. Reconstitution of a core chromatin remodeling complex from SWI/SNF subunits. Molecular Cell. 1999;3:247–253. doi: 10.1016/s1097-2765(00)80315-9. [DOI] [PubMed] [Google Scholar]
- 10.Martens JA, Winston F. Recent advances in understanding chromatin remodeling by Swi/Snf complex. Curr. Opin. Genet. Dev. 2003;13:136–142. doi: 10.1016/s0959-437x(03)00022-4. [DOI] [PubMed] [Google Scholar]
- 11.Chen J, Archer TK. Regulating SWI/SNF subunit levels via protein-protein interactions and proteasomal degradation: BAF155 and BAF170 limit expression of BAF57. Mol. Cell. Biol. 2005;25:9016–9027. doi: 10.1128/MCB.25.20.9016-9027.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Matsuoka S, et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316:1160–1166. doi: 10.1126/science.1140321. [DOI] [PubMed] [Google Scholar]
- 13.Chai B, Huang J, Cairns BR, Laurent BC. Distinct roles for the RSC and Swi/Snf ATP-dependent chromatin remodelers in DNA double-strand break repair. Genes Dev. 2005;19:1656–1661. doi: 10.1101/gad.1273105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Park JH, et al. Mammalian SWI/SNF complexes facilitate DNA double-strand break repair by promoting gamma-H2AX induction. Embo. J. 2006;25:3986–3997. doi: 10.1038/sj.emboj.7601291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sung P, Klein H. Mechanism of homologous recombination: mediators and helicases take on regulatory functions. Nature Rev. Mol. Cell. Biol. 2006;7:739–750. doi: 10.1038/nrm2008. [DOI] [PubMed] [Google Scholar]
- 16.Lieber MR, Ma Y, Pannicke U, Schwarz K. Mechanism and regulation of human non-homologous DNA end-joining. Nature Rev. Mol. Cell Biol. 2003;4:712–720. doi: 10.1038/nrm1202. [DOI] [PubMed] [Google Scholar]
- 17.Pierce AJ, Johnson RD, Thompson LH, Jasin M. XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev. 1999;13:2633–2638. doi: 10.1101/gad.13.20.2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lin SY, Rai R, Li K, Xu ZY, Elledge SJ. BRIT1/MCPH1 is a DNA damage responsive protein that regulates the Brca1-Chk1 pathway, implicating checkpoint dysfunction in microcephaly. Proc. Natl. Acad. Sci. USA. 2005;102:15105–15109. doi: 10.1073/pnas.0507722102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Scully R, Livingston DM. In search of the tumour-suppressor functions of BRCA1 and BRCA2. Nature. 2000;408:429–432. doi: 10.1038/35044000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu X, Lee J, Stern DF. Microcephalin is a DNA damage response protein involved in regulation of CHK1 and BRCA1. J. Biol. Chem. 2004;279:34091–34094. doi: 10.1074/jbc.C400139200. [DOI] [PubMed] [Google Scholar]
- 21.Wood JL, Liang Y, Li K, Chen J. Microcephalin/MCPH1 associates with the Condensin II complex to function in homologous recombination repair. J. Biol. Chem. 2008;283:29586–29592. doi: 10.1074/jbc.M804080200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nakanishi K, et al. Human Fanconi anemia monoubiquitination pathway promotes homologous DNA repair. Proc. Natl. Acad. Sci. SA. 2005;102:1110–1115. doi: 10.1073/pnas.0407796102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ziv Y, et al. Chromatin relaxation in response to DNA double-strand breaks is modulated by a novel ATM-and KAP-1 dependent pathway. Nat. Cell. Biol. 2006;8:870–876. doi: 10.1038/ncb1446. [DOI] [PubMed] [Google Scholar]
- 24.Boyer LA, Latek RR, Peterson CL. The SANT domain: a unique histone-tail-binding Nature Rev. Mol. Cell. Biol. 2004;5:1–6. doi: 10.1038/nrm1314. [DOI] [PubMed] [Google Scholar]
- 25.Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003;300:1542–1548. doi: 10.1126/science.1083430. [DOI] [PubMed] [Google Scholar]
- 26.Sartori AA, et al. Human CtIP promotes DNA end resection. Nature. 2007;450:509–514. doi: 10.1038/nature06337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Narlikar GJ, Fan H, Kingston RE. Cooperation between complexes that regulate chromatin structure and transcription. Cell. 2002;108:475–487. doi: 10.1016/s0092-8674(02)00654-2. [DOI] [PubMed] [Google Scholar]
- 28.Peterson CL, Workman JL. Promoter targeting and chromatin remodeling by the SWI/SNF complex. Curr. Opin Genet. Dev. 2000;10:187–192. doi: 10.1016/s0959-437x(00)00068-x. [DOI] [PubMed] [Google Scholar]
- 29.Goodarzi AA, et al. ATM signaling facilitates repair of DNA double-strand breaks associated with heterochromatin. Molecular Cell. 2008;31:167–177. doi: 10.1016/j.molcel.2008.05.017. [DOI] [PubMed] [Google Scholar]
- 30.Rass U, Ahel I, West SC. Defective DNA repair and neurodegenerative disease. Cell. 2007;130:991–1004. doi: 10.1016/j.cell.2007.08.043. [DOI] [PubMed] [Google Scholar]
- 31.Roberts CWM, Orkin SH. The SWI/SNF complex-chromatin and cancer. Nature Rev. Cancer. 2004;4:133–142. doi: 10.1038/nrc1273. [DOI] [PubMed] [Google Scholar]
- 32.Yu X, Ghini CC, He M, Mer G, Chen J. The BRCT domain is a phospho-protein binding domain. Science. 2003;302:639–642. doi: 10.1126/science.1088753. [DOI] [PubMed] [Google Scholar]
- 33.Pandita TK, et al. Ionizing radiation activates the ATM kinase throughout the cell cycle. Oncogene. 2000;19:1386–1391. doi: 10.1038/sj.onc.1203444. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.