

Abstract

Metal-free, nucleophilic activation of a B–B bond has been exploited in the development of a highly efficient method for conjugate additions of commercially available bis(pinacolato)diboron to cyclic or acyclic α,β-unsaturated carbonyls. Reactions are readily catalyzed by 2.5–10 mol % of a simple N-heterocyclic carbene (NHC). A variety of cyclic and acyclic unsaturated ketones and esters can serve as substrates. Transformations deliver β-boryl carbonyls bearing tertiary as well as quaternary B-substituted carbons in up to >98% yield. Preliminary studies indicate that, although related Cu–NHC-catalyzed reactions are equally efficient, the metal-free variant is more functional group tolerant; in contrast to the Cu-catalyzed reactions, the metal-free processes proceed readily in the presence of a terminal alkyne and do not promote concomitant diboration of an aldehyde. Representative functionalization of the resulting boron enolates demonstrates the strong influence of the Lewis acidic B of the β-boronate.
Development of metal-free catalytic processes is critical to advancement of modern chemical synthesis; such protocols are valuable even in cases where a metal-catalyzed variant is available. The two reaction classes often proceed by mechanistically distinct pathways and can give rise to complementary reactivity and selectivity patterns. It is thus noteworthy that C–B bond formation has remained exclusively in the domain of metal-based catalysis.1 Pt-,2 Rh-,3 Ni-4 and Cu-catalyzed 5 conjugate additions of diborons 6 with unsaturated esters have been disclosed; these transformations are largely or entirely limited to reactions of acyclic substrates (with achiral and chiral catalysts, respectively). In only three cases is the formation of quaternary B-substituted carbons reported (all with acyclic enones).4 Herein, we introduce a method for efficient C–B bond formation through transformations that are catalyzed by 2.5–10 mol % of a readily available N-heterocyclic carbene (NHC). Cyclic and acyclic α,β-unsaturated ketones or esters serve as substrates; β-boryl carbonyls with tertiary or quaternary B-substituted carbons are obtained in up to >98% yield. We illustrate that boron conjugate addition under metal-free conditions delivers reactivity and site-selectivity levels not attainable through the use of a Cu-catalyzed variant.5
The present investigations are based on the principle that a nucleophilic NHC7 might associate with a Lewis acidic boron of commercially available bis(pinacolato)diboron (1) (Scheme 1).8 The resulting electronic reorganization9,10 could lead to activation of the B–B bond, promoting reaction with an appropriate electrophilic site through the general pathway in Scheme 1.
Scheme 1.

Proposed Model for Bis(pinacolato)diboron Activation and Conjugate Addition to an Enone
To initiate our studies, we subjected cyclohexenone and bis(pinacolato)diboron (1) to a premixed solution of commercially available imidazolinium salt 2 and NaOt-Bu (10 mol % of each) in THF at 22 °C. As shown in entry 1 of Table 1, 66% conversion to the desired boronate 6b is achieved in 12 h. Use of unsaturated 3 leads to a more efficient process (entry 2, 92% conv); the sterically more demanding 4 promotes 45% conversion to 6b under the same conditions (entry 3, Table 1). Complete reaction is obtained when the carbene derived from Cy-substituted imidazolium salt 5 serves as the catalyst (>98% conv, entry 4).11 The activity of the latter carbene is substantially superior to those corresponding to 2–4: with only 2.5 mol % 5 (vs 10 mol % in Table 1) formation of 6 is complete within one hour (vs 12 h). The findings in entries 5–7 of Table 1, indicating no reaction with 10 mol % NaOt-Bu, PPh3 or PCy3 in 12 hours, underline the exceptional catalytic activity of the NHC catalysts. A relatively sluggish conjugate addition is observed with triphenylphosphine oxide (entry 8), suggesting an alternative and potentially effective avenue for catalyst development.
Table 1.
Activation of Bis(pinacolato)diboron and Conjugate Additions with Various Catalystsa
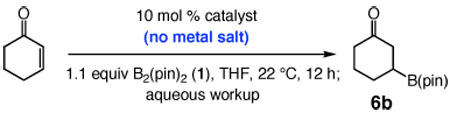 | |||
|---|---|---|---|
| entry | heterocyclic salt or phosphine | base; mol % | conv (%)b |
| 1 | 2 | NaOt-Bu; 10 | 66 |
| 2 | 3 | NaOt-Bu; 10 | 92 |
| 3 | 4 | NaOt-Bu; 10 | 45 |
| 5 | none | NaOt-Bu; 10 | <2 |
| 6 | PPh3 | none | <2 |
| 7 | PCy3 | none | <2 |
| 8 | OPPh3 | none | 50 |
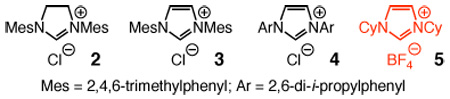 | |||
Mes = 2,4,6-trimethylphenyl; Ar = 2,6-di-i-propylphenyl
Performed under N2 atm.
Conversion to product determined by analysis of 400 MHz 1H NMR spectra of unpurified mixtures.
The following experiments were carried out to gauge the validity of the proposed scenario regarding NHC activation of 1 (Scheme 1): (1) Theoretical studies (DFT)12 of a complex arising from association of the carbene derived from 5 and 1 were performed. The partial atomic charges (Scheme 2) indicate that complexation of an NHC with 1 results in polarization of B–B bond and a diminished electrophilic character of both B atoms; the unassociated boron bears a lower positive charge than the one with which the NHC interacts (electron density of the B–B bond is strongly polarized towards the unbound B center). Carbene association leads to weakening of the B–B bond, as evidenced by an increase in this bond length (1.703 Å in B2(pin)2 vs 1.749 Å in the NHC•1 complex). (2) Coordination of the highly effective carbene derived from 5 with diboron 1 was investigated by 11B NMR spectroscopy; complete disappearance of the signal for 1 is observed within 5 min (22 °C). Consistent with the aforementioned hypothesis, significant upfield shifts are observed for boron atom signals (Scheme 2), with one being more strongly shielded (from δ 30.1 to δ 4.5 and 6.3 for the sp3- and sp2-hybridized B centers, respectively). The same experiment with the less effective carbene derived from 4 leads to the appearance of signals at δ –1.8 and 8.2 (∼20% conv in 1 h; additional transformation not observed after 12 h).
Scheme 2.

DFT Calculations and 11B NMR Studies on 1 and the Derived NHC Complexa
aDFT calculations were performed on a complex with the carbene derived from imidazolium salt 5. Spectroscopic studies were performed with the carbene derived from 4-5. See Supporting Information for additional details.
One hour and 2.5 mol % of the NHC catalyst is sufficient for >98% conversion of five-, six-, seven- and eight-membered ring enones to 6a–6d in 89–93% yield (entries 1–4, Table 2). Transformations in entries 5–6 (Table 2) illustrate that addition proceeds readily in cases where the β carbon of the unsaturated carbonyl is sterically congested. Efficient formation of boronates 8–9 demonstrates that tertiary C–B bonds are generated by facile catalytic additions to trisubstituted cyclic enones. The low and moderate diastereoselectivity in which 8 and 9 are obtained (1.6:1 and 7.1:1, respectively) are likely kinetically controlled, arising from aqueous quench of the boron enolates. β-Boryl lactone 10 (entry 9, Table 2) is isolated in 95% yield; higher catalyst loading (5 mol % 5) and longer time are required due to diminished reactivity of the unsaturated lactone (24 vs 1 h).
Table 2.
Synthesis of Tertiary B-Substituted Carbons by NHC-Catalyzed Boron Conjugate Additions to Cyclic Enonesa
| Entry | product | yield (%)b | |
|---|---|---|---|
| 1 | 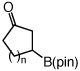 |
n = 1 (6a) | 93 |
| 2 | n = 2 (6b) | 91 | |
| 3 | n = 3 (6c) | 93 | |
| 4 | n = 4 (6d) | 89 | |
| 5 | 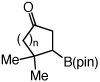 |
n = 1 (7a) | 97 |
| 6 | n = 2 (7b) | 98 | |
| 7c | 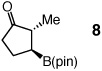 |
88 | |
| 8d | 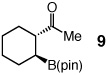 |
90 | |
| 9e | 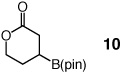 |
95 | |
Performed under N2 atm with 2.5 mol % 5, 22 °C, 1.0 h; >98% conv in all cases.
Yield of purified products.
dr = 1.6:1.
dr = 7.1:1.
Performed with 5 mol % 5, 24 h.
Efficient formation of B-substituted quaternary carbon centers underlines one of the more noteworthy aspects of the present protocol. With 2.5 mol % catalyst, 11 and 12 are obtained in 96% and 91% yield, respectively (Scheme 3). The catalytic process can be used to obtain significant quantities of β-boroketones: reaction with 0.5 gram (4.54 mmol) of β-methylcyclohexenone delivers 0.94 g of 11 after purification (87% yield). Synthesis of 13, with two additional methyl units, expected to retard the rate of reaction, proceeds to 73% conversion in 12 hours (69% yield). The limitation of the method is manifested by the relatively inefficient formation of lactone 14; a comparison of the conversion level (45%) and yield of the isolated product (44%), however, illustrates that the transformation proceeds with minimal byproduct formation. Reaction of the commercially available, enantiomerically pure bicyclic enone 15 (Scheme 3), delivering boronate 16 in 78% yield as a single diastereomer (X-ray analysis), represents another process involving a particularly congested β-substituted enone.
Scheme 3.

Synthesis of Quaternary B-Substituted Carbons by NHC-Catalyzed Boron Conjugate Additions to Cyclic Enones
NHC-catalyzed transformations can be performed with acyclic substrates (Scheme 4). Synthesis of 18 (92% yield), bearing a B-substituted quaternary carbon, requires 5 mol % 5 at 0 °C13 for 6 h. Formation of 19 proceeds in >98% yield via a tetrasubstituted boron-enolate. Catalytic generation of 20, isolated in 70% yield and 7.2:1 diastereomeric ratio, illustrates that additions with acyclic substrates can proceed with useful stereoselectivity levels. Synthesis of β-boryl ester 21 (93% yield) shows that the metal-free protocol can be used to obtain products accessed by the recently disclosed Cu-catalyzed procedures.5
Scheme 4.

NHC-Catalyzed Boron Conjugate Additions to Acyclic Enonesa
aPerformed at 22 °C (except for 17→18); >98% conv in all cases, except 88% for 20 (conv to the desired product by 400 MHz 1H NMR). Yields of purified products. dr = diastereomeric ratio.
To identify some of the special attributes of the metal-free method, we briefly investigated the corresponding Cu-catalyzed process, likely the most advanced variant promoted by a metal complex.5 With CuOt-Bu and the carbene from 5, additions proceed as efficiently as with NHC-catalyzed transformations; two examples are shown in Scheme 5. Unlike certain phosphine-catalyzed processes,5h and in contrast to reports regarding NHC– Cu-catalyzed additions,5g MeOH is not required for efficient transformation. Additions promoted by a Cu complex, similar to the NHC-catalyzed processes, can furnish the boron-enolate products (vs protonated ketones with MeOH). This advantage likely arises from higher reactivity of Cu-enolates bearing a strong σ-donor NHC (e.g., i in Scheme 5 vs the less Lewis basic P-based ligands), allowing for facile reaction with 1.
Scheme 5.

Comparison of Metal-Free with NHC–Cu-Catalyzed Processes
Initial studies indicate that the NHC-catalyzed process can be more functional group tolerant than the Cu-catalyzed variant. Whereas the presence of one equiv of 1-hexyne leads to complete inhibition of the Cu-catalyzed reaction (eq 1), 14 the metal-free transformation proceeds to 85% conversion (81% yield). Association of NHC–Cu–B(pin) with π-Lewis acidic alkyne likely leads to inhibition of the metal complex’s catalytic activity. Thus, at least in some cases, site-selective C–B formation and boron enolate generation by conjugate addition to an alkyne-bearing enone can only be achieved through Cu-free catalysis.
 |
(1) |
The NHC-catalyzed reactions furnish molecules not easily accessible by alternative protocols. Oxidation of 11 delivers the corresponding tertiary alcohol in 94% yield.15 As illustrated in eq 2, when addition to cyclohexenone is performed in the presence of benzaldehyde, the resulting boron enolate participates in an aldol addition, affording 22 as a single diastereomer (85% yield). The outcome of the tandem conjugate addition/aldol process, established by X-ray crystallography, indicates that reaction occurs from the sterically more hindered boron-enolate face (C–C bond formation syn to boronate). Aldehyde approach might be directed through carbonyl association16 with the adjacent C-bound boron. Generation of 22 is unaccompanied by products resulting from benzoin condensation, a Stetter-type process7 or aldehyde diboration (<2%).5i When the transformation in eq 2 is performed with 2.5 mol % CuOt-Bu, in addition to 22 (31%; >98:<2 dr), there is 19% benzaldehyde diboration (and 26% of an unidentified byproduct).Error! Bookmark not defined. A more efficient aldol addition can be achieved if the aldehyde is introduced subsequent to the Cu-catalyzed process (to avoid its diboration). The above findings, however, illustrate that boron addition to an aldehyde-containing enone would proceed with high efficiency and site-selectivity only under the Cu-free conditions. Studies to establish the basis for the chemoselectivity furnished by the NHC catalyst are in progress.
 |
(2) |
A C–B bond can be converted to a C–O, C–N17 or a C–C17b,18 bond by various procedures, including catalytic cross-coupling reactions; the present protocol thus allows access to an assortment of valuable molecules. Development of the enantioselective variants and elucidation of the mechanistic details are underway.
Supplementary Material
Experimental procedures and spectral, analytical data for all reaction products (PDF). This material is available on the web: http://www.pubs.acs.org
Acknowledgments
Financial support was provided by the NIH (GM-57212) and the NSF (CHE-0715138). K-s. L. and A. R. Z. are Schering-Plough and LaMattina Graduate Fellows, respectively. We thank Dr. B. Li and K. Mandai for securing X-ray structures. Mass spectrometry facilities (Boston College) are supported by NSF (DBI-0619576).
References
- 1.For metal-catalyzed C–B forming reactions (not conjugate additions), see: Borohydride addition to alkenes: Beletskaya I, Pelter A. Tetrahedron. 1997;53:4957–5026. Cross-coupling with organohalides: Ishiyama T, Ishida K, Miyaura N. Tetrahedron. 2001;57:9813–9816. Diboron addition to alkenes: Burks HE, Morken JP. Chem. Commun. 2007:4717–4725. doi: 10.1039/b707779c.
- 2.(a) Lawson YG, Lesley MJG, Marder TB, Norman NC, Rice CR. Chem. Commun. 1997:2051–2052. [Google Scholar]; (b) Abu Ali H, Goldberg I, Sbrenik M. Organometallics. 2001;20:3962–3965. [Google Scholar]; (c) Bell NJ, Cox AJ, Cameron NR, Evans JSO, Marder TB, Duin MA, Elsevier CJ, Baucherel X, Tulloch AAD, Tooze RP. Chem. Commun. 2004:1854–1855. doi: 10.1039/b406052k. [DOI] [PubMed] [Google Scholar]
- 3.Kabalka GW, Das BC, Das S. Tetrahedron Lett. 2002;43:2323–2325. [Google Scholar]
- 4.Hirano K, Yorimitsu H, Oshima K. Org. Lett. 2007;9:5031–5033. doi: 10.1021/ol702254g. [DOI] [PubMed] [Google Scholar]
- 5. Ito H, Yamanaka H, Tateiwa J-i, Hosomi A. Tetrahedron Lett. 2000;41:6821–6825. Mun S, Lee J-E, Yun J. Org. Lett. 2006;8:4887–4889. doi: 10.1021/ol061955a. Lee J-E, Kwon J, Yun J. Chem. Commun. 2008:733–734. doi: 10.1039/b716697d. For boron conjugate additions with stoichiometric amounts of Cu salts, see: Takahashi K, Ishiyama T, Miyaura N. J. Organomet. Chem. 2001;625:47–53. Kabalka GW, Das BC, Das S. Tetrahedron Lett. 2001;42:7145–7146. For enantioselective versions, see: Lee J-E, Yun J. Angew. Chem., Int. Ed. 2007;47:145–147. doi: 10.1002/anie.200703699. Lillo V, Prieto A, Bonet A, Díaz-Requejo MM, Ramírez J, Pérez PJ, Fernández E. Organometallics. 2009;28:659–662. Sim H-S, Feng X, Yun J. Chem., Eur. J. 2009;15:1939–1943. doi: 10.1002/chem.200802150. Laitar DS, Tsui EY, Sadighi JP. J. Am. Chem. Soc. 2006;128:11036–11037. doi: 10.1021/ja064019z.
- 6.For metal-catalyzed element–element additions to unsaturated C–C bnds, see: Beletskaya I, Moberg C. Chem. Rev. 2006;106:2320–2354. doi: 10.1021/cr050530j.
- 7.For a recent review on N-heterocyclic carbenes as catalysts, see: Enders D, Niemeir O, Henseler A. Chem. Rev. 2007;107:5606–5655. doi: 10.1021/cr068372z.
- 8.(a) Chase PA, Stephan DW. Angew. Chem., Int. Ed. 2008;47:7433–7437. doi: 10.1002/anie.200802596. [DOI] [PubMed] [Google Scholar]; (b) Ueng S-H, Brahmi MM, Derat E, Fensterbank L, Lacóte E, Malacria M, Curran DP. J. Am. Chem. Soc. 2008;130:10082–10083. doi: 10.1021/ja804150k. [DOI] [PubMed] [Google Scholar]
- 9.Denmark SE, Beutner GL. Angew. Chem., Int. Ed. 2008;47:1560–1638. doi: 10.1002/anie.200604943. [DOI] [PubMed] [Google Scholar]
- 10.For direct activation of Grignard reagents with NHC complexes, see: Lee Y, Hoveyda AH. J. Am. Chem. Soc. 2006;128:15604–15605. doi: 10.1021/ja067456m.
- 11.Control experiments indicate that the change in the counterion of the imidazolinium and imidazolium salts is not responsible for variation in activity.
- 12.For details of DFT calculations and 11B NMR studies, see the Supporting Information.
- 13.The lower reaction temperature for synthesis of 18 (0 °C vs 22 °C for formation of 19–21) is due to formation of unidentified byproducts at 22 °C.
- 14.Products from boron-copper addition to 1-hexyne are not observed. See: Lee Y, Hoveyda AH. J. Am. Chem. Soc. 2009;131:3160–3161. doi: 10.1021/ja809382c.
- 15.See the Supporting Information for details.
- 16.Hoveyda AH, Evans DA, Fu GC. Chem. Rev. 1993;93:1307–1370. [Google Scholar]
- 17.(a) Fernandez E, Maeda K, Hooper MW, Brown JM. Chem. Eur. J. 2000;6:1840–1846. doi: 10.1002/(sici)1521-3765(20000515)6:10<1840::aid-chem1840>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]; (b) Hupe E, Marek I, Knochel P. Org. Lett. 2002;4:2861–2863. doi: 10.1021/ol0262486. [DOI] [PubMed] [Google Scholar]
- 18.(a) Chen A, Ren L, Crudden CM. J. Org. Chem. 1999;64:9704–9710. [Google Scholar]; (b) Moran WJ, Morken JP. Org. Lett. 2006;8:2413–2415. doi: 10.1021/ol060735u. [DOI] [PubMed] [Google Scholar]; (c) Molander GA, Gormisky PE. J. Org. Chen. 2008;73:7481–7485. doi: 10.1021/jo801269m. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental procedures and spectral, analytical data for all reaction products (PDF). This material is available on the web: http://www.pubs.acs.org