

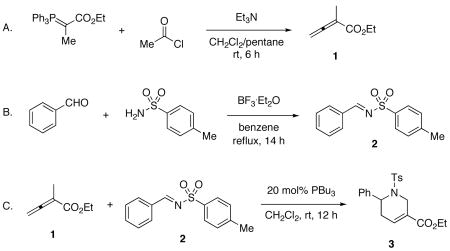
1. Procedure
A. Ethyl 2-methylbuta-2,3-dienoate (1)
A flame-dried, 500-mL, two-necked, round-bottomed flask equipped with an egg-shaped magnetic stir bar, a rubber septum, and an argon inlet is charged with ethyl 2-(triphenylphosphoranylidene)propionate (27.18 g, 75.0 mmol) (Note 1), triethylamine (10.5 mL, 75.3 mmol) (Note 6), anhydrous dichloromethane (125 mL) (Note 7), and pentane (125 mL) (Note 8) and the mixture is stirred under argon. Acetyl chloride (5.3 mL, 75 mmol) (Note 9) is added via syringe over a period of 1 h using a syringe pump; during the addition the flask is placed in a 25 °C water bath to control the heat evolved during the reaction. During the addition the reaction turns cloudy, then a yellow-orange precipitate forms. Stirring is continued for an additional 6 h after which time the heterogeneous mixture is filtered through a Büchner funnel connected to a vacuum from a water aspirator. The precipitate is washed with pentane (50 mL) and the filtrate is concentrated to ca. 50 mL (Note 10) at 0 °C on a rotary evaporator (20–25 mmHg). Pentane (150 mL) and an egg-shaped magnetic stir bar are added to the residue-containing flask and the mixture is stirred vigorously for 30 min. The white precipitate (triphenylphosphine oxide) is removed by filtering the mixture through a Büchner funnel and washing the precipitate with pentane (50 mL). The filtrate is concentrated to ca. 30 mL at 0 °C on a rotary evaporator (20–25 mmHg). The residue is filtered again to remove the precipitate (triphenylphosphine oxide) and the precipitate is rinsed with pentane (10 mL). The filtrate is concentrated to ca. 20 mL as described above (Note 11). The residue is transferred to a 25-mL one-necked, round-bottomed flask along with an egg-shaped magnetic stir bar and purified by distillation (54–55 °C, 25–26 mmHg) (Note 12) to afford 5.73 g (61%) of 1 (Note 13) as a colorless oil.
B. (E)-N-Benzylidene-4-methylbenzenesulfonamide (2)
A flame-dried, 500-mL, one-necked, round-bottomed flask equipped with an egg-shaped magnetic stir bar is charged with p-toluenesulfonamide (10.25 g, 59.9 mmol) (Note 14), benzaldehyde (6.7 mL, 66 mmol) (Note 15), benzene (300 mL) (Note 16) and BF3 · Et2O (0.75 mL, 6.0 mmol) (Note 17). The flask is equipped with a Dean–Stark trap, which is attached to a reflux condenser and a gas adapter under argon and the solution is refluxed (oil bath temperature: 90–95 °C) (Note 18) for 14 h (Note 19). The yellow solution is cooled to room temperature and concentrated using a rotary evaporator (20–25 mmHg) at 35 °C to afford a brown solid. Ethyl acetate (50 mL) (Note 4) and hexane (100 mL) (Note 20) are added to the flask; a reflux condenser and gas adapter are attached and the solution is refluxed under Ar (oil bath temperature: 85–90 °C). Upon dissolution of the solid (ca. 5 min) the stir bar is removed, the solution is allowed to cool to room temperature and then cooled to − 20 °C in a freezer overnight. The resulting solid is collected by filtering through a Büchner funnel connected to a vacuum and washing with hexane (100 mL) to provide 14.27 g (92%) of 2 (Note 21) as an off-white solid.
C. Ethyl 6-phenyl-1-tosyl-1,2,5,6-tetrahydropyridine-3-carboxylate (3)
A flame-dried, 1-L, one-necked, round-bottomed flask equipped with an egg-shaped magnetic stirring bar and a rubber septum is charged with 2 (7.78 g, 30.0 mmol) and tri-n-butylphosphine (1.48 mL, 5.93 mmol) (Note 22). The flask is purged with argon and anhydrous dichloromethane (600 mL) (Note 7) is added via cannula under argon; the mixture is stirred for 5 min. Ethyl 2-methylbuta-2,3-dienoate (1) (4.61 g, 36.5 mmol) is added to the flask via syringe over a period of 5 min. After stirring the light-yellow solution at room temperature for 13 h (Note 23), the reaction mixture is concentrated by rotary evaporation (20–25 mmHg) at 35 °C to afford a yellow residue, which is transferred to a 100-mL, one-necked, pear-shaped flask equipped with an egg-shaped magnetic stirring bar. Chloroform (20 mL) (Note 24) and hexane (20 mL) (Note 20) are added to the flask, a reflux condenser is attached, and the solution is heated to reflux under Ar (oil bath temperature: 85–90 °C). When the solid is dissolved, the stir bar is removed, the solution is allowed to cool to room temperature and then cooled to − 20 °C in a freezer overnight. The resulting solid is collected by filtering the slurry through a Büchner funnel connected to a vacuum. The solid is washed with hexane (100 mL) to provide 8.77 g (76% yield) of 3 (Note 25) as a yellow solid. The filtrate is concentrated by rotary evaporation (20–25 mmHg) at 35 °C to give an orange residue, which, when purified using silica gel flash column chromatography (Note 26), provides an additional 2.13 g (18% yield) of 3 as a white solid.
2. Notes
Ethyl 2-(triphenylphosphoranylidene)propionate was purchased by the checkers from Alfa Aesar under the name (1-ethoxycarbonylethylidene)- triphenylphosphorane (97%) and was used as received. The submitters prepared this reagent from ethyl 2-bromopropanoate (Note 2) and triphenylphosphine (Note 3) using the following procedure (note that this procedure was not checked): A 500-mL, one-necked, round-bottomed flask equipped with an egg-shaped magnetic stirring bar is charged with triphenylphosphine (52.46 g, 200.0 mmol), ethyl acetate (130 mL) (Note 4), and ethyl 2-bromopropanoate (26.03 mL, 200.0 mmol). A reflux condenser is attached to the flask and the solution is heated under reflux (oil bath temperature: 75–80 °C) under argon for 24 h. The resulting white precipitate is collected through suction filtration on a Büchner funnel and washed with ethyl acetate (100 mL). The salt is dissolved in dichloromethane (500 mL) (Note 5) and the solution is transferred to a 1-L separation funnel. Aqueous sodium hydroxide solution (2 M, 200 mL) is added and, after vigorously shaking, the organic and aqueous layers are separated. The aqueous phase is extracted with dichloromethane (100 mL). The combined organic phases are washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated at 30 °C under rotary evaporation (20–25 mmHg); drying under vacuum (0.1–0.2 mmHg) affords 54.51 g (75.2%) of ethyl 2-(triphenylphosphoranylidene)propionate as a light-yellow solid, mp 156–158 °C.
Ethyl 2-bromopropionate (99%) was purchased from Aldrich Chemical Company, Inc., and was used as received.
Triphenylphosphine (99%) was purchased from Acros Chemical Company, Inc., and was used as received.
Ethyl acetate (99.9%) was purchased from Fisher Scientific and was used as received.
Dichloromethane (99.9%) was purchased from Fisher Scientific and was used as received.
Triethylamine (99.5%) was purchased from Aldrich Chemical Company, Inc. and was used as received.
Dichloromethane, supplied by Fisher Scientific, was purified by passing over activated alumina using the Sol-Tek ST-002 solvent purification system. The submitters distilled dichloromethane from calcium hydride.
Pentane (99.7%) was purchased from Fisher Scientific and was used as received.
Acetyl chloride (98%) was purchased from Aldrich Chemical Company, Inc. and was used as received.
A precipitate formed toward the end of the rotary evaporation process. Over-concentration of the solvent causes some loss of the product.
A small amount of precipitate (triphenylphophine oxide) was observed in the residue.
Due to the volatility of ethyl 2-methylbuta-2,3-dienoate (1) the condenser was cooled using a recirculating pump placed in an ice/water bath. Additionally the pressure was lowered gradually from 60 mmHg to 25 mmHg to minimize bumping.
The physical properties are as follows: IR (thin film) 3063, 2984, 1969, 1944, 1712, 1369, 1278, 1222, 1123, 1027, 853, 778, 615 cm −1; 1H NMR (500 MHz, CDCl3) δ: 1.29 (t, J = 7.0 Hz, 3 H), 1.88 (t, J = 3.0 Hz, 3 H), 4.21 (q, J = 7.0 Hz, 2 H), 5.07 (q, J = 3.0 Hz, 2 H); 13C NMR (125 MHz, CDCl3) δ: 14.2, 14.7, 61.0, 77.8, 95.5, 167.6, 214.0. MS (EI) m/z (%) 126 (14) [M]+, 98 (47%), 86 (47%), 84 (75), 53 (100); HR-EI calcd for C7H10O2 126.0681, found 126.0680. Anal. Calcd for C7H10O2: C, 66.65; H, 7.99. Found: C, 66.08; H, 8.04. The product did not analyze correctly for carbon (0.57% difference from theory for the checkers, 1.08% difference for the submitters). The product is homogeneous in all other respects.
p-Toluenesulfonamide (98%) was purchased from Aldrich Chemical Company, Inc., and was used as received. The material contained ≤ 2% o-toluenesulfonamide by 1H NMR. The absence of observable multiplets between 8.04-8.01 ppm and 7.51-7.46 ppm and singlet at 2.70 indicated the material contained less than 2% o-toluenesulfonamide.
Benzaldehyde (99.5%) was purchased from Aldrich Chemical Company, Inc., and was used as received.
Benzene, supplied by EMD Chemicals, Inc., was distilled from calcium hydride.
Boron trifluoride-diethyl etherate (36%) was purchased from Aldrich Chemical Company, Inc., and was used as received.
Over time a white solid collected in the Dean-Stark trap and at the bottom of the reflux condenser. The solid was analyzed by 19F NMR (282 MHz, DMSO) and the spectrum shows a single resonance at − 148.2 ppm.
The submitters report that the product decomposes slightly on E. Merck silica gel thin layer chromatography plates (60F-254), so 1H NMR spectroscopic analysis of the crude reaction mixture is used to monitor the reaction. Concentrating a portion (ca. 0.5 mL) of the crude reaction mixture at 35 °C using rotary evaporation affords a residue that is dissolved in CDCl3 (purchased from Cambridge Isotope Laboratories, Inc.). Disappearance of the signal for the NH2 protons (ca. 5.0 ppm) in the 1H NMR spectrum indicates that the reaction is complete.
Hexane (99.9%) was purchased from Fisher Scientific and was used as received.
The physical properties are as follows: mp 112–113 °C; IR (thin film) 1598, 1572, 1450, 1321, 1157, 1088, 782, 757 cm−1; 1H NMR (500 MHz, CDCl3) δ: 2.45 (s, 3 H), 7.36 (d, J = 8.5 Hz, 2 H), 7.50 (t, J = 8.0 Hz, 2 H), 7.62 (tt, J = 7.5 and 1.5 Hz, 1 H), 7.89–7.94 (m, 4 H), 9.04 (s, 1 H); 13C NMR (125 MHz, CDCl3) δ: 21.7, 128.1, 129.1, 129.8, 131.3, 132.3, 134.9, 135.1, 144.6, 170.1; MS (ESI) m/z 282 [M + Na]+; HR-ESIMS calcd for C14H13NO2SNa [M + Na]+ 282.0565, found 282.0552. Anal. Calcd for C14H13NO2S: C, 64.84; H, 5.05; N, 5.40. Found: C, 64.79; H, 5.01; N, 5.43.
Tri-n-butylphosphine (97%) was purchased from Aldrich Chemical Company, Inc., and was used as received.
The disappearance of starting material 2 is monitored using TLC (Note 19): Rf = 0.37 (EtOAc/hexane, 1:4) for 2. The spots are visualized under UV light and p-anisaldehyde stain.
Chloroform, supplied by EMD Chemical, Inc., was distilled from calcium chloride.
The physical properties are as follows: mp 149–150 °C; IR (thin film) 2981, 1709, 1659, 1598, 1495, 1449, 1340, 1264, 1161, 1100, 958, 727 cm−1; 1H NMR (500 MHz, CDCl3) δ: 1.29 (t, J = 7.0 Hz, 3 H), 2.43 (s, 3 H), 2.58–2.50 (m, 1 H), 2.69 (ddd, J = 19.0, 5.5, 2.5 Hz, 1 H), 3.50–3.43 (m, 1 H), 4.19 (q, J = 7.0 Hz, 2 H), 4.50 (d, J = 19.0 Hz, 1 H), 5.37 (d, J = 7.0 Hz, 1 H), 7.05–7.04 (m, 1 H), 7.32–7.27 (m, 7 H), 7.71 (d, J = 8.5 Hz, 2 H); 13C NMR (100 MHz, CDCl3) δ: 14.1, 21.5, 26.9, 39.5, 52.1, 60.8, 127.0, 127.2, 127.6, 127.8, 128.6, 129.7, 136.1, 137.4, 138.1, 143.4, 164.6. MS (ESI) m/z 408 [M + Na]+; HR-ESIMS calcd for C21H24NO4SNa [M + Na]+ 408.1245, found 408.1208. Anal. Calcd for C21H23NO4S: C, 65.43; H, 6.01; N, 3.63. Found: C, 64.99; H, 5.92; N, 3.61.
Flash column chromatography is performed using a 4-cm-wide, 25-cm-high column packed with E. Merck silica gel 60 (230–400 mesh, 100 g). The column is packed by slurrying the silica with ethyl acetate, dichloromethane, and hexane (1:1:10), loading the residue with dichloromethane, and eluting with ethyl acetate, dichloromethane, and hexane (1:1:10). The collected fractions are analyzed using TLC (Note 19), eluting with ethyl acetate, dichloromethane and hexane (1:1:10; Rf = 0.21 for 3). The spots are visualized using UV light and p-anisaldehyde stain. Concentration of the product-containing fractions using rotary evaporation (20–25 mmHg) at 35 °C and then drying under vacuum (0.06 mmHg) provides 3.
Safety and Waste Disposal Information
All hazardous materials should be handled and disposed of in accordance with “Prudent Practices in the Laboratory”; National Academy Press; Washington, DC, 1995.
3. Discussion
During the past decade, catalysis employing nucleophilic organic molecules, such as N-heterocyclic carbenes, amines, and phosphines, has garnered tremendous attention. This interest is due in part to the spectacular expansion of the scope of nucleophilic catalysis beyond its classical examples, such as acyloin condensations and the Morita–Baylis–Hillman (MBH) reactions. In particular, the use of activated allenes, instead of alkenes, as reactants in MBH reactions in the presence of nucleophilic tertiary phosphines has led to the development of many processes for the construction of carbocycles and heterocycles.2 Typically, phosphine-catalyzed MBH reactions of allenes are operationally simple and do not require aqueous work-up because the two reactants and the phosphine catalyst are all organic; the products may be purified through flash column chromatography after evaporation of the reaction solvent. Among these reactions, Lu’s [3+2] cycloaddition, in which the allene reacts with an alkene or imine to provide a cyclopentene or pyrroline, is particularly noteworthy.3 As the first example of phosphine-catalyzed allene cycloaddition, Lu’s reaction has been applied in the total syntheses of several natural products,4 and a highly enantioselective variant has been established.5 Our research group has also been engaged in the development of nucleophilic phosphine-catalyzed reactions of allenoates, and this method has served to demonstrate the ever-expanding versatility of allene/phosphine catalysis.6 One of the most broadly useful reactions is the phosphine-catalyzed [4+2] annulation of α-alkyl allenoates and imines to form tetrahydropyridines.7 This reaction, like Lu’s [3+2] reaction, has been applied in natural product syntheses,8,9 and an enantioselective variant of the reaction employing Gladiali’s phosphepine has provided products with up to 99% ee.9 Recently, we disclosed an all-carbon variant of the [4+2] reaction, which we used to synthesize cyclohexenes from α-alkyl allenoates and activated olefins.10
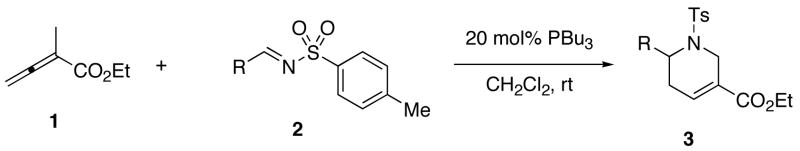
Using the procedure described herein, highly functionalized tetrahydropyridines are easily synthesized in high yield from N-tosylimines and 2-methyl-2,3-butadienoates in the presence of catalytic amounts of PBu3.7 Table 1 lists several examples of these products. All of the non-acidic aryl N-tosylimines tested, except for one bearing a nitro substituent on the aryl group, provided products in yields exceeding 90% (Table 1, entries 1–9). The salicyl and 2-pyrrolyl N-tosylimines did not provide their expected products (Table 1, entries 10 and 12), presumably because their acidic protons quenched the key β-phosphonium dienolate zwitterionic intermediate 4 in the catalytic cycle described in Scheme 1. This problem is easily overcome when the phenol and pyrrole moieties are protected as silyl ethers and BOC carbamates, respectively (Table 1, entries 11 and 13). Naphthyl and other heteroaryl imines are also viable substrates (Table 1, entries 14–16). Among the alkyl N-tosylimines examined, only the non-enolizable N-tosylpivalaldimine 2o afforded the desired product (Table 1, entries 17 and 18).
Table 1.
Synthesis of Tetrahydropyridines 3 From Ethyl 2-Methyl-2,3-butadienoate and N-Tosylaldiminesa
 | |||
|---|---|---|---|
| entry | R | product | yield (%)b |
| 1 | Ph (2) | 3 | 94c |
| 2 | 4-MeOC6H4 (2a) | 3a | 99 |
| 3 | 4-MeC6H4 (2b) | 3b | 95 |
| 4 | 3-ClC6H4 (2c) | 3c | 96 |
| 5 | 2-ClC6H4 (2d) | 3d | 93 |
| 6 | 4-FC6H4 (2e) | 3e | 95 |
| 7 | 4-NCC6H4 (2f) | 3f | 98 |
| 8 | 2-F3CC6H4 (2g) | 3g | 98 |
| 9 | 4-O2NC6H4 (2h) | 3h | 86 |
| 10 | 2-HOC6H4 (2m) | 3i | 0 |
| 11 | 2-TBSOC6H4 (2n) | 3j | 93 |
| 12 | 2-pyrrolyl (2l) | 3k | 0 |
| 13 | N-Boc-2-pyrrolyl (2m) | 3l | 99 |
| 14 | 1-naphthyl (2i) | 3m | 96 |
| 15 | 2-furyl (2j) | 3n | 97 |
| 16 | 4-pyridyl (2k) | 3o | 92d |
| 17 | tert-butyl (2o) | 3p | 86e |
| 18 | n-propyl (2p) | 3q | 0f |
1.0 mmol scale.
Isolated yields.
30 mmol scale.
30 mol% PBu3 was used.
3 equiv of Na2CO3 was added.
The imine decomposed to the corresponding aldehyde and p-toulenesulfonamide.
Scheme 1.

Proposed mechanism
In the proposed mechanism, the nucleophilic phosphine adds to ethyl 2-methyl-2,3-butadienoate (1), forming the resonance-stabilized zwitterions 4. Because of steric congestion at the α-carbon atom, the zwitterion 4 undergoes addition to the imine at its γ-carbon atom, producing the phosphonium amide 5. A proton transfer converts the amide anion in 5 to the vinylogous ylide 6; another proton transfer transforms the ylide 6 into the allylic phosphonium amide 7. The ready conjugate addition of the amide anion to the α,β-unsaturated ester and subsequent β-elimination of the phosphine provides the cyclized product 3. Based on pKa values, we suspected that the proton transfer from the sulfonamide 5 to the ylide 6 would be the rate-determining step.11 Therefore, increasing the acidity of the β ′-carbon atom appears to accelerate the overall reaction. In fact, the reaction between 2-(4-cyanobenzyl)-2,3-butadienoate (8b) and N-tosylbenzaldimine (2) reached completion within 30 min (cf. 9 h for the corresponding reaction of 2-methyl-2,3-butadienoate), producing the expected product in 99% yield with high diastereoselectivity (dr 98:2), favoring the syn product.
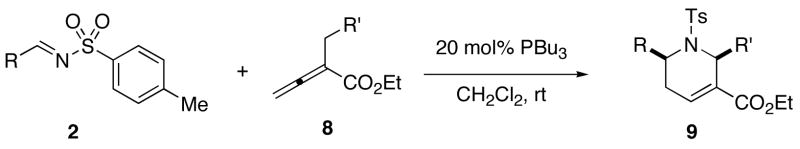
Table 2 lists examples of successful [4+2] reactions performed using 2-arylmethyl-2,3-butadienoates and N-tosylimines as substrates. With N-tosyl benzaldimine as the imine reaction partner, the yields and diastereoselectivities were excellent (Table 2, entries 1–4), except for the system in which 2-ortho-tolylmethyl-2,3-butadienoate was used (dr 88:12; Table 2, entry 5). A variety of electron-withdrawing and -donating substituents are tolerated in both the imine and allenoate substrates (Table 2, entries 6–11). A nitro substituent diminishes the reaction efficiency slightly (Table 2, entry 7). An ortho substituent in the imine substrate also results in diminished diastereoselectivities (Table 2, entries 8 and 11) and/or a lower product yield (entry 8).
Table 2.
Synthesis of Tetrahydropyridines 9 From Ethyl 2-Arylmethyl-2,3-butadienaoates and N-Tosylaldiminesa
 | |||||
|---|---|---|---|---|---|
| entry | R | R ′ | product | yield (%)b | drc |
| 1d | Ph (2) | Ph (8a) | 9a | 92 | 98:2 |
| 2 | Ph (2) | 4-NCC6H4 (8b) | 9b | 99 | 98:2 |
| 3 | Ph (2) | 3-MeOC6H4 (8c) | 9c | 99 | 98:2 |
| 4 | Ph (2) | 2-FC6H4 (8d) | 9d | 99 | 97:3 |
| 5 | Ph (2) | 2-MeC6H4 (8e) | 9e | 82 | 88:12 |
| 6 | 4-MeOC6H4 (2a) | Ph (8a) | 9f | 99 | 97:3 |
| 7 | 4-O2NC6H4 (2h) | Ph (8a) | 9g | 90 | 95:5 |
| 8 | 2-F3CC6H4 (2g) | 4-NCC6H4 (8b) | 9h | 80 | 90:10 |
| 9 | 3-ClC6H4 (2c) | 4-NCC6H4 (8b) | 9i | 99 | 98:2 |
| 10 | 4-MeC6H4 (2b) | 3-MeOC6H4 (8c) | 9j | 99 | 98:2 |
| 11 | 2-ClC6H4 (2d) | 3-MeOC6H4 (8c) | 9k | 96 | 83:17 |
1.0 mmol scale.
Isolated yields.
Diastereoisomeric ratio, determined using 1H NMR spectroscopy.
30 mmol scale.
In summary, the formation of multisubstituted tetrahydropyridines from readily available sulfonylimines and α-alkylallenoates in the presence of catalytic amounts of tributylphosphine is an operationally simple and mild reaction.
Biographies
 Ohyun Kwon was born in South Korea in 1968. She received her BS and MS in chemistry (with Eun Lee) from Seoul National University in 1991 and 1993, respectively. She came to the US in 1993 and obtained her Ph.D. (with Samuel J. Danishefsky) from Columbia University in 1998. After a postdoctoral stint in the laboratory of Stuart L. Schreiber at Harvard University, Ohyun Kwon started her independent career as an Assistant Professor at University of California, Los Angeles, in 2001. Her research involves the development of phosphine-catalyzed reactions and their applications in natural product synthesis and chemical biology.
Ohyun Kwon was born in South Korea in 1968. She received her BS and MS in chemistry (with Eun Lee) from Seoul National University in 1991 and 1993, respectively. She came to the US in 1993 and obtained her Ph.D. (with Samuel J. Danishefsky) from Columbia University in 1998. After a postdoctoral stint in the laboratory of Stuart L. Schreiber at Harvard University, Ohyun Kwon started her independent career as an Assistant Professor at University of California, Los Angeles, in 2001. Her research involves the development of phosphine-catalyzed reactions and their applications in natural product synthesis and chemical biology.
 Kui Lu was born in Jiangxi Province, China, in 1981. He received his BS degree from China Agricultural University in 2002, and then a Ph.D. degree in organic chemistry from Peking University, China, in 2007 under the supervision of Professors Zhen Yang and Jiahua Chen. He is currently working as a postdoctoral fellow in Professor Kwon’s group at the University of California, Los Angeles, focusing on the development of new chiral phosphines and phosphine-catalyzed reactions.
Kui Lu was born in Jiangxi Province, China, in 1981. He received his BS degree from China Agricultural University in 2002, and then a Ph.D. degree in organic chemistry from Peking University, China, in 2007 under the supervision of Professors Zhen Yang and Jiahua Chen. He is currently working as a postdoctoral fellow in Professor Kwon’s group at the University of California, Los Angeles, focusing on the development of new chiral phosphines and phosphine-catalyzed reactions.
 Matthew Davis was born in 1981 in Park Forest, Illinois. In 2004 he received his B.S. degree in chemistry from Hope College in Holland, Michigan. He is currently pursuing graduate studies at the University of Pittsburgh, under the guidance of Prof. Kay Brummond. His research currently focuses on expanding the scope of the Rh(I)-catalyzed cyclocarbonylation reaction of allene-ynes.
Matthew Davis was born in 1981 in Park Forest, Illinois. In 2004 he received his B.S. degree in chemistry from Hope College in Holland, Michigan. He is currently pursuing graduate studies at the University of Pittsburgh, under the guidance of Prof. Kay Brummond. His research currently focuses on expanding the scope of the Rh(I)-catalyzed cyclocarbonylation reaction of allene-ynes.
Appendix Chemical Abstracts Nomenclature; (Registry Number)
Ethyl 2-(triphenylphosphoranylidene)propionate: Propanoic acid, 2-(triphenylphosphoranylidene)-, ethyl ester; (5717-37-3)
Ethyl 2-methylbuta-2,3-dienoate: 2,3-Butadienoic acid, 2-methyl-, ethyl ester; (5717-41-9)
(E)-N-Benzylidene-4-methylbenzenesulfonamide: Benzenesulfonamide, 4-methyl-N-(phenylmethylene)-, [N(E)]-: (51608-60-7)
p-Toluenesulfonamide: Benzenesulfonamide, 4-methyl-; (70-55-3)
Benzaldehyde; (100-52-7)
BF3 · Et2O; (109-63-7)
Ethyl 6-phenyl-1-tosyl-1,2,5,6-tetrahydropyridine-3-carboxylate: 3-Pyridinecarboxylic acid, 1,2,5,6-tetrahydro-1-[(4-methylphenyl)- sulfonyl]-6-phenyl-, ethyl ester; (528853-66-9)
Tri-n-butylphosphine: Phosphine, tributyl-; (998-40-3)
References
- 1.Department of Chemistry and Biochemistry, University of California, Los Angeles, 607 Charles E. Young Drive East, Los Angeles, California 90095–1569.
- 2.For reviews on phosphine-catalyzed reactions, see: Lu X, Zhang C, Xu Z. Acc Chem Res. 2001;34:535. doi: 10.1021/ar000253x.Valentine DH, Hillhouse JH. Synthesis. 2003;3:317.Methot JL, Roush WR. Adv Synth Catal. 2004;346:1035.Lu X, Du Y, Lu C. Pure Appl Chem. 2005;77:1985.Nair V, Menon RS, Sreekanth AR, Abhilash N, Biji AT. Acc Chem Res. 2006;39:520. doi: 10.1021/ar0502026.Denmark SE, Beutner GL. Angew Chem, Int Ed. 2008;47:1560. doi: 10.1002/anie.200604943.Ye LW, Zhou J, Tang Y. Chem Soc Rev. 2008;37:1140. doi: 10.1039/b717758e.
- 3.(a) Zhang C, Lu X. J Org Chem. 1995;60:2906. [Google Scholar]; (b) Xu Z, Lu X. Tetrahedron Lett. 1999;40:549. [Google Scholar]; (c) Du Y, Lu X, Yu Y. J Org Chem. 2002;67:8901. doi: 10.1021/jo026111t. [DOI] [PubMed] [Google Scholar]; (d) Zhu XF, Henry CE, Kwon O. Tetrahedron. 2005;61:6276. [Google Scholar]; (e) Lu X, Lu Z, Zhang X. Tetrahedron. 2006;62:457. [Google Scholar]; (f) Xia Y, Liang Y, Chen Y, Wang M, Jiao L, Huang F, Liu S, Li Y, Yu ZX. J Am Chem Soc. 2007;129:3470. doi: 10.1021/ja068215h. [DOI] [PubMed] [Google Scholar]; (g) Mercier E, Fonovic B, Henry C, Kwon O, Dudding T. Tetrahedron Lett. 2007;48:3617. [Google Scholar]
- 4.(a) Du Y, Lu X. J Org Chem. 2003;68:6463. doi: 10.1021/jo034281f. [DOI] [PubMed] [Google Scholar]; (b) Wang JC, Krische MJ. Angew Chem, Int Ed. 2003;42:5855. doi: 10.1002/anie.200352218. [DOI] [PubMed] [Google Scholar]; (c) Pham TQ, Pyne SG, Skelton BW, White AH. J Org Chem. 2005;70:6369. doi: 10.1021/jo050827h. [DOI] [PubMed] [Google Scholar]
- 5.(a) Zhu G, Chen Z, Jiang Q, Xiao D, Cao P, Zhang X. J Am Chem Soc. 1997;119:3836. [Google Scholar]; (b) Wilson JE, Fu GC. Angew Chem, Int Ed. 2006;45:1426. doi: 10.1002/anie.200503312. [DOI] [PubMed] [Google Scholar]; (c) Cowen BJ, Miller SJ. J Am Chem Soc. 2007;129:10988. doi: 10.1021/ja0734243. [DOI] [PubMed] [Google Scholar]; (d) Fang YQ, Jacobsen EN. J Am Chem Soc. 2008;130:5660. doi: 10.1021/ja801344w. [DOI] [PubMed] [Google Scholar]; (e) Voituriez A, Panossian A, Fleury-Bregeot N, Retailleau P, Marinetti A. J Am Chem Soc. 2008;130:14030. doi: 10.1021/ja806060a. [DOI] [PubMed] [Google Scholar]
- 6.Zhu XF, Henry CE, Wang J, Dudding T, Kwon O. Org Lett. 2005;7:1387. doi: 10.1021/ol050203y.Zhu XF, Schaffner AP, Li RC, Kwon O. Org Lett. 2005;7:2977. doi: 10.1021/ol050946j.Dudding T, Kwon O, Mercier E. Org Lett. 2006;8:3643. doi: 10.1021/ol061095y.Castellano S, Fiji HDG, Kinderman SS, Watanabe M, de Leon P, Tamanoi F, Kwon O. J Am Chem Soc. 2007;129:5843. doi: 10.1021/ja070274n.Zhu XF, Henry CE, Kwon O. J Am Chem Soc. 2007;129:6722. doi: 10.1021/ja071990s.Henry CE, Kwon O. Org Lett. 2007;9:3069. doi: 10.1021/ol071181d.Creech GS, Kwon O. Org Lett. 2008;10:429. doi: 10.1021/ol702462w.Creech GS, Zhu XF, Fonovic B, Dudding T, Kwon O. Tetrahedron. 2008;64:6935. doi: 10.1016/j.tet.2008.04.075.For phosphine-catalyzed reactions of acetylenes, see: Sriramurthy V, Barcan GA, Kwon O. J Am Chem Soc. 2007;129:12928. doi: 10.1021/ja073754n.
- 7.Zhu XF, Lan J, Kwon O. J Am Chem Soc. 2003;125:4716. doi: 10.1021/ja0344009. [DOI] [PubMed] [Google Scholar]
- 8.Tran YS, Kwon O. Org Lett. 2005;7:4289. doi: 10.1021/ol051799s. [DOI] [PubMed] [Google Scholar]
- 9.Wurz RP, Fu GC. J Am Chem Soc. 2005;127:12234. doi: 10.1021/ja053277d. [DOI] [PubMed] [Google Scholar]
- 10.Tran YS, Kwon O. J Am Chem Soc. 2007;129:12632. doi: 10.1021/ja0752181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.The values of pKa for benzenesulfonamide and the methyltriphenylphosphonium ion in DMSO are 16.1 and 22.4, respectively. These values were obtained from the “Bordwell pKa Table” at http://www.chem.wisc.edu/areas/reich/pkatable; the original references are: Bordwell FG, Fried HE, Hughes DL, Lynch TY, Satish AV, Whang YE. J Org Chem. 1990;55:3330.Zhang XM, Bordwell FG. J Am Chem Soc. 1994;116:968.