

Abstract
The synthetic peptide, R2N-COCH2OCH2CO-Gly-Gly-Gly-Pro-Gly-Gly-Gly-OR’, was shown to be selective for Cl- over K+ when R is n-octadecyl and R’ is benzyl. Nineteen heptapeptides have now been prepared in which the N-terminal and C-terminal residues have been varied. All of the N-terminal residues are dialkyl but the C-terminal chains are esters, 2° amides, or 3° amides. The compounds having varied N-terminal anchors and C-terminal benzyl groups are as follows: 1, R = n-propyl; 2, R = n-hexyl; 3, R = n-octyl; 4, R = n-decyl; 5, R = n-dodecyl; 6, R = n-tetradecyl; 7, R = n-hexadecyl; 8, R = n-octadecyl. Compounds 9-19 have R = n-octadecyl and C-terminal residues as follows: 9, OR’ = OCH2CH3; 10, OR’ = OCH(CH3)2; 11, OR’ = O(CH2)6CH3; 12, OR’ = OCH2-c-C6H11; 13, OR’ = O(CH2)9CH3; 14, OR’ = O (CH2)17CH3; 15, NR’2 = N[(CH2)6CH3]2; 16, NHR’ = NH(CH2)9CH3; 17, NR’2 = N[(CH2)9CH3]2; 18, NHR’ = NH(CH2)17CH3; 19, NR’2 = N[(CH2)17CH3]2. The highest anion transport activities were observed as follows. For the benzyl esters whose N-terminal residues were varied, i.e. 1-8, compound 3 was most active. For the C18 anchored esters 10-14, n-heptyl ester 11 was most active. For the C18 anchored, C-terminal amides 15-19, di-n-decylamide 17 was most active. It was concluded that both the C- and N-terminal anchors were important for channel function in the bilayer but that activity was lost unless only one of the two anchoring groups was dominant.
Introduction
Cell biologists, neuroscientists, and others within the biological community have extensively studied channel proteins for more than a century. Much about their properties and behavior has been well characterized and recorded. Indeed, several monographs are available that describe channel function.1 During the past decade, the field has been revolutionized by solid state studies that have revealed the structures of cation,2,3 anion,4 water,5 and pressure-sensitive channels.6 The importance of this work is apparent from the award of the 2003 Nobel Prize in Chemistry to Agre7 and MacKinnon.8
During the past two decades, our understanding of what compounds may function as channels has also expanded. For example, Reusch and coworkers showed in 19889 that bacteria used a poly-α-hydroxybutyrate/calcium polyphosphate channel. Sophisticated function by such a simple chemical structure was greeted initially by skepticism, but classical total synthesis by Seebach and coworkers conclusively demonstrated both structure and function.10
Synthetic peptides that show ion channel activity have been constructed de novo. Montal and coworkers demonstrated channel-like activity from compounds that they called synporins.11 These template-assembled synthetic proteins (TASP, four-helix bundles) showed channel activity.12 At about the same time, DeGrado and coworkers reported the design of simple peptides (“minimalist proteins”)13 that functioned as cation-conducting channels.14
Tabushi reported the first channel compound that was designed by chemical conceptualization rather than from peptides. It used a cyclodextrin head group and was designed to penetrate only one bilayer of the phospholipid membrane. Transmembrane transport of cobalt cation was demonstrated.15 Other early synthetic ion channels were reported by Fyles,16 Lehn,17 Menger,18 Nolte,19 Kobuke,20 and by our group.21 Among these, when transport was demonstrated, it involved cations. A number of other examples, including those reported by Voyer,22 Matile,23 and Koert,24 have appeared since and the expanding progress in this field has been reviewed.25,26,27,28 The field of synthetic, anion-conducting channels has lagged well behind the development of synthetic cation channels. Of course, this has also been true of anion complexation by synthetic host molecules.29 To our knowledge, chloride or anion transport activity is limited to only a few synthetic examples: Tomich’s synthetic peptides,30 the oligophenoxyacetamides,31 our membrane-anchored heptapeptides,32 and Matile’s recent anion channel.33 We now report that within the anion-channel-forming synthetic framework we have developed, the C-terminal and N-terminal anchors significantly influence the rates and selectivity of ion transport through a phospholipid bilayer membrane.
Results and Discussion
A phospholipid-mimetic anchor
Our design of the synthetic cation channel compounds we call hydraphiles was based on the notion that a single molecular span would transcend both leaflets of the phospholipid bilayer and lead to a functional channel.34 In contrast to this crown ether-based design, the formation of pores from the antifungals amphotericin and nystatin suggested that a single, membrane-leaflet span might produce a functional channel.35 We considered that a simple or primitive channel must have been much like a phospholipid membrane monomer. This demanded a combination of hydrophobic anchor chains and a mimic of the acyl glycerol “midpolar regime” of bilayer lipids. Both challenges could be met by reaction of a dialkylamine with diglycolic anhydride. Thus, when dioctadecylamine was heated (reflux, 48 h) with diglycolic anhydride in toluene, only (C18H37)2NCOCH2OCH2COOH was isolated (87% after crystallization). This readily accessible subunit provides both the hydrophobic chains and the carbonyl groups required to mimic the portion of a phospholipid that is well within the bilayer structure. It is also versatile as the chain lengths (R1 and R2) may be varied and may be either the same or different. The anchor unit, R1R2NCOCH2OCH2COOH, can be attached to the N-terminal residue of any peptide by standard coupling methods.
Design of the peptide sequence
The peptide sequence chosen was based in part on the observation that a proline is conserved in the presumed selectivity filter of the ClC family of proteins. The sequence is typically G-X-X-P. The sequence Gly-Gly-Gly-Pro (GGGP) in our design meets this condition. Three additional glycines incorporated at the C-terminal end of GGGP give a symmetrical heptapeptide, GGGPGGG. Molecular models suggest that the C- and N-terminal ends of this bent heptapeptide will be separated by 7-8 Å, depending on overall flexibility. The V-shaped opening is therefore wide enough to accommodate a hydrated chloride anion, which is calculated to be ∼6.5 Å in diameter.36 A model of H2N-Gly-Gly-Gly-Pro-Gly-Gly-Gly-OCH3 in the tube metaphor, created by energy minimization in Gaussian 98W, is shown in Figure 1.
Figure 1.
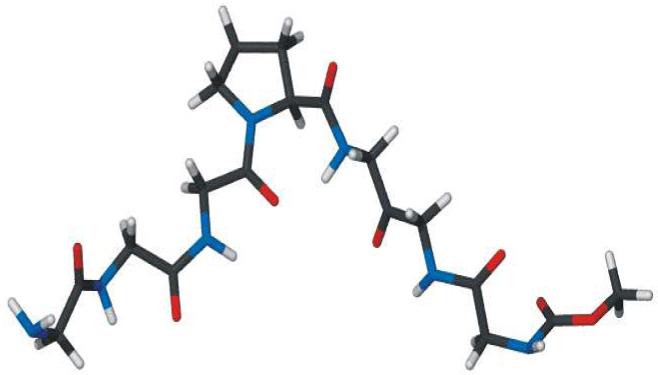
Molecular model of H2N-Gly-Gly-Gly-Pro-Gly-Gly-Gly-OCH3 obtained by energy minimization in Gaussian 98W.
The range of compounds studied
Carboxyl groups are typically ionized at physiologic pH. If not substituted, the free carboxyl group in GGGPGGG would make the C-terminus of the molecule very polar. The C-terminal ester, initially benzyl, was therefore intended to “cap” the heptapeptide. Ultimately, variations in the C-terminal ester were prepared that included both esters and amides. It was anticipated that a C-terminal amide would differ from an ester in stability, polarity, and steric and conformational features. Nineteen synthetic Gly-Gly-Gly-Pro-Gly-Gly-Gly derivatives were prepared for this report. The C- and N-terminal groups were varied. The model shown above was simplified to a C-terminal methyl ester and N-terminal amine to simplify the calculation. The molecular weight of the model shown (not synthesized) is 471 Da. The simplest structure prepared for this study is 1 (C3-benzyl), which has an N-terminal dipropylamine residue and a C-terminal benzyl ester (mw 746 Da). The largest ester residue, 14 (C18-octadecyl), has a molecular weight of 1328 Da. The corresponding bis(octadecyl)amide, 16, has a weight of 1579 Da. The range of molecular weights for these heptapetides is therefore two-fold. The structures are identified in Table 1.
Table 1.
C-Terminal derivatives of R2NCOCH2OCH2CONH-Gly-Gly-Gly-Pro-Gly-Gly-Gly-OR’ (NHR’ or NR’2)
| Cpd. No. | Short name used in text | N-Terminal group (NHR’ or NR’2) | C-Terminal group, OR’ or NRR’ |
|---|---|---|---|
| 1 | C3-benzyl | [CH3(CH2)2]2N | OCH2C6H5 |
| 2 | C6-benzyl | [CH3(CH2)5]2N | OCH2C6H5 |
| 3 | C8-benzyl | [CH3(CH2)7]2N | OCH2C6H5 |
| 4 | C10-benzyl | [CH3(CH2)9]2N | OCH2C6H5 |
| 5 | C12-benzyl | [CH3(CH2)11]2N | OCH2C6H5 |
| 6 | C14-benzyl | [CH3(CH2)13]2N | OCH2C6H5 |
| 7 | C16-benzyl | [CH3(CH2)15]2N | OCH2C6H5 |
| 8 | C18-benzyl | [CH3(CH2)17]2N | OCH2C6H5 |
| 9 | C18-ethyl | [CH3(CH2)17]2N | OCH2CH3 |
| 10 | C18-isopropyl | [CH3(CH2)17]2N | OCH(CH3)2 |
| 11 | C18-heptyl | [CH3(CH2)17]2N | O(CH2)6CH3 |
| 12 | C18-cyclohexylmethyl | [CH3(CH2)17]2N | OCH2C6H11 |
| 13 | C18-decyl | [CH3(CH2)17]2N | O(CH2)9CH3 |
| 14 | C18-octadecyl | [CH3(CH2)17]2N | O(CH2)17CH3 |
| 15 | C18-diheptylamide | [CH3(CH2)17]2N | N[(CH2)6CH3]2 |
| 16 | C18-decylamide | [CH3(CH2)17]2N | NH(CH2)9CH3 |
| 17 | C18-didecylamide | [CH3(CH2)17]2N | N[(CH2)9CH3]2 |
| 18 | C18-octadecylamide | [CH3(CH2)17]2N | NH(CH2)17CH3 |
| 19 | C18-dioctadecylamide | [CH3(CH2)17]2N | N[(CH2)17CH3]2 |
Hydrophobic/hydrophilic balance
The graph in Figure 2 shows the calculated octanol/water partition coefficients (logP) for 1-8. The partition coefficient P is an estimate of the affinity a compound has for a bilayer or nonpolar environment (represented by octanol). Equal (i.e. 1/1) distribution between the two phases would give a partition coefficient of 1, the log10 of which is 0. We expected all of these compounds to favor octanol, as the graph clearly shows. Increasing the N-terminal chain length further favors solubility in the organic phase as shown by the positive slopes of both lines in Figure 2. The data presented were obtained from the AlogP website (http://146.107.217.178/lab/alogps/index.html).
Figure 2.
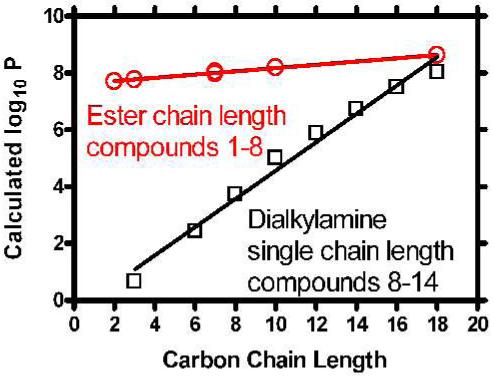
Calculated octanol:water partition coefficients (logP) for (top panel) 1-8 and (lower panel) 8-14.
The upper line in Figure 2 shows the effect of changing the C-terminal residue while maintaining octadecyl N-terminal chains (1-8). Note that in the latter case, heptyl, cyclohexylmethyl, and benzyl all possess 7 carbons and essentially overlap in the graph. Hydrophobicity changes greatly in compounds 8-14, in which the N-terminal alkyl chains are varied and the C-terminal group is always benzyl. The “chain length” designation in the graph of Figure 2 reflects each of the two chain lengths accounting for the steep slope. In both cases, the calculated log P values were fitted by a straight line having r2 ≥ 0.98. In short, these calculations confirm expectations about the hydrophobicity properties of these molecules.
Synthesis of 1-19
The compounds prepared for this study were constructed in essentially the four steps shown in Scheme 1. The first stage is the addition of a dialkylamine to diglycolic anhydride (see above). Compounds 8-19, each of which has twin octadecyl chains at its N-terminus, were begun in this way. Compounds 1-7 were prepared similarly but the procedure was varied to account for the lower boiling points of the smaller dialkylamines. For example, the reaction of diglycolic anhydride and dipropylamine used refluxing THF rather than toluene (see Experimental Section for additional details). Isolated yields for R2NCOCH2OCH2COOH derivatives were in the range 75-100%.
Scheme 1.
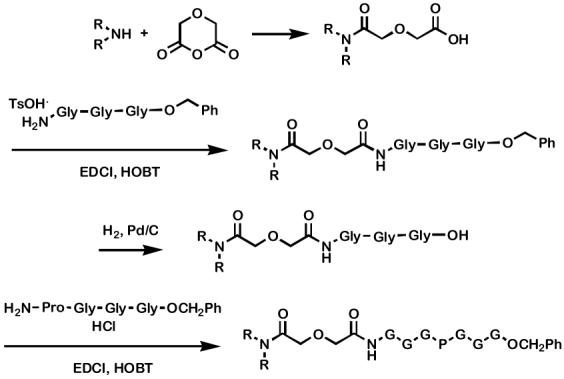
Once formed, R2NCOCH2OCH2COOH was coupled with TsOH·H2N-Gly-Gly-Gly-OCH2Ph and mediated by Me2N(CH2)3N=C=NEt·HCl (EDCI) and N-hydroxybenzotriazole (HOBT) under standard conditions. The resulting tripeptide was debenzylated by hydrogenolysis (H2, Pd/C) to give R2NCOCH2OCH2CO-Gly-Gly-Gly-OH. A second coupling, using EDCI and HOBT with HCl·H-Pro-Gly-Gly-Gly-OCH2Ph, gave the amphiphilic heptapeptide ester. This approach was used for the preparation of 1-8. Esters 9-12 were prepared by coupling R2NCOCH2OCH2CONH-GGG-OH with the H-PGGG-OR’ fragment. Decyl and octadecyl ester derivatives 13 and 14 were prepared by coupling R2NCOCH2OCH2CONH-GGGPGGG-OH withR’OH. Hydrogenolysis of benzyl ester 1 gave the free acid, which was coupled (DCCI, DMAP) to an alcohol or amine to afford 9-19. Standard coupling conditions (EDCI, HOBT, Et3N in CH2Cl2) between [CH3(CH2)17]2NCOCH2OCH2CO-Gly-Gly-Gly-Pro-Gly-Gly-Gly-OH and the alkyl or dialkylamine gave the 15-19 in 30-40% yield.
Assay of channel activity
The success of a channel design is demonstrated only by function. Methods used previously in our lab and by others include fluorescence measurements of a pH sensitive dye,16 competition with proton transport,37,38 and NMR methods.39 Planar bilayer conductance measurements are labor intensive but provide direct information about both transport and selectivity.30a We have used NMR methods to study Na+ transport40 in the crown ether-based hydraphile cation channels.32 Direct NMR observation of 35Cl and 37Cl, both of which have spin 3/2, are less useful for this assay. We therefore elaborated a method previously described by one of us41 and by others42 that uses chloride-selective electrodes to measure chloride efflux from liposomes. The liposomes required for these studies were prepared from 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) and 1,2-dioleoyl-sn-glycero-3-phosphate (DOPA) (7:3 w/w) as previously reported.41 The size distribution was assayed by light scattering after passing through a 200 nm filter and found to be narrow and slightly larger than the specified filter size.
Chloride release was monitored directly by using an Accumet Chloride Combination Electrode. The electrode was introduced into the liposome suspension and the transporter was added, typically as a ∼9 mM solution in 2-propanol. In no case was more than 20 μL of 2-propanol added to the liposome suspension. When monitoring was complete, the vesicles were lysed by addition of 2% aqueous Triton X100 (100 μL). The data were normalized to this final value.
Chloride ion release as a function of the N-terminal anchor chain length
Figure 3 shows the results obtained for chloride release from phospholipid vesicles mediated by compounds 1-8. The period of observation was at least 1500 seconds and each trace is the average of three separate experiments. The method assesses the ability of the channel to insert into the bilayer and induce ion release. Once a functional channel inserts in the bilayer, the liposomes empty within a few seconds.41 The black trace just below the “C14” label shows data for the C18-benzyl channel (8, “SCMTR”).32 As the anchor chain is shortened, the release of chloride increases in a systematic way until the twin anchor chains pass n-octyl (3). The chloride release traces for C14-benzyl (6) and C3-benzyl (1) are nearly superimposed on those for C16-benzyl (7) and C18-benzyl (8).
Figure 3.
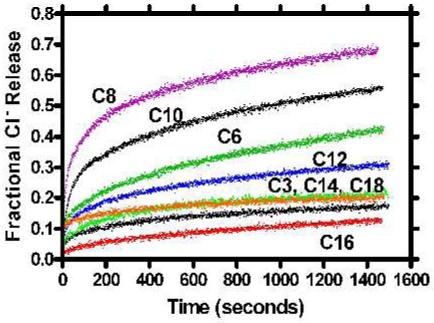
Fractional chloride ion release from 200 nm phospholipid vesicles mediated by compounds 1-8.
It seems counterintuitive that a decrease in anchor chain length would lead to an increase in ion transport. Such an enhancement in transport would be more reasonable if ion selectivity, pore structure, and stoichiometry were affected by the differences in chain length. We do not have direct evidence to show whether or not changes in pore stoichiometry occur, but we have determined ion selectivity for C10-benzyl (4) and C18-benzyl (8) channels. The latter (8) is >10-fold selective for Cl- over K+ but the former transports both chloride and cations with equal efficacy. Thus, the higher release rates may be attributed to a less selective, and higher conducting, pore.
Carboxyfluorescein release from vesicles
Anion release from the compounds studied above was confirmed by use of the fluorescent dye carboxyfluorescein (CF). In this technique, vesicles are formed in the presence of the dye. The external solution is then exchanged by using a size exclusion column to remove CF. The concentration of dye within the vesicles is high enough that self-quenching occurs. Addition of 8 or its relatives leads to release of the dye. As release proceeds, the concentration of CF is low enough in the external medium that self-quenching does not occur and the dye’s fluorescence is readily detectable. Quantitative measurement of the fluorescence at λ = 520 nm permitted the release rate to be determined.
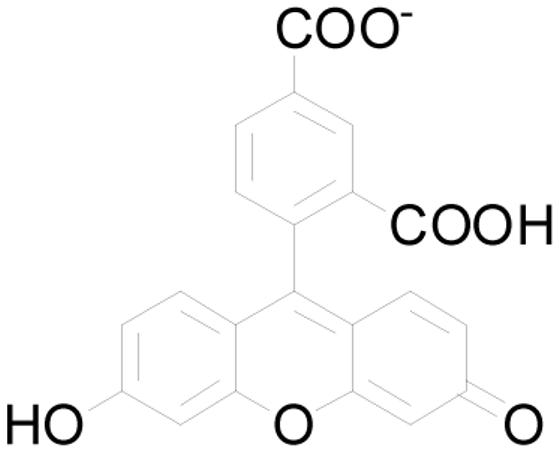
Carboxyfluorescein is larger than hydrated chloride. Judging from CPK and computer models, CF is ∼10 Å × 10 Å. Its thickness depends on the position of the dicarboxyphenyl group with respect to three fused rings. An opening formed by dimerization of the heptapeptides would have to open relatively little (i.e. “breathe”) to permit its passage.
The N-terminal anchor chain-dependent release of CF is shown in Figure 4. The ionophores that induced the behavior shown are, from the top trace to the bottom, 3 (C8-benzyl), 4 (C10-benzyl), 5 (C12-benzyl), 6 (C14-benzyl), 7 (C16-benzyl), and 8 (C18-benzyl). All are benzyl esters and their respective N-terminal, twin alkyl chain lengths are each 8, 10, 12, 14, 16, and 18 carbon atoms.
Figure 4.
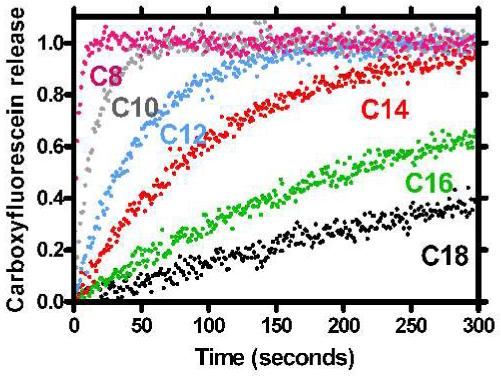
Carboxyfluorescein release from 200 nm phospholipid vesicles mediated by compounds 3-8.
Although many of the same compounds are involved, there are several differences between these results and those shown in Figure 3. First, this study is not as extensive as that presented in Figure 3, which shows results for 1-8. Figure 4 records results only for compounds 3-8. Figure 3 records the release of Cl-, monitored over a period of 1500 s. The analysis used a chloride-selective electrode. The data in Figure 4 were obtained by fluorescence of CF over a period of ∼6000 s, but data for only the first 300 s are reported. The Cl- and CF experiments were done using different ratios of lipid to ionophore and CF release reached a plateau very rapidly. In the chloride release experiments, the typical lipid concentration was 0.31 mM and the compound was added to a final concentration of 65 μM. In the CF dequenching experiments, an excess of ionophore was present ([lipids] = 3.5 μM, [compound] = 10 μM). The lipid:ionophore ratios in the experiments that monitored Cl- or CF release are different. The experimental methods use different concentrations of ionophore and inorganic or organic anion. The detection involves an ion-selective electrode in one case and fluorescence in the other. These differences are required to obtain both reproducible results over a broad concentration range and good signal to noise ratios. Notwithstanding the differences, the trends apparent for the release of the Cl- or CF anion are remarkably similar.
An additional set of studies was undertaken with the C10-benzyl (4) and C18-benzyl (8) channels. The concentration dependence of the CF release from liposomes mediated by 4 and by 8 was studied. As before, the CF fluorescence was monitored at λ = 520 nm. The concentration range studied for 8 was 12.3-190 μM and for the more active C10-benzyl (4), it was 0.17-16.7 μM. The variation in release rates over a 10-fold concentration range permitted calculation of stoichiometry. In both cases (4 and 8), the Hill analysis reported a pore stoichiometry of ∼2. In addition, dextran blocking43 of the pore sizes in both cases suggested a value near 8 Å. Data are shown in Figure 5.
Figure 5.
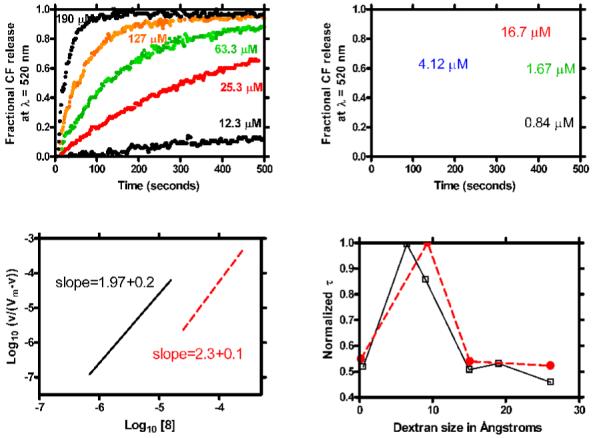
Data for the pore stoichiometry and sizes obtained from carboxyfluorescein release from liposomes mediated by (left panels) 8 and (right panels) 4.
The heptapeptide C-terminal ester group
The original design for a chloride-selective channel envisioned a long chain hydrocarbon anchor, a connector unit that mimicked the phospholipid’s midpolar regime, and a heptapeptide. The structure of the heptapeptide was suggested by conserved sequences in the ClC family of proteins. The first compound to be prepared was terminated by a benzyl residue, primarily to protect the ionizable carboxyl group. The ester residue was varied in order to determine whether or not it had an effect on ion transport efficacy. Heptapeptides having ethyl (9), 2-propyl (10), n-heptyl (11), cyclohexylmethyl (12), n-decyl (13), and n-octadecyl (14) were prepared in addition to the benzyl ester (8) already in hand.
The graph in Figure 6 plots the fractional chloride release for C18-ester derivatives 8-14. Surprisingly, the n-heptyl ester engenders the greatest chloride release. The remaining compounds fall into two groups. The cyclohexylmethyl (12) and n-decyl (13) esters show nearly identical chloride release behavior. The remaining compounds, the ethyl (9), 2-propyl (10), n-octadecyl (14) and benzyl esters (8), clearly show chloride release but it is modest at best. Our initial demonstration of chloride channel activity was with C18-benzyl ester 8, which is among the least active compounds we have explored.
Figure 6.
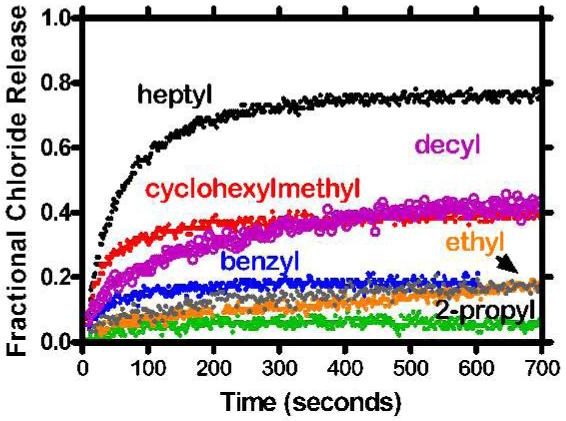
Fractional chloride release from 200 nm phospholipid vesicles mediated by (C18) 2NCOCH2OCH2CON-GGGPGGG-OR, i.e., compounds 8-14
There are three surprises in these data. First, the n-heptyl ester (11) is by far the most active transporter of chloride. In terms of structure, polarity, or hydrophobicity, it seems to be in the middle of this family rather than at either extreme. The n-octadecyl ester constitutes the chain length and hydrophobic extreme and its low activity is the second surprise. Third, we anticipated that the cyclohexylmethyl (12) and benzyl (8) esters, being similar in size, shape, rigidity, and hydrophobicity, would be much more similar than they are. The cyclohexylmethyl ester is significantly more active than is benzyl, its isostere. It is less active than heptyl and more active than benzyl, although all three compounds have the same number of carbon atoms.
In an attempt to confirm that n-heptyl ester 11 did not constitute an anomaly, we undertook two different experimental studies. First, we studied the concentration dependence of its chloride release. In addition, we examined the release of carboxyfluorescein mediated by C18-heptyl ester (11). The release of chloride ion was assayed over a concentration range of 34-134 μM or about a fourfold change in concentration. Carboxyfluorescein was studied over a 14-fold range from 1.95-27.8 μM. The data are shown in Figure 7.
Figure 7.

Anion transport by C18-heptyl ester (11). Upper panel: Fractional chloride ion release by 11 at (from bottom to top) 34, 50, 67, 84, 101, 117, and 134 μM. Lower panel: Fractional carboxyfluorescein release by 11 at (from bottom to top) 1.95, 4.85, 9.80, and 27.8 μM.
The two graphs shown in the upper and lower panels of Figure 7 demonstrate the concentration dependent release of Cl- and CF ions, respectively. In both cases, the release of ions increases with the concentration of 11. The experiments are not directly comparable, however. Indeed, this is a concentration-dependent study. In the 65 μM concentration case, the weight ratio of lipid to ionophore is 2.6:1 in the Cl- release experiments and 1:20 in the fluorescence experiments. Further, the concentrations of 11 differ significantly owing to the higher concentration required for electrochemical detection compared to the fluorescence method.
C-Terminal secondary and tertiary amides as secondary anchors
Five amide compounds were examined in this part of the study. In all cases, the N-terminal anchors were twin octadecyl chains: 15-19. The results of the ester studies suggested that the shortest C-terminal chains were likely to be inactive. Further, it appeared that a C-terminal residue of “intermediate” length might lead to the highest activity. A variable that is difficult to address is the importance of 2° vs. 3° amides at the C-terminus. Binding and transport studies conducted with lariat ether carriers having sidearms terminated by esters, 1°- or 2°-amides showed significant differences, especially between the two classes of amides.44 We thus prepared two, 2° C-terminal amides: C18-decylamide (16) and C18-octadecylamide (18). We prepared three 3°, C-terminal amides and C18-diheptylamide (15) C18-didecylamide (17), and C18-dioctadecylamide (19). These five compounds were prepared by coupling the C-terminal acid, 182-[DGA]-GGGPGGG-OH, with diheptylamine (→15), decylamine (→16), didecylamine (→17), octadecylamine (→18), and dioctadecylamine (→19). Standard conditions (EDCI, HOBT, CH2Cl2) were used and yields were 32-42% for the final products. The amides were typically white solids with melting points in the 100-200 °C range.
Release of chloride ion from liposomes was assayed with a chloride-selective electrode as described above for the esters. Data are shown in Figure 8 for 15-19. The top curve represents chloride ion release from vesicles by C18-didecylamide, 17. It is by far, the most active of the five compounds tested. The remaining four compounds are less active but appear to fall into two “groups” of two each. By far, the lowest Cl- transport activity is observed for the compounds having octadecyl C-terminal groups (18, 19), whether one or two chains are present. More active, but less active than 17, are the 2°-decyl (16) and 3° diheptyl (15) heptapeptides.
Figure 8.
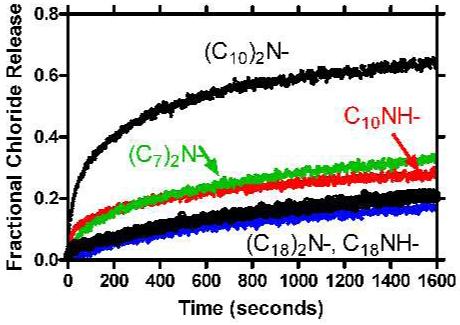
Fractional chloride release from 200 nm phospholipid vesicles mediated by (C18) 2NCOCH2OCH2CON-GGGPGGG-NR1R2, i.e., compounds 15-19.
The data set for the amides is less extensive than for the esters. Nevertheless, the C7 and C10 amides (15-17) proved to be much more active than the 2° or 3° C18 amides (18, 19). A similar trend was observed for the esters, C18-decyl (13) being more active than C18-octadecyl (14). Of course the highest activity in the ester case was observed for the heptyl ester (11), with the cyclohexylmethyl ester (12) about as active as decyl (13). The C18-diheptylamide did not exhibit peak activity in the amide series, but it is clear that shorter chain lengths are favored at the C-terminal end of the peptide when the N-terminal chains are octadecyl.
General observations about the structural requirements for function
The membrane-anchored heptapeptides were designed to incorporate three structural features. The membrane anchor comprises the twin-hydrocarbon tails that were designed to mimic the fatty acyl chains of phospholipids. The midpolar regime of phospholipids is mimicked in this design by the diglycolic acid residue, which places carbonyl groups in a position similar to that occupied by the glyceryl ester carbonyl groups. The heptapeptide was designed to serve the combined purpose of amphiphilic headgroup and selectivity filter. The C-terminal ester was incorporated to prevent the carboxyl from ionizing and potentially controlling the selectivity of the ionophore. It now seems clear that while the C-terminal ester group prevents ionization, it also serves as a “secondary” anchor system when two octadecyl chains are present at the molecule’s N-terminus.
All of the evidence accumulated to date is in concert with channel formation by dimerization of heptapeptides such as 8. The pore size of such a dimer should be 7-8 Å. Both an examination of molecular models and dextran sizing experiments comport with this. Molecular models further suggest that the two heptapeptides “fit” together better when organized more or less as mirror images (see Figure 9) but this does not constitute evidence. We believe that the octadecyl chains play the dominant organizational role within the bilayer to form the pore. The octadecyl chains are long enough to span only one bilayer. This suggests that the known reorganization of lipid headgroups in the adjacent leaflet occurs to complete formation of the pore.45
Figure 9.

Schematic representation of two molecules of (C18)2NCOCH2OCH2CON-GGGPGGG-OR in one bilayer leaflet.
The ionophoretic activity data reported in this paper suggest that the C-terminal residue in (C18)2NCOCH2OCH2CON-GGGPGGG-OR, -NHR, or -NR1R2 plays a“secondary” anchoring role. Whether the C-terminal residue is ester or amide, the greatest transport function is achieved when the N-terminal bis(octadecyl)amine derivatives possess an intermediate chain length at its C-terminus. We speculate that shorter chains do not sufficiently anchor the heptapeptide and that longer chains interfere with the dimer organization of the structure illustrated in Figure 9. Studies are currently underway to covalently link heptapeptides to assess the validity of this postulate.
Conclusions
We presented above details of the design and preparation of a novel family of membrane-active heptapeptides. The ability of these compounds to transport anions (chloride or carboxyfluorescein) through the phospholipid bilayers of synthetic liposomes was studied. Transport was found to depend on both the C- and N-terminal “anchor” chains. When a benzyl group was present at the C-terminus, anion transport increased as N-terminal chain length decreased from C18 to C8 and then declined. When the N-terminal chain was bis(octadecyl), anion transport was most effective when the C-terminal “secondary anchor” was intermediate in length. Although not all C-terminal chain lengths were examined, peak activity was observed when the ester residue was n-heptyl (compound 11) and when the amide residue was bis(decyl) (compound 17). These differences cannot be attributed to hydrophilic/hydrophobic balance. We surmise that the presence of four long anchor chains prevents the dimer organization shown schematically in Figure 9. Additional studies are underway to probe this issue.
Acknowledgement
We thank the NIH for support of this work through GM 63190 and for a Chemistry-Biology Interface training grant that supported MEW.
Appendix
Experimental section
General
1H-NMR were recorded at 300 MHz in CDCl3 (13C-NMR at 75 MHz) and are reported in ppm ( ) downfield from internal (CH3)4Si unless otherwise indicated. Infrared spectra were calibrated against the 1601 cm-1 band of polystyrene. Optical rotations were measured on a Perkin-Elmer Model 241 Polarimeter in a glass microcell (100 mm path length, 1 mL volume) with a Na gas discharge lamp as the light source. Melting points were determined on a Thomas Hoover apparatus in open capillaries and are uncorrected. Thin layer chromatographic (TLC) analyses were performed on aluminum oxide 60 F-254 neutral (Type E) with a 0.2 mm layer thickness or on silica gel 60 F-254 having a 0.2 mm layer thickness. Preparative chromatography columns were packed with activated aluminum oxide (MCB 80-325 mesh, chromatographic grade, AX 611) or with Kieselgel 60 (70-230 mesh).
All reactions were conducted under dry N2 unless otherwise stated. All reagents were the best (non-LC) grade commercially available and were distilled, recrystallized, or used without further purification, as appropriate. Molecular distillation temperatures refer to the oven temperature of a Kugelrohr apparatus. Combustion analyses were performed by Atlantic Microlab, Inc., Atlanta, GA, and are reported as percents. Where water is factored into the analytical data, spectral evidence is presented for its presence.
General procedure 1
A solution of di-n-alkylamine (1.0 equiv) and diglycolic anhydride (1.1 equiv) was refluxed in THF (15 mL/g) for 48 h. The solvent was evaporated and the crude product was dissolved in CHCl3, washed with 10% aq HCl, and the organic layer was evaporated to dryness. The residue was recrystallized from diethyl ether to give Alk2-[DGA]OH.
General procedure 2
To a 0 °C solution of R1COOH (1.0 equiv) in CH2Cl2 (30 mL/g) were added 1-(3-dimethylaminopropyl)-3-ethyl carbodiimide hydrochloride (EDCI, 1.1 equiv), 1-hydroxybenzotriazole (HOBT, 1.1 equiv), H2N-R2 tosylate or hydrochloride (1.0 equiv) and Et3N (2 equiv). After stirring at room temperature (rt) for 48 h, the reaction was washed with 5% aq citric acid (20 mL), 5% aq NaHCO3 (20mL), and H2O (20 mL). The organic phase was dried (MgSO4), evaporated, and the residue was crystallized or chromatographed over silica gel to give R1CONHR2.
General procedure 3
RCOOCH2Ph (1.0 equiv) in EtOH (30 mL/g), 10% Pd/C (0.1 g/g of RCOOCH2Ph) was shaken under 60 psi H2 pressure for 3 h in a Parr apparatus. Filtration of the hot mixture through a celite pad and evaporation of the solvent gave a product that was used in the next step without further purification.
General procedure 4
Boc-protected peptide (1 equiv) was dissolved in dioxane (2 mL/g) and cooled to 0 °C. A 4N HCl solution in dioxane (4 mL/g) was added and the reaction stirred for 1.5 hours at 0 °C. The solvent was evaporated and the residue was dried at the high vacuum pump. The product was then immediately used in the next step with no further purification.
[CH3(CH2)2]2NCOCH2OCH2CO-Gly-Gly-Gly-Pro-Gly-Gly-Gly-OCH2C6H5, “C3-benzyl,” 1
Dipropylcarbamoylmethoxyacetic acid (32-[DGA]-OH)
General procedure 1. Quantities used: di-n-propylamine (2.0 g, 19.8 mmol) and diglycolic anhydride (2.5 g, 21.7 mmol). Product: white solid (3.2 g, 75%), mp 55-6 °C. 1H-NMR: 0.91 (6H, m, CH3), 1.59 (4H, m, CH3CH2CH2N), 3.07 (2H, t, J = 7.8 Hz, CH3CH2CH2N), 3.23 (2H, t, J = 7.8 Hz, CH3CH2CH2N), 4.20 (2H, s, COCH2O), 4.40 (2H, s, COCH2O). 13C-NMR: 11.1, 11.2, 20.6, 21.7, 48.4, 71.2, 73.0, 170.8, 171.8.
32-[DGA]-GGG-OCH2Ph
General procedure 2. Quantities used: 32-[DGA] OH (0.5 g, 2.3 mmol) and H2N-GGG-OCH2Ph tosylate (1.0 g, 2.3 mmol). Crude product chromatographed over silica gel (eluant CHCl3:CH3OH 97:3 v/v) to give 32-[DGA]-GGG-OCH2Ph (0.87 g, 79%) as a light oil. 1H-NMR: 0.89 (6H, m, CH3), 1.55 (4H, m, CH3CH2CH2N), 3.04 (2H, t, J = 7.5 Hz, CH3CH2CH2N), 3.23 (2H, t, J = 7.5 Hz, CH3CH2CH2N), 3.98-4.06 (6H, m, Gly CH2), 4.09 (2H, s, COCH2O), 4.33 (2H, s, COCH2O), 5.14 (2H, s, PhCH2O), 7.04 (1H, t, J = 5.7 Hz, NH), 7.32 (5H, m, Ar-H), 7.95 (1H, t, J = 5.7 Hz, Gly NH), 8.22 (1H, t, J = 5.7 Hz, Gly NH). 13C-NMR: 11.2, 11.3, 20.7, 21.9, 41.1, 43.1, 48.0, 48.4, 67.0, 69.8, 72.0, 128.2, 128.4, 128.6, 135.3, 168.7, 169.5, 169.55, 169.9, 171.5. IR (KBr): cm-1 1748 (COOR), 1652 (CO-N).
32-[DGA]-GGG-OH
General procedure 3. Quantities used: 32-[DGA]-GGG-OCH2Ph (0.82 g, 1.7 mmol). Product: white solid (0.66 g, 100%), mp 177-8 °C, that was used in the next step without purification.
32-[DGA]-GGGPGGG-OCH2Ph
General procedure 2. Quantities used: 32-[DGA]-GGG-OH (0.3 g, 0.8 mmol) and PGGG-OCH2Ph·HCl (0.32 g, 0.8 mmol). Crude product recrystallized from acetone to leave a white solid (0.42 g, 74%), mp 111-2 °C. 1H-NMR: 0.86 (6H, m, CH3), 1.51 (4H, m, CH3CH2CH2N), 1.85-2.20 (4H, m, Pro NCH2CH2CH2), 2.12 (H2O), 3.03 (2H, t, J = 7.5 Hz, CH3CH2CH2N), 3.22 (2H, t, J = 7.5 Hz, CH3CH2CH2N), 3.40-3.45 (1H, m, Pro NCH2CH2CH2), 3.55-4.07 (13H, m, Pro NCH2CH2CH2, Gly CH2), 4.11 (2H, s, COCH2O), 4.27 (2H, s, COCH2O), 4.32 (1H, bs, Pro CH), 5.11 (2H, d, J = 5.1 Hz, PhCH2O), 7.32 (5H, s, Ar-H), 7.54 (2H, m, Gly NH), 7.93 (2H, bt, Gly NH), 8.24 (2H, m, Gly NH). 13C-NMR: 11.1, 11.3, 20.7, 21.9, 25.0, 29.0, 41.2, 41.7, 42.6, 42.8, 43.4, 46.7, 47.7, 48.3, 61.1, 67.0, 69.4, 71.2, 128.1, 128.3, 128.5, 135.3, 168.5, 170.0, 170.1, 170.2, 170.7, 170.9, 173.5. Anal. Calcd. for C34H50N8O11·1.5H2O: C 52.77, H 6.90, N 14.48. Found: C 52.86, H 7.05, N 14.12.
[CH3(CH2)5]2NCOCH2OCH2CO-Gly-Gly-Gly-Pro-Gly-Gly-Gly-OCH2C6H5, “C6-benzyl,” 2
Dihexylcarbamoylmethoxyacetic acid (62-[DGA]-OH)
General procedure 1. Quantities used: di-n-hexylamine (2.1 mL, 9.5 mmol) and diglycolic anhydride (1.00 g, 8.6 mmol). Product: pale yellow oil (2.60 g, 81%). 1H-NMR: 0.88 (6H, m, CH3), 1.28 (12H, m, NCH2CH2(CH2)3CH3), 1.54 (4H, m, NCH2CH2(CH2)3CH3), 3.07 (2H, t, J = 7.8 Hz, NCH2), 3.34 (2H, t, J = 7.5 Hz, NCH2), 4.20 (2H, s, COCH2O), 4.38 (2H, s, COCH2O). 13C-NMR: 13.8, 13.9, 22.4, 26.3, 26.4, 27.2, 28.4, 31.3, 31.4, 46.7, 46.8, 71.1, 72.9, 170.7, 172.1.
62-[DGA]-GGG-OCH2Ph
General procedure 2. Quantities used: 62-[DGA] (0.50 g, 1.7 mmol) and GGG-OCH2Ph tosylate (0.75 g, 1.7 mmol). Crude product was chromatographed over silica gel (eluant CHCl3:CH3OH 95:5) to give 62-[DGA]-GGG-OCH2Ph (0.86 g, 92%) as a light oil. 1H-NMR: 0.86 (6H, m, CH3), 1.24 (12H, m, NCH2CH2(CH2)3CH3), 1.47 (4H, m, NCH2CH2(CH2)3CH3), 3.02 (2H, t, J = 7.5 Hz, NCH2), 3.22 (2H, t, J = 7.8 Hz, NCH2), 3.93-4.00 (6H, m, Gly CH2), 4.06 (2H, s, COCH2O), 4.26 (2H, s, COCH2O), 5.09 (2H, s, OCH2Ph), 7.30 (6H, m, NH, ArH), 7.92 (1H, t, J = 6.0 Hz, NH), 8.22 (1H, t, J = 6.0 Hz, NH). 13C-NMR: 13.96, 14.0, 22.5, 26.5, 26.7, 27.5, 28.8, 31.4, 31.5, 41.2, 43.0, 46.3, 46.8, 67.0, 69.7, 71.8, 128.4, 128.6, 128.8, 135.5, 168.7, 169.9, 170.0, 170.2, 171.5.
62-[DGA]-GGG-OH
General procedure 3. Quantities used: 62-[DGA]-GGG-OCH2Ph (0.58 g, 1.0 mmol). Product: white solid (0.38 g, 77%), mp 121-123 °C. 1H-NMR: 0.88 (6H, m, CH3), 1.25 (12H, m, NCH2CH2(CH2)3CH3), 1.49 (4H, m, NCH2CH2(CH2)3CH3), 3.05 (2H, t, J = 7.8 Hz, NCH2), 3.24 (2H, t, J = 7.8 Hz, NCH2), 3.96- 4.18 (8H, m, Gly CH2, COCH2O), 4.31 (2H, s, COCH2O), 7.09 (1H, bt, NH), 7.91 (1H, t, J = 5.7 Hz, NH), 8.25 (1H, t, J = 6.3 Hz, NH). 13C-NMR: 13.9, 14.1, 22.5, 26.5, 26.6, 27.5, 28.7, 31.4, 31.5, 41.1, 42.9, 46.3, 46.8, 61.3, 69.7, 71.8, 168.7, 169.8, 170.1, 170.2, 171.5.
62-[DGA]-GGGPGGG-OCH2Ph
General procedure 2. Quantities used: 62-[DGA]-GGG-OH (0.11 g, 0.2 mmol) and PGGG-OCH2Ph·HCl (0.10 g, 0.2 mmol). Crude product was chromatographed over silica gel (eluant CHCl3:CH3OH 9:1) to give a white solid (0.15 g, 78%), mp 129-30 °C. 1H-NMR: 0.87 (6H, m, CH3), 1.26 (12H, m, NCH2CH2(CH2)3CH3), 1.48 (4H, m, NCH2CH2(CH2)3CH3), 1.85-2.2 (4H, m, Pro NCH2CH2CH2), 3.06 (2H, t, J = 7.8 Hz, NCH2), 3.26 (2H, t, J = 7.5 Hz, NCH2), 3.40-3.58 (2H, m, Pro NCH2CH2CH2), 3.7-4.16 (14 H, m, Gly CH2, COCH2O), 4.27 (2H, s, COCH2O), 4.33 (1H, m, Pro CH), 5.13 (2H, dd, OCH2Ph), 7.34 (5H, m, ArH), 7.48 (2H, m, NH), 7.90 (2H, m, NH), 8.02 (1H, t, J = 6.0 Hz, NH), 8.32 (1H, t, J = 6.0 Hz, NH). 13C-NMR: 14.18, 14.23, 22.7, 25.4, 26.7, 26.9, 27.7, 29.0, 29.2, 31.6, 31.7, 41.5, 42.0, 43.0, 43.2, 43.7, 46.1, 46.5, 47.1, 61.5, 67.3, 69.7, 71.7, 128.5, 128.6, 128.8, 135.6, 168.6, 168.9, 170.2, 170.4, 170.5, 171.0, 171.3, 173.7. Anal. Calcd. for C40H62N8O11·0.5H2O: C 57.19, H 7.56, N 13.33. Found: C 56.91, H 7.71, N 13.12.
[CH3(CH2)7]2NCOCH2OCH2CO-Gly-Gly-Gly-Pro-Gly-Gly-Gly-OCH2C6H5, “C8-benzyl,” 3
Dioctylcarbamoylmethoxyacetic acid (82-[DGA]-OH)
General procedure 1. Quantities used: di-n-octylamine (2.0 g, 8.3 mmol) and diglycolic anhydride (1.0 g, 9.1 mmol). Product: white solid (2.1 g, 73%), mp 45-6 °C. 1H-NMR: 0.86 (6H, m, CH3), 1.26 (20H, m, CH3(CH2)5CH2CH2N), 1.53 (4H, m, CH3(CH2)5CH2CH2N), 3.07 (2H, t, J = 7.8 Hz, CH3(CH2)5CH2CH2N), 3.32 (2H, t, J = 7.8 Hz, CH3(CH2)5CH2CH2N), 4.19 (2H, s, COCH2O), 4.37 (2H, s, COCH2O). 13C-NMR: 14.0, 22.5, 25.8, 26.7, 26.9, 27.3, 28.5, 29.0, 29.1, 29.2, 31.6, 31.7, 46.7, 46.8, 71.1, 72.8, 170.5, 171.8.
82-[DGA]-GGG-OCH2Ph
General procedure 2. Quantities used: 82-[DGA] (0.5 g, 1.4 mmol) and GGG-OCH2Ph tosylate (0.63 g, 1.4 mmol). Crude product was chromatographed over silica gel (eluant CHCl3:CH3OH 98:2) to give 82-[DGA]-GGG-OCH2Ph (0.76 g, 88%) as a colorless oil. 1H-NMR: 0.86 (6H, m, CH3), 1.25 (20H, m, CH3(CH2)5CH2CH2N), 1.50 (4H, m, CH3(CH2)5CH2CH2N), 3.05 (2H, t, J = 7.5 Hz, CH3(CH2)5CH2CH2N), 3.25 (2H, t, J = 7.5 Hz, CH3(CH2)5CH2CH2N), 3.97-4.05 (6H, m, Gly CH2), 4.10 (2H, s, COCH2O), 4.31 (2H, s, COCH2O), 5.14 (2H, s, PhCH2O), 7.09 (1H, t, J = 5.1 Hz, NH), 7.34 (5H, m, Ar-H), 7.89 (1H, , t, J = 6.3 Hz, Gly NH), 8.27 (1H, t, J = 6.0 Hz, Gly NH). 13C-NMR: 14.0, 22.56, 22.59, 26.8, 27.0, 27.5, 28.8, 29.1, 29.2, 29.3, 31.7, 31.8, 41.1, 43.1, 46.4, 46.8, 67.0, 71.9, 128.2, 128.4, 128.6, 135.3, 168.5, 169.5, 169.6, 169.9, 171.4.
82-[DGA]-GGG-OH
General procedure 3. Quantities used: 82-[DGA]-GGG-OCH2Ph (1.0 g, 1.62 mmol). Product: white solid (0.85 g, 100%), mp 125-7 °C. This product was used directly in the subsequent reaction.
82-[DGA]-GGGPGGG-OCH2Ph
General procedure 2. Quantities used: 82-[DGA]-GGG-OH (0.3 g, 0.6 mmol) and PGGG-OCH2Ph·HCl (0.23 g, 0.6 mmol). Crude product crystallized from CH3OH to leave a white solid (0.25 g, 50%), mp 116-7 °C. 1H-NMR: 0.85 (6H, m, CH3), 1.24 (20H, m, CH3(CH2)5CH2CH2N), 1.47 (4H, m, CH3(CH2)5CH2CH2N), 1.85-2.20 (4H, m, Pro NCH2CH2CH2), 3.03 (2H, t, J = 7.5Hz, CH3(CH2)5CH2CH2N), 3.24 (2H, t, J = 7.5Hz, CH3(CH2)5CH2CH2N), 3.40-3.45 (1H, m, Pro NCH2CH2CH2), 3.75-4.07 (13H, m, Pro NCH2CH2CH2, Gly CH2), 4.08 (2H, s, COCH2O), 4.25 (2H, s, COCH2O), 4.33 (1H, bs, Pro CH), 5.11 (2H, d, J = 5.1 Hz, PhCH2O), 7.32 (5H, s, Ar-H), 7.58 (2H, bt, Gly NH), 7.93 (1H, bt, Gly NH), 7.95 (1H, bt, Gly NH), 8.26 (1H, m, Gly NH), 8.31 (1H, m, Gly NH). 13C-NMR: 14.2, 22.7, 25.3, 27.0, 27.2, 27.7, 29.0, 29.2, 29.3, 29.4, 29.5, 31.88, 31.94, 41.4, 42.0, 42.9, 43.0, 43.6, 46.4, 47.0, 61.3, 67.2, 69.2, 69.5, 71.4, 128.3, 128.5, 128.7, 135.6, 168.5, 168.9, 170.31, 170.38, 170.40, 170.6, 171.0, 171.1, 173.8. Anal. Calcd. for C44H70N8O11·0.5H2O: C 58.98, H 7.99, N 12.50. Found: C 59.04, H 7.95, N 12.63.
[CH3(CH2)9]2NCOCH2OCH2CO-Gly-Gly-Gly-Pro-Gly-Gly-Gly-OCH2C6H5, “C10-benzyl,” 4
This compound was prepared as previously described.30d
[CH3(CH2)11]2NCOCH2OCH2CO-Gly-Gly-Gly-Pro-Gly-Gly-Gly-OCH2C6H5, “C12-benzyl,” 5
Didodecylcarbamoylmethoxyacetic acid (122-[DGA]-OH)
General procedure 1. Quantities used: di-n-dodecylamine (1.0 g, 2.8 mmol) and diglycolic anhydride (0.3 g, 2.6 mmol). Product: white solid (1.20 g, 90%), mp 59-60 °C. 1H-NMR: 0.88 (6H, m, CH3), 1.26 (36H, m, CH3(CH2)9CH2CH2N), 1.54 (4H, m, CH3(CH2)9CH2CH2N), 3.07 (2H, t, J = 7.8 Hz, CH3(CH2)9CH2CH2N), 3.34 (2H, t, J = 7.2 Hz, CH3(CH2)9CH2CH2N), 4.20 (2H, s, COCH2O), 4.38 (2H, s, COCH2O). 13C-NMR: 14.1, 22.6, 26.8, 26.9, 27.4, 28.6, 29.2, 29.3, 29.5, 31.9, 46.8, 71.2, 73.1, 170.3, 170.6.
122-[DGA]-GGG-OCH2Ph
General procedure 2. Quantities used: 122-[DGA]-OH (0.17 g, 0.4 mmol) and TsOH·H2N-GGG-OCH2Ph (0.16 g, 0.4 mmol). Crude product was chromatographed (silica gel, eluant CHCl3:CH3OH 95:5) to give 122-[DGA]-GGG-OCH2Ph (0.15 g, 58%) as a colorless oil. 1H-NMR: 0.88 (6H, m, CH3), 1.25 (36H, m, CH3(CH2)9CH2CH2N), 1.51 (4H, m, CH3(CH2)9CH2CH2N), 3.05 (2H, t, J = 7.8 Hz, CH3(CH2)9CH2CH2N), 3.26 (2H, t, J = 7.5 Hz, CH3(CH2)9CH2CH2N), 3.98-4.06 (6H, m, Gly CH2), 4.10 (2H, s, COCH2O), 4.31 (2H, s, COCH2O), 5.14 (2H, s, PhCH2O), 7.18 (1H, bs, NH), 7.34 (5H, m, Ar-H), 7.94 (1H, t, J = 5.7 Hz, NH), 8.27 (1H, t, J = 6.0 Hz, NH). 13C-NMR: 14.0, 22.5, 26.7, 26.9, 27.4, 28.6, 29.2, 29.3, 29.4, 29.5, 31.8, 41.0, 43.0, 46.3, 46.7, 66.9, 69.6, 71.8, 128.3, 128.4, 128.6, 135.3, 168.6, 169.7, 169.8, 170.1, 171.6.
122-[DGA]-GGG-OH
General procedure 3. Quantities used: 122-[DGA]-GGG-OCH2Ph (0.15 g, 0.2 mmol). Product: white solid (0.116 g, 100%), mp 122-3°C. 1H-NMR (CD3OD): 0.95 (6H, t, J = 6.6 Hz, CH3), 1.23 (36H, m, CH3(CH2)9CH2CH2N), 1.45 (4H, m, CH3(CH2)9CH2CH2N), 3.11 (2H, t, J = 7.2 Hz, CH3(CH2)9CH2CH2N), 3.19 (2H, t, J = 7.2 Hz, CH3(CH2)9CH2CH2N), 3.72-3.80 (6H, m, Gly CH2), 3.97 (2H, s, COCH2O), 4.28 (2H, s, COCH2O), 8.17 (2H, m, NH), 8.25 (1H, t, J = 6.0 Hz, NH). 13C-NMR (CD3OD): 13.9, 22.1, 26.2, 26.4, 27.1, 28.3, 28.7, 29.0, 31.3, 41.6, 41.8, 45.1, 46.1, 68.8, 70.3, 168.3, 169.2, 169.4, 169.7, 171.4.
122-[DGA]-GGGPGGG-OCH2Ph
General procedure 2. Quantities used: 122-[DGA]-GGG-OH (0.10 g, 0.2 mmol) and PGGG-OCH2Ph·HCl (0.075 g, 0.2 mmol). Crude product was chromatographed (silica gel, CHCl3:CH3OH 95:5) and crystallized from CH3OH to leave a white solid (0.093 g, 60%), mp 111-2 °C. 1H-NMR: 0.87 (6H, t, J = 6.3 Hz, CH3), 1.25 (36H, m, CH3(CH2)9CH2CH2N), 1.48 (4H, m, CH3(CH2)9CH2CH2N), 1.85-2.20 (4H, m, Pro NCH2CH2CH2), 3.08 (2H, t, J = 7.5 Hz, CH3(CH2)9CH2CH2N), 3.24 (2H, t, J = 7.5 Hz, CH3(CH2)9CH2CH2N), 3.48 (1H, m, Pro NCH2CH2CH2), 3.59 (1H, m, Pro NCH2CH2CH2), 3.77-4.17 (14H, Gly CH2, COCH2O), 4.28 (2H, s, COCH2O), 4.34 (1H, bs, Pro CH), 5.14 (2H, d, J = 3.9 Hz, PhCH2O), 7.34 (5H, s, Ar-H), 7.46 (2H, m, Gly NH), 7.87 (3H, m, Gly NH), 8.37 (1H, bs, Gly NH). 13C-NMR: 14.2, 22.7, 25.3, 27.0, 27.2, 27.7, 29.0, 29.2, 29.3, 29.4, 29.5, 31.88, 31.94, 41.4, 42.0, 42.9, 43.0, 43.6, 46.4, 47.0, 61.3, 67.2, 69.2, 69.5, 71.4, 128.3, 128.5, 128.7, 135.6, 168.5, 168.9, 170.31, 170.38, 170.40, 170.6, 171.0, 171.1, 173.8. Anal. Calcd. for C52H86N8O11·H2O: C 61.39, H 8.72, N 11.01. Found: C 61.39, H 8.67, N 10.93.
[CH3(CH13)2]2NCOCH2OCH2CO-Gly-Gly-Gly-Pro-Gly-Gly-Gly-OCH2C6H5, “C14-benzyl,” 6
Di-n-tetradecylamine
A solution of tetradecylamine (5.0 g, 21.5 mmol), DMAP (0.4 g, 3.3 mmol) and NEt3 (5.0 mL) in toluene (60 mL) was cooled to 0 °C and a solution of myristoyl chloride (5.5 g, 21.6 mmol) in toluene (50 mL) was added dropwise. The reaction was stirred at rt (2 da), the solvent was removed, and the residue crystallized twice from ethanol and dried in vacuo to constant weight. This product (8.4 g, 19.8 mmol) was dissolved in anhydrous THF and a suspension of LiAlH4 (1.5 g, 39.5 mmol) in THF was carefully added. The reaction was heated (reflux, 18 h), then cooled (rt) and poured into 10% aq Na2SO4 (300 mL), CHCl3 (300 mL) was added, and the mixture was stirred and filtered. The solid was washed with chloroform, the filtrates were combined, and the organic phase was washed with brine and evaporated. The residue was crystallized from EtOH to give HN((CH2)13CH3)2(7.35 g, 83%), mp 59-60 °C. 1H-NMR: 0.87 (6H, t, J = 6.6 Hz, CH3), 1.25 (44H, m, CH3(CH2)11CH2CH2N), 1.47 (4H, m, CH3(CH2)11CH2CH2N), 2.57 (4H, t, J = 7.2 Hz, CH3(CH2)11CH2CH2N). 13C-NMR: 14.3, 22.9, 27.6, 29.6, 29.8, 29.9, 30.4, 32.1, 50.4.
Ditetradecylcarbamoylmethoxyacetic acid (142-[DGA]-OH)
General procedure 1. Quantities used: di-n-tetradecylamine (7.00 g, 17.1 mmol) and diglycolic anhydride (2.0 g, 17.2 mmol). Product: white solid (7.12 g, 79%), mp 72-4 °C. 1H-NMR: 0.87 (6H, t, J = 7.2 Hz, CH3), 1.25 (44H, m, CH3(CH2)11CH2CH2N), 1.54 (4H, m, CH3(CH2)11CH2CH2N), 3.07 (2H, t, J = 7.5 Hz, CH3(CH2)11CH2CH2N), 3.34 (2H, t, J = 7.8 Hz, CH3(CH2)11CH2CH2N), 4.20 (2H, s, COCH2O), 4.38 (2H, s, COCH2O). 13C-NMR: 14.2, 22.8, 26.9, 27.1, 27.5, 28.7, 29.2, 29.4, 29.5, 29.6, 29.7, 29.8, 32.0, 46.9, 47.0, 71.1, 72.8, 170.6, 172.1.
142-[DGA]-GGG-OCH2Ph
General procedure 2. Quantities used: 142-[DGA] (0.98 g, 1.9 mmol) and GGG-OCH2Ph tosylate (0.84 g, 1.9 mmol). Crude product was chromatographed (silica gel, CHCl3:CH3OH 95:5) to give a colorless oil (1.12 g, 76%). 1H-NMR: 0.86 (6H, t, J = 6.9 Hz, CH3), 1.23 (44H, m, CH3(CH2)11CH2CH2N), 1.48 (4H, m, CH3(CH2)11CH2CH2N), 3.04 (2H, t, J = 8.1 Hz, CH3(CH2)11CH2CH2N), 3.24 (2H, t, J = 7.8 Hz, CH3(CH2)11CH2CH2N), 3.96-4.02 (6H, m, Gly CH2), 4.08 (2H, s, COCH2O), 4.28 (2H, s, COCH2O), 5.12 (2H, s, PhCH2O), 7.30 (6H, bs, NH, Ar-H), 7.92 (1H, , t, J = 5.7 Hz, Gly NH), 8.26 (1H, t, J = 6.0 Hz, Gly NH). 13C-NMR: 14.2, 22.8, 27.0, 27.2, 27.7, 28.9, 29.5, 29.7, 29.8, 32.0, 41.3, 43.2, 46.5, 47.0, 67.1, 69.8, 71.9, 128.3, 128.5, 128.7, 135.5, 168.6, 169.7, 169.8, 170.1, 171.4.
142-[DGA]-GGG-OH
General procedure 3. Quantities used: 142-DGA-[GGG]-OCH2Ph (1.10 g, 1.4 mmol). A white solid (0.87 g, 89 %), mp 125-7 °C. 1H-NMR (CDCl3-CD3OD 9:1): 0.72 (6H, t, J = 6.6 Hz, CH3), 1.13 (44H, m, CH3(CH2)11CH2CH2N), 1.40 (4H, m, CH3(CH2)11CH2CH2N), 2.96 (2H, t, J = 7.5 Hz, CH3(CH2)11CH2CH2N), 3.16 (2H, t, J = 7.2 Hz, CH3(CH2)11CH2CH2N), 3.80-3.83 (6H, m, Gly CH2), 3.96 (2H, s, COCH2O), 4.15 (2H, s, COCH2O). 13C-NMR (CDCl3:CD3OD 9:1): 13.9, 22.6, 26.7, 26.9, 27.4, 28.7, 29.0, 29.2, 29.5, 29.6, 31.8, 40.9 (bs), 42.5, 46.3, 46.9, 68.9, 70.8, 168.5, 170.2, 171.4, 171.6.
142-[DGA]-GGGPGGG-OCH2Ph
General procedure 2. Quantities used: 142-[DGA]-GGG-OH (0.80 g, 1.2 mmol) and PGGG-OCH2Ph·HCl (0.55 g, 1.3 mmol). Crude product was chromatographed (silica gel, CHCl3:CH3OH 98:2 9:1) to give a solid, which was crystallized from CH3OH (0.92 g, 76%), m.p 127-9 °C. 1H-NMR: 0.85 (6H, t, J = 6.6 Hz, CH3), 1.23 (44H, m, CH3(CH2)11CH2CH2N), 1.46 (4H, m, CH3(CH2)11CH2CH2N), 1.85-2.15 (4H, m, Pro NCH2CH2CH2), 3.04 (2H, t, J = 6.9Hz, CH3CH2(CH2)11CH2N), 3.23 (2H, t, J = 7.2Hz, CH3CH2(CH2)11CH2N), 3.46 (2H, m, Pro NCH2CH2CH2), 3.72-4.10 (14H, Gly CH2, COCH2O), 4.24 (2H, s, COCH2O), 4.32 (1H, bs, Pro CH), 5.10 (2H, d, J = 6.3 Hz, PhCH2O), 7.31 (5H, s, Ar-H), 7.55 (2H, m, Gly NH), 7.90 (2H, m, Gly NH), 8.23 (2H, bt, Gly NH). 13C-NMR: 14.2, 22.8, 25.2, 27.0, 27.2, 27.7, 29.0, 29.1, 29.4, 29.5, 29.6, 29.7, 32.0, 41.3, 41.9, 42.8, 43.0, 43.6, 46.4, 47.0, 61.2, 67.1, 69.5, 71.4, 128.3, 128.4, 128.7, 135.6, 168.4, 168.7, 170.19, 170.25, 170.3, 170.4, 170.9, 171.0, 173.7. Anal. Calcd. for C56H94N8O11·H2O: C 62.66, H 9.01, N 10.43. Found: C 62.76, H 8.83, N 10.38.
CH3(CH15)2]2NCOCH2OCH2CO-Gly-Gly-Gly-Pro-Gly-Gly-Gly-OCH2C6H5, “C16-benzyl,” 7
Di-n-hexadecylamine
The procedure used for di-n-tetradecylamine was employed using hexadecylamine and palmitoyl chloride (65%), mp 79-80 °C. 1H-NMR: 0.87 (6H, t, J = 7.2 Hz, CH3), 1.25 (52H, m, CH3(CH2)13CH2CH2N), 1.48 (4H, m, CH3(CH2)13CH2CH2N), 2.58 (4H, t, J = 7.5 Hz, CH3(CH2)13CH2CH2N). 13C-NMR: 14.3, 22.9, 27.6, 29.6, 29.8, 29.9, 30.3, 32.1, 50.3.
Di-n-hexadecylcarbamoylmethoxyacetic acid (162-[DGA]-OH)
General procedure 1. Quantities used: di-n-hexadecylamine (8.5 g, 18.4 mmol) and diglycolic anhydride (2.14 g, 17.2 mmol). The product was a white solid (8.60 g, 80%), mp 79-81 °C. 1H-NMR: 0.87 (6H, t, J = 6.3 Hz, CH3), 1.24 (52H, m, CH3(CH2)13CH2CH2N), 1.53 (4H, m, CH3(CH2)13CH2CH2N), 3.07 (2H, t, J = 7.8 Hz, CH3(CH2)13 CH2CH2N), 3.33 (2H, t, J = 7.5 Hz, CH3(CH2)13CH2CH2N), 4.19 (2H, s, COCH2O), 4.37 (2H, s, COCH2O). 13C-NMR: 14.3, 22.9, 27.0, 27.1, 27.6, 28.8, 29.4, 29.5, 29.6, 29.7, 29.8, 29.9, 32.1, 47.0, 71.3, 73.0, 170.7, 172.0.
162-[DGA]-GGG-OCH2Ph
General procedure 2. Quantities used: 162-[DGA] (1.10 g, 1.9 mmol) and GGG-OCH2Ph tosylate (0.85 g, 1.9 mmol). Crude product was chromatographed (silica gel, CHCl3:CH3OH 95:5) to give the product (1.28 g, 81%), a colorless oil. 1H-NMR: 0.85 (6H, t, J = 6.9 Hz, CH3), 1.23 (44H, m, CH3(CH2)13CH2CH2N), 1.48 (4H, m, CH3(CH2)13CH2CH2N), 3.03 (2H, t, J = 7.8 Hz, CH3(CH2)13CH2CH2N), 3.23 (2H, t, J = 7.5 Hz, CH3(CH2)13CH2CH2N), 3.94-4.01 (6H, m, Gly CH2), 4.07 (2H, s, COCH2O), 4.27 (2H, s, COCH2O), 5.10 (2H, s, PhCH2O), 7.30 (6H, bs, NH, Ar-H), 7.92 (1H, t, J = 6.0 Hz, NH), 8.23 (1H, t, J = 6.0 Hz, NH). 13C-NMR: 14.2, 22.8, 27.0, 27.1, 27.7, 28.9, 29.4, 29.5, 29.6, 29.8, 32.0, 41.2, 43.1, 46.5, 46.9, 67.0, 69.7, 71.8, 128.3, 128.4, 128.7, 135.4, 168.5, 169.7, 169.8, 170.0, 171.3.
162-[DGA]-GGG-OH
General procedure 3. Quantities used: 162-[DGA]-GGG-OCH2Ph (1.25 g, 1.5 mmol). Product: white solid (1.0 g, 90 %), mp 188-90 °C. 1H-NMR (CDCl3:CD3OD 4:1): 0.74 (6H, t, J = 6.9 Hz, CH3), 1.12 (52H, m, CH3(CH2)13CH2CH2N), 1.41 (4H, m, CH3(CH2)13CH2CH2N), 2.97 (2H, t, J = 7.5 Hz, CH3(CH2)13CH2CH2N), 3.16 (2H, t, J = 7.2 Hz, CH3(CH2)13CH2N), 3.79 (2H, s, Gly CH2), 3.80 (2H, s, Gly CH2), 3.83 (2H, s, Gly CH2), 3.90 (2H, s, COCH2O), 4.16 (2H, s, COCH2O). 13C-NMR (CDCl3-CD3OD 4:1): 13.9, 22.6, 26.8, 26.9, 27.4, 28.7, 29.22, 29.25, 29.4, 29.5, 29.56, 29.60, 31.8, 41.3 (bs), 42.50, 42.55, 46.3, 46.9, 68.9, 70.7, 168.5, 170.1, 170.2, 171.4, 172.3.
162-[DGA]-GGGPGGG-OCH2Ph
General procedure 2. Quantities used: 162-[DGA]-GGG-OH (0.95 g, 1.3 mmol) and PGGG-OCH2Ph·HCl (0.57 g, 1.4 mmol). The crude material was chromatographed (silica gel, CHCl3:CH3OH 98:2 9:1) and crystallized from CH3OH to give a white solid (1.0 g, 72%), mp 131-3 °C. 1H-NMR: 0.84 (6H, t, J = 6.3 Hz, CH3), 1.22 (52H, m, CH3(CH2)13CH2CH2N), 1.46 (4H, m, CH3(CH2)13CH2CH2N), 1.85-2.15 (4H, m, Pro NCH2CH2CH2), 3.03 (2H, t, J = 7.5Hz, CH3CH2(CH2)13CH2N), 3.22 (2H, t, J = 6.9 Hz, CH3CH2(CH2)13CH2N), 3.46 (2H, m, Pro NCH2CH2CH2), 3.71-4.10 (14H, Gly CH2, COCH2O), 4.24 (2H, s, COCH2O), 4.32 (1H, bs, Pro CH), 5.09 (2H, d, J = 6.3 Hz, PhCH2O), 7.30 (5H, s, Ar-H), 7.55 (2H, m, NH), 7.90 (2H, m, NH), 8.21 (2H, bt, NH). 13C-NMR: 14.2, 22.8, 25.2, 27.0, 27.2, 27.7, 29.0, 29.1, 29.4, 29.5, 29.7, 29.8, 32.0, 41.3, 41.9, 42.8, 43.0, 43.6, 46.4, 46.9, 47.0, 61.2, 67.1, 69.6, 71.4, 128.3, 128.4, 128.7, 135.6, 168.4, 168.6, 170.21, 170.24, 170.30, 170.34, 170.8, 171.0, 173.7. Anal. Calcd. for C60H102N8O11·0.5H2O: C 64.32, H 9.27, N 9.99. Found: C 64.50, H 9.28, N 10.00.
[CH3(CH2)17]2NCOCH2OCH2CO-Gly-Gly-Gly-Pro-Gly-Gly-Gly-OCH2C6H5, “C18-benzyl,” 8
This compound was prepared as described in detail previously.31d
182-[DGA]-GGGPGGG-OH
General procedure 3. Quantities used: 182-[DGA]-GGGPGGG-OCH2Ph (1.0 g, 0.9 mmol). Product: white solid (0.87 g, 94%), mp 167 °C dec. 1H-NMR: 0.84 (6H, t, J = 6.9 Hz, CH3), 1.22 (60H, m, CH3(CH2)15CH2CH2N), 1.50 (4H, bs, CH3(CH2)15CH2CH2N), 1.80-2.20 (4H, m, Pro NCH2CH2CH2), 3.06 (2H, t, J = 7.5 Hz, CH3(CH2)16CH2N), 3.26 (2H, t, J = 7.5 Hz, CH3(CH2)16CH2N), 3.00-4.40 (19H, Pro NCH2CH2CH2, Gly CH2, COCH2O, Pro CH), 7.50 (1H, t, J = 6.0 Hz, NH), 7.69 (1H, t, J = 6.0 Hz, NH), 7.90-8.05 (2H, m, NH), 8.32 (1H, bs, NH). 13C-NMR: 14.1, 22.7, 25.1, 26.9, 27.1, 27.6, 28.8, 29.3, 29.4, 29.6, 29.7, 31.9, 41.6, 41.9, 42.6, 42.8, 43.4, 46.4, 47.0, 61.2, 69.0, 70.9, 168.6, 168.9, 170.4, 170.8, 171.0, 171.3, 172.6, 173.7. IR (CHCl3): cm-1 3306, 3083, 2918, 2850, 1730, 1658, 1651, 1646, 1540, 1467, 1412, 1378, 1338, 1241, 1130, 1030, 909, 722.
[CH3(CH2)17]2NCOCH2OCH2CO-Gly-Gly-Gly-Pro-Gly-Gly-Gly-OCH2CH3, “C18-ethyl,” 9
TsOH·GGG-OEt
Triglycine (2.0 g, 10.6 mmol), p-TsOH (2.2 g, 11.6 mmol), EtOH (10 mL, 0.17 mol), and toluene (50 mL) were heated at reflux for 4 da (water removed by Dean-Stark trap) and the solvent was then evaporated. The residue was dissolved in CH3OH and Et2O was added until cloudy. Cooling overnight (4 °C) precipitated a white solid (3.4 g, 83%), mp 140-2 °C. 1H-NMR (CDCl3:CD3OD 9:1): 1.05 (3H, t, J = 7.2 Hz, CH2CH3), 2.18 (3H, s, Ar-CH3), 3.65 (2H, s, Gly CH2), 3.70 (2H, s, Gly CH2), 3.77 (2H, s, Gly CH2), 3.96 (2H, q,J = 7.2 Hz, CH2CH3), 6.99 (2H, d,J = 8.1 Hz, ArH), 7.50 (2H, d,J = 8.1 Hz, ArH). 13C-NMR (CDCl3:CD3OD 9:1): 12.7, 20.9, 40.8, 40.9, 42.5, 42.6, 61.3, 125.5, 128.8, 140.7, 141.2, 167.1, 167.2, 170.1.
Boc-PGGG-OEt
General procedure 2. Quantities used: Boc-L-Proline (0.35g, 1.6 mmol) and GGG-OEt tosylate (0.63 g, 1.6 mmol). The crude material was chromatographed (silica gel, CHCl3:CH3OH 95:5 9:1) to give the final product (0.60 g, 88%) as a colorless oil. 1H-NMR: 1.18 (3H, t, J = 6.6 Hz, CH2CH3), 1.35 (9H, s, C (CH3)3), 1.75-2.15 (4H, m, Pro NCH2CH2CH2), 3.40 (2H, m, Pro NCH2CH2CH2), 3.75-4.00 (7H, m, Gly CH2, Pro CH), 4.09 (2H, q,J = 7.2 Hz, CH2CH3), 7.32 (2H, bs, NH), 7.65 (2H, bs, NH), 7.92 (2H, bs, NH). 13C-NMR: 14.2, 24.7, 28.4, 29.8, 41.3, 43.0, 43.3, 47.3, 60.7, 61.3, 77.4, 80.7, 155.6, 169.7, 169.8, 170.3, 174.1
PGGG-OEt·HCl
General procedure 4. Quantities used: Boc-PGGG-OEt (0.23 g, 0.5 mmol). The product was used immediately in the subsequent reaction.
182-[DGA]-GGGPGGG-OEt
General procedure 2. Quantities used: 182-[DGA]-GGG-OH (0.43 g, 0.5 mmol) and PGGG-OEt·HCl (0.19 g, 0.5 mmol). The crude material was chromatographed (silica gel, CHCl3:CH3OH 95:5 9:1) to give the final product (0.22 g, 38%) as a white solid, mp 127-9 °C. 1H-NMR: 0.84 (9H, t, J = 6.9 Hz, CH3), 1.22 (60H, m, CH3(CH2)15CH2CH2N), 1.48 (4H, m, CH3(CH2)15CH2CH2N), 1.85-2.21 (4H, m, Pro NCH2CH2CH2), 3.05 (2H, t, J = 7.5Hz, CH3CH2(CH2)15CH2N), 3.23 (2H, t, J = 7.2 Hz, CH3CH2(CH2)15CH2N), 3.53 (1H, m, Pro NCH2CH2CH2), 3.60 (1H, m, Pro NCH2CH2CH2), 3.73-4.15 (16H, Gly CH2, COCH2O, OCH2CH3), 4.26 (2H, s, COCH2O), 4.35 (1H, m, Pro CH), 7.53 (1H, bt, NH), 7.59 (1H, bt, NH), 7.91 (2H, m, NH), 8.25 (2H, bt, NH). 13C-NMR: 14.2, 22.8, 25.3, 27.0, 27.2, 27.8, 29.0, 29.2, 29.5, 29.6, 29.7, 29.8, 32.0, 41.4, 42.0, 42.8, 43.0, 43.6, 46.4, 47.0, 61.3, 61.5, 69.5, 71.4, 168.5, 168.8, 170.3, 170.5, 170.9, 171.1, 173.7. Anal. Calcd. for C59H108N8O11·H2O: C 63.07, H 9.87, N 9.97. Found: C 63.01, H 9.79, N 10.11.
[CH3(CH2)17]2NCOCH2OCH2CO-Gly-Gly-Gly-Pro-Gly-Gly-Gly-OCH2(CH3)2, “C18-isopropyl,” 10
HCl·GGG-OCH(CH3)2
GGG (1.0 g, 5.3 mmol) was suspended in 2-PrOH (10 mL), cooled (5 °C), SOCl2 (0.8 mL, 11.0 mmol) was added dropwise, and the mixture was heated under reflux (4 da). After evaporation of solvent, toluene (20 mL) was added and evaporated. The residue was dried at high vacuum and crystallized from CH3OH:Et2O (20 mL:40 mL) to give white crystals which were crystallized a second time to afford pure product (0.87 g, 61%), mp 185-186 °C. 1H NMR (CD3OD): 1.25 (6H, d, J = 6.3 Hz, OCH(CH3)2), 3.76 (2H, s, Gly CH2), 3.92 (2H, s, Gly CH2), 4.00 (2H, s, Gly CH2), 5.04 (1H, m, OCH(CH3)2. 13C-NMR (CD3OD): 22.1, 41.7, 42.4, 43.2, 70.4, 168.0, 170.9, 171.9. IR (nujol): cm-1 3330, 3232, 2954, 2924, 2854, 1732, 1703, 1646, 1789, 1573, 1548, 1489, 1461, 1411, 1377, 1219, 1153, 1102, 1033, 961, 911, 820, 723.
Cbz-PGGG-OCH(CH3)2
General procedure 2. Quantities used: Cbz-l-Proline (0.48 g, 1.9 mmol), and HCl·GGG-O-CH(CH3)2 (0.5 g, 1.9 mmol). The crude material was chromatographed (silica gel, CH3OH:CH2Cl2 98:2 85:5) to give colorless crystals (0.74 g, 86%), mp 93-95 °C. 1H-NMR: 1.21 (6H, d, J = 6.3 Hz, OCH(CH3)2), 1.80-2.10 (4H, m, Pro NCH2CH2CH2), 3.45-3.60 (2H, m, Pro NCH2CH2CH2), 3.80-4.10 (6H, Gly CH2), 4.24 (1H, t, J = 6.3 Hz, Pro CH), 4.90-5.20 (3H, OCH(CH3)2, PhCH2O), 7.08 (1H, t, J = 6.0 Hz, NH), 7.33 (5H, Ar-H), 7.44 (1H, t, J = 6.0 Hz, NH), 7.75 (1H, t, J = 6.0 Hz, NH). 13C-NMR:: 21.7, 24.7, 29.5, 41.4, 42.9, 43.3, 47.1, 61.0, 67.5, 69.0, 127.7, 128.1, 128.5, 136.2, 156.0, 169.1, 169.5, 169.9, 173.3. IR (CHCl3): cm-1 3312, 3066, 2981, 2938, 2882, 1744, 1669, 1541, 1420, 1376, 1359, 1210, 1108, 1031, 984, 947, 920, 772, 734, 699, 669.
PGGG-OCH(CH3)2
General procedure 3. Quantities used: Cbz-PGGG-OCH(CH3)2 (0.36 g, 0.8 mmol). Product: white solid (0.25 g, 100%), mp 144-145 °C. 1H-NMR (CD3OD): 1.251 (6H, d, J = 6.3 Hz, OCH(CH3)2), 1.75-2.30 (4H, m, Pro NCH2CH2CH2), 3.00-3.15 (2H, m, Pro NCH2CH2CH2), 3.85-3.95 (7H, Gly CH2, and Pro CH), 5.02 (1H, m, OCH(CH3)2). 13C-NMR (CD3OD): 22.1, 26.7, 31.7, 42.4, 43.4, 43.7, 48.0, 61.6, 70.4, 170.9, 172.2, 176.7. IR (nujol): cm-1 3177, 2922, 2855, 2726, 1740, 1647, 1542, 1461, 1377, 1304, 1206, 1170, 1028, 967, 770, 722.
182-[DGA]-GGGPGGG-OCH(CH3)2
General procedure 2. Quantities used: 182-[DGA]-GGG-OH (0.27 g, 0.3 mmol) and PGGG-OCH(CH3)2 (0.11 g, 0.3mmol). Crude product was crystallized (2 CH3OH) to give a white solid (0.26 g, 70%), mp 129-131 °C. 1H-NMR: 0.87 (6H, t, J = 6.9 Hz, CH3(CH2)15CH2CH2N), 1.20-1.30 (66H, CH3(CH2)15CH2CH2N, OCH(CH3)2), 1.51 (2H, m, CH3(CH2)15CH2CH2N), 1.95-2.25 (4H, m, Pro NCH2CH2CH2), 3.08 (2H, t, J = 7.5 Hz, CH3(CH2)15CH2CH2N), 3.27 (2H, t, J = 7.5 Hz, CH3(CH2)15CH2CH2N), 3.75-4.40 (19H, Pro NCH2CH2CH2, Gly CH2, Pro CH, and COCH2O), 5.01 (1H, m, OCH(CH3)2), 7.33 (1H, bs, NH), 7.50 (1H, bs, NH), 7.87 (3H, bs, NH), 8.34 (1H, bs, NH). 13C-NMR: 14.1, 21.8, 22.7, 25.2, 26.9, 27.1, 27.6, 28.9, 29.0, 29.3, 29.4, 29.5, 29.6, 29.7, 31.9, 41.5, 42.0, 43.0, 43.5, 46.3, 47.0, 61.3, 69.2, 69.6, 71.6, 168.4, 168.8, 169.5, 170.0, 170.1, 170.3, 170.6, 171.2, 173.2. IR (CHCl3): cm-1 3308, 2920, 2851, 1744, 1652, 1540, 1467, 1376, 1338, 1209, 1129, 1109, 1031, 722. Anal. Calcd. for C60H110N8O11·H2O: C63.35, H 9.92, N 9.85. Found: C 63.40, H 9.92, N 9.52.
[CH3(CH2)17]2NCOCH2OCH2CO-Gly-Gly-Gly-Pro-Gly-Gly-Gly-O(CH2)6CH3, “C18-heptyl,” 11
TsOH·GGG-OC7H15
GGG (2.0 g, 10.6 mmol) and p-TsOH·H2O (2.2 g, 11.6 mmol) were added to 1-heptanol (12 g, 103.3 mmol) and toluene (40 mL). The mixture was heated to reflux and water was removed azeotropically (Dean-Stark trap, 3 h). The mixture was cooled (rt), evaporated, and crystallized from CH3OH:Et2O (20 mL:60 mL) to give 4.32 g (89%) of a white solid, mp 149-150 °C. 1H-NMR (CD3OD): 0.91 (3H, t, J = 7.2 Hz, CH3(CH2)4CH2CH2O), 1.32 (8H, m, CH3(CH2)4CH2CH2O), 1.64 (2H, bs, CH3(CH2)4CH2CH2O), 2.37 (3H, s, Ar-CH3), 3.65-3.75 (2H, m, Gly CH2), 3.93 (2H, s, Gly CH2), 3.98 (2H, s, Gly CH2), 4.11 (2H, t, J = 6.6 Hz, CH3(CH2)4CH2CH2O), 7.24 (2H, d, J = 8.1 Hz, Ar-H), 7.71 (2H, d, J = 8.1 Hz, Ar-H). 13C-NMR (CD3OD): 14.5, 21.4, 23.8, 27.0, 29.8, 30.2, 33.0, 41.7, 42.1, 43.2, 66.6, 127.1, 130.0, 141.9, 143.6, 168.1, 171.4, 171.9. IR (nujol): cm-1 3336, 3263, 3110, 2922, 2853, 2719, 1740, 1706, 1685, 1651, 1594, 1547, 1499, 1461, 1412, 1377, 1368, 1313, 1232, 1214, 1175, 1128, 1037, 1014, 912, 816, 738, 724, 684.
Cbz-PGGG-OC7H15
General procedure 2. Quantities used: Cbz-L-Proline (1.08 g, 4.3 mmol) and TsOH·GGG-OC7H15 (2.0 g, 4.4 mmol). The crude product was chromatographed (silica gel, CH3OH:CHCl3 98:2 95:5) to give colorless crystals (2.01 g, 90%), mp 94-96 °C. 1H-NMR: 0.87 (3H, t, J = 6.9 Hz, CH3(CH2)4CH2CH2O), 1.27 (8H, m, CH3(CH2)4CH2CH2O), 1.60 (2H, bs, CH3(CH2)4CH2CH2O), 1.75-2.20 (4H, m, Pro NCH2CH2CH2), 3.45-3.60 (2H, m, Pro NCH2CH2CH2), 3.75-4.00 (6H, Gly CH2), 4.07 (2H, t, J = 6.6 Hz, CH3(CH2)4CH2CH2O), 4.24 (1H, t, J = 6.3 Hz, Pro CH), 5.02-5.17 (2H, m, PhCH2O), 7.00 (1H, bs, NH), 7.21 (1H, bs, NH), 7.33 (5H, m, Ar-H), 7.68 (1H, bs, NH). 13C-NMR: 14.0, 22.5, 25.7, 28.5, 28.8, 29.5, 31.6, 41.2, 43.0, 43.4, 47.1, 61.0, 65.5, 67.6, 127.8, 128.2, 128.5, 136.2, 156.0, 169.4, 169.7, 171.7, 173.3. IR (CHCl3): cm-1 3310, 3068, 2955, 2931, 2858, 1749, 1669, 1541, 1419, 1360, 1206, 1125, 1091, 1030, 986, 919, 770, 731, 698.
PGGG-OC7H15
General procedure 3. Quantities used: Cbz-PGGG-OC7H15(1.0 g, 1.9 mmol). Product: a white, waxy solid (0.74 g, 100%). 1H NMR (CD3OD): 0.91 (3H, t, J = 7.2 Hz, CH3(CH2)4CH2CH2O), 1.32 (8H, m, CH3(CH2)4CH2CH2O), 1.65 (2H, bs, CH3(CH2)4CH2CH2O), 1.75-2.20 (4H, m, Pro NCH2CH2CH2), 2.70-3.00 (2H, m, Pro NCH2CH2CH2), 3.70-4.05 (7H, Gly CH2, Pro CH), 4.13 (2H, t, J = 6.6 Hz, CH3(CH2CH2O). 13C-NMR (CD3OD): 14.5, 22.3, 23.8, 26.1, 26.8, 27.0, 27.1, 27.4, 29.8, 30.2, 31.9, 33.0, 42.1, 43.5, 43.7, 43.8, 61.7, 64.3, 66.5, 171.0, 171.4, 172.0, 178.2. IR (nujol): cm-1 3284, 3177, 2921, 2855, 1740, 1653, 1461, 1377, 1155, 1077, 1029, 967, 892, 846, 770, 723.
182-[DGA]-GGGPGGG-OC7H15
General procedure 2. Quantities used: 182-[DGA]-GGG-OH (0.16 g, 0.2 mmol) and PGGG-OC7H15 (0.08 g, 0.2 mmol). Crude product was chromatographed (silica gel, CH3OH:CHCl3 9:1 85:15) to give a white solid (0.14 g, 61%), mp 99-101 °C. 1H NMR: 0.85-0.90 (9H, CH3(CH2)15CH2CH2N, CH3(CH2)4CH2CH2O), 1.24 (68H, m, CH3(CH2)15CH2CH2N, CH3(CH2)4CH2CH2O), 1.49 (4H, bs, CH3(CH2)15CH2CH2N), 1.60 (2H, bs, CH3(CH2)4CH2CH2O), 1.80-2.20 (4H, m, Pro NCH2CH2CH2), 3.05 (2H, t, J = 7.5 Hz, CH3(CH2)15CH2CH2N), 3.24 (2H, t, J = 7.5 Hz, CH3(CH2)15CH2CH2N), 3.45-3.75 (2H, m, Pro NCH2CH2CH2), 3.85-4.10(16H, Gly CH2, CH3(CH2)4CH2CH2O, COCH2O), 4.27 (2H, s, COCH2O), 4.36 (1H, t, J = 6.6 Hz, Pro CH), 7.65 (2H, bs, NH), 7.89 (1H, bs, NH), 8.03 (1H, bs, NH), 8.26 (1H, bs, NH), 8.42 (1H, bs, NH). 13C-NMR: 14.0, 14.1, 22.6, 22.7, 25.1, 25.8, 26.9, 27.1, 27.6, 28.5, 28.8, 28.9, 29.1, 29.3, 29.4, 29.5, 29.6, 29.7, 31.7, 31.9, 41.2, 42.0, 42.8, 42.9, 43.4, 46.3, 61.3, 65.6, 69.4, 71.2, 168.5, 168.9, 170.2, 170.3, 170.4, 170.7, 171.2, 173.3. IR (CHCl3): cm-1 3308, 2920, 2851, 1744, 1652, 1540, 1467, 1376, 1338, 1209, 1129, 1109, 1031, 722. Anal. Calcd. for C64H118N8O11·H2O: C 64.40, H 10.13, N 9.39. Found: C 64.36, H 10.11, N 9.21.
[CH3(CH2)17]2NCOCH2OCH2CO-Gly-Gly-Gly-Pro-Gly-Gly-Gly-O-CH2-c-C6H11, “C18-cyclohexylmethyl,” 12
TsOH·GGG-OCH2C6H11
GGG (2.0 g, 10.6 mmol) and p-TsOH·H2O (2.2 g, 11.6 mmol) were added to cyclohexylmethanol (13.3 g, 116.4 mmol) and toluene (40 mL). The mixture was heated and water removed (reflux, Dean-Stark trap, 3h). The mixture was cooled to rt and evaporated, the residue was crystallized from CH3OH:Et2O methanol-ether (1:20 v:v) to give a white solid (4.80 g, 99%) mp 161-162 °C. 1H-NMR (CD3OD): 0.90-1.05 (2H, m, OCH2C6H11), 1.15-1.40 (3H, OCH2C6H11), 1.60-1.80 (6H, m, OCH2C6H11), 2.37 (3H, s, ArCH3), 3.77 (2H, 3, Gly CH2), 3.92-3.96 (4H, s, OCH2C6H11, CH2 Gly CH2), 3.98 (2H, s, Gly CH2), 7.24 (2H, d, J = 8.1 Hz, Ar-H), 7.71 (2H, d, J = 8.1 Hz, Ar-H). 13C-NMR (CD3OD): 21.4, 26.9, 27.6, 30.8, 38.6, 41.8, 42.0, 42.1, 43.2, 71.4, 127.1, 130.0, 141.9, 168.1, 171.4, 171.9. IR (nujol): 3307, 2954, 2923, 2854, 1746, 1710, 1656, 1547, 1460, 1404, 1377, 1203, 1127, 1035, 1011, 984, 814, 722, 685 cm-1.
BocPGGG-OCH2C6H11
General procedure 2. Quantities used: Boc-L-Proline (0.47 g, 2.2 mmol), and TsOH·GGG-O-CH2C6H11 (1.0 g, 2.2 mmol). Crude product was chromatographed (silica gel, eluant, CHCl3:CH3OH 98:2 95:5) which afforded a white solid (1.04 g, 99%), mp 69-70 °C. 1H-NMR: 0.85-1.00 (2H, m, OCH2C6H11), 1.05-1.35 (3H, OCH2C6H11), 1.41 (9H, s, C(CH3)3), 1.55-1.75 (6H, m, OCH2C6H11), 1.80-2.20 (4H, m, Pro NCH2CH2CH2), 3.35-3.50 (2H, m, Pro NCH2CH2CH2), 3.80-4.10 (8H, Gly CH2, OCH2C6H11), 4.15 (1H, t, J = 6.3 Hz, Pro CH), 7.10 (1H, bs, NH), 7.28 (1H, bs, NH), 7.82 (1H, bs, NH). 13C-NMR: 24.6, 25.6, 26.2, 28.3, 28.5, 29.5, 36.9, 41.1, 43.0, 43.3, 45.9, 47.3, 60.7, 70.4, 80.8, 155.6, 169.4, 169.7, 169.9, 173.7. IR: cm-1 3312, 3080, 2978, 2930, 2854, 1751, 1669, 1541, 1479, 1451, 1408, 1367, 1240, 1199, 1165, 1132, 1091, 1033, 983, 923, 889, 856, 755, 667.
PGGG-OCH2Ph·HCl
General procedure 4. Quantities used: Boc-PGGG-OCH2C6H11 (0.2 g, 0.4 mmol). The product was used in subsequent reaction withoutfurther purification.
182-[DGA]-GGGPGGG-OCH2C6H11
General procedure 2. Quantities used: 182-[DGA]-GGG-OH (0.16 g, 0.2 mmol), and HCl·PGGG-OCH2C6H11 (0.08 g, 0.2 mmol). Crude product was chromatographed (silica gel, CHCl3:CH3OH 9:1 85:15) to give the product as a white solid (0.14 g, 61%), mp 100-102 °C. 1H NMR: 0.87 (8H, CH3(CH2)15CH2CH2N, OCH2C6H11), 1.00-1.35 (63H, CH3(CH2)15CH2CH2N, OCH2C6H11), 1.47 (4H, bs, CH3(CH2)15CH2CH2N), 1.50-1.60 (6H, m, OCH2C6H11), 1.80-2.20 (4H, m, Pro NCH2CH2CH2), 3.05 (2H, t, J = 7.5 Hz, CH3(CH2)15CH2CH2N), 3.24 (2H, t, J = 7.5 Hz, CH3(CH2)15CH2CH2N), 3.45-3.75 (2H, m, Pro NCH2CH2CH2), 3.85-4.25(18H, Gly CH2, OCH2C6H11, COCH2O), 4.27 (2H, s, COCH2O), 4.36 (1H, t, J = 6.6 Hz, Pro CH), 7.70 (2H, bs, NH), 7.90 (1H, bs, NH), 8.10 (1H, bs, NH), 8.42 (2H, bs, NH). 13C-NMR: 14.1, 22.7, 25.2, 25.6, 26.3, 26.9, 27.1, 27.6, 28.9, 29.0, 29.3, 29.4, 29.5, 29.6, 29.7, 37.0, 41.1, 42.0, 43.0, 43.5, 46.4, 47.0, 61.3, 69.6, 70.5, 71.5, 168.5, 168.9, 170.1, 170.2, 170.3, 170.4, 170.7, 171.1, 173.2 ppm. IR (CHCl3): cm-1 3328, 3085, 2920, 2851, 1745, 1727, 1652, 1601, 1541, 1467, 1412, 1378, 1341, 1243, 1204, 1130, 1031, 909, 722. Anal. Calcd. for C64H116N8O11·H2O: C64.50, H 9.98, N 9.40. Found: C 64.34, H 9.93, N 9.47.
[CH3(CH2)17]2NCOCH2OCH2CO-Gly-Gly-Gly-Pro-Gly-Gly-Gly-O(CH2)9CH3, “C18-decyl,” 13
182-[DGA]-GGGPGGG-OC10H21
182-[DGA]-GGGPGGG-OH (0.20 g, 0.2 mmol) was suspended at 0° C in a mixture of CH2Cl2 (25 mL), DCCI (0.03 g, 0.2 mmol), DMAP (0.01 g, 0.1 mmol), and 1-decanol (0.03 g, 0.2 mmol). After stirring at rt for 48 h, the solvent was evaporated and the residue was chromatographed (silica gel, CHCl3:CH3OH 9:1) to give a white solid (0.13 g, 58%), mp 137-8 °C. 1H-NMR: 0.80 (9H, m, CH3), 1.17 (74H, m, NCH2CH2(CH2)15CH3, OCH2CH2(CH2)7CH3), 1.42-1.54 (6H, m, NCH2CH2(CH2)15CH3, OCH2CH2(CH2)7CH3), 1.85-2.2 (4H, m, Pro NCH2CH2CH2), 3.00 (2H, t, J = 7.5 Hz, NCH2CH2(CH2)15CH3), 3.19 (2H, t, J = 7.5 Hz, NCH2CH2(CH2)15CH3), 3.45-3.60 (2H, m, Pro NCH2CH2CH2), 3.7-4.1 (16 H, m, Gly CH2, COCH2O, OCH2CH2(CH2)7CH3), 4.22 (2H, s, COCH2O), 4.28 (1H, m, Pro CH), 7.34 (1H, t, J = 6.0 Hz, NH), 7.45 (1H, t, J = 6.0 Hz, NH), 7.81 (2H, m, NH), 7.98 (1H, t, J = 6.0 Hz, NH), 8.23 (1H, t, J = 6.0 Hz, NH). 13C-NMR: 14.3, 22.8, 25.3, 26.0, 27.1, 27.2, 27.8, 28.7, 29.0, 29.3, 29.48, 29.52, 29.6, 29.7, 29.8, 29.9, 32.1, 41.4, 42.0, 42.9, 43.0, 43.1, 46.5, 47.1, 61.4, 65.8, 69.6, 71.5, 168.5, 168.9, 170.3, 170.4, 170.5, 170.6, 170.9, 171.2, 173.7 ppm. Anal. Calcd. for C67H124N8O11·0.5H2O: C 65.60, H 10.27, N 9.13. Found: C 65.53, H 10.24, N 9.03.
[CH3(CH2)17]2NCOCH2OCH2CO-Gly-Gly-Gly-Pro-Gly-Gly-Gly-O(CH2)17CH3, “C18-octadecyl,” 14
182-[DGA]-GGGPGGG-OC18H37
182-[DGA]-GGGPGGG-OH (0.20 g, 0.2 mmol), DCCI (0.04 g, 0.3 mmol), DMAP (0.01 g, 0.1 mmol), and 1-octadecanol (0.06 g, 0.2 mmol) were suspended in CH2Cl2 (25 mL) at 0° C. After stirring at rt for 48 h, the solvent was evaporated and the residue was crystallized from CH3OH to give a white solid (0.20 g, 81%), mp 144-5 °C. 1H-NMR: 0.80 (9H, m, CH3), 1.18 (90H, m, NCH2CH2(CH2)15CH3, OCH2CH2(CH2)15CH3), 1.43 (4H, m, NCH2CH2(CH2)15CH3), 1.54 (2H, bt, OCH2CH2(CH2)15CH3), 1.8-2.2 (4H, m, Pro NCH2CH2CH2), 3.00 (2H, t, J = 7.5 Hz, NCH2CH2(CH2)15CH3), 3.19 (2H, t, J = 7.5 Hz, NCH2CH2(CH2)15CH3), 3.42-3.65 (2H, m, Pro NCH2CH2CH2), 3.7-4.1 (16 H, m, Gly CH2, COCH2O, OCH2CH2(CH2)15CH3), 4.22 (2H, s, COCH2O), 4.28 (1H, m, Pro CH), 7.32 (1H, bt, NH), 7.44 (1H, bt, NH), 7.81 (2H, m, NH), 7.85 (1H, bt, NH), 8.27 (1H, t, J = 6.0 Hz, NH). 13C-NMR: 14.3, 22.9, 25.4, 26.1, 27.1, 27.3, 27.8, 28.7, 29.1, 29.2, 29.6, 29.7, 29.8, 29.9, 30.0, 32.1, 41.4, 42.2, 43.1, 43.2, 43.7, 46.5, 47.1, 47.2, 61.5, 65.9, 69.8, 71.8, 168.6, 169.1, 170.3, 170.37, 170.44, 170.6, 170.9, 171.4, 173.5. Anal. Calcd. for C75H140N8O11·H2O: C 66.83, H 10.62, N 8.31. Found: C 66.81, H 10.63, N 8.02.
[CH3(CH2)17]2NCOCH2OCH2CO-Gly-Gly-Gly-Pro-Gly-Gly-Gly-NH (CH2)9CH3, “C18-decylamide” 15
182-[DGA]-GGGPGGG-NHC10H21
General procedure 2. Quantities used: 182-[DGA]-GGGPGGG-OH (0.25 g, 0.2 mmol), and decylamine (0.04 g, 0.2 mmol). The crude product was chromatographed (silica gel, CHCl3:CH3 95:5 9:1) to give a white solid (0.10 g, 36%), mp 174-176 °C. 1H-NMR: 0.86 (9H, t, J = 7.2 Hz, CH3), 1.25 (74H, m, CH3(CH2)15CH2CH2N and CH3(CH2)7CH2CH2NH), 1.47 (6H, bs, CH3(CH2)15CH2CH2N and CH3(CH2)7CH2CH2NH), 1.90-2.30 (4H, m, Pro NCH2CH2CH2), 3.07 (2H, t, J = 7.8 Hz, CH3(CH2)16CH2N), 3.15 (2H, t, J = 6.9 Hz, CH3(CH2)8CH2NH), 3.26 (2H, t, J = 7.5 Hz, CH3(CH2)16CH2N), 3.50-3.60 (1H, m, Pro NCH2CH2CH2), 3.65-3.73 (1H, m, Pro NCH2CH2CH2), 3.75-4.20 (14H, m, Gly CH2, COCH2O), 4.28 (2H, s, COCH2O), 4.35 (1H, t, J = 7.5 Hz, Pro NCH), 7.18 (1H, J = 6.0 Hz, NH), 7.58 (1H, J = 6.0 Hz, NH), 7.85 (1H, J = 6.0 Hz, NH), 7.93 (1H, J = 6.0 Hz, NH), 7.95 (1H, J = 6.0 Hz, NH) 8.17 (1H, J = 6.0 Hz, NH), 8.29 (1H, bs, Gly NH). 13C-NMR: 14.10, 22.7, 25.1, 26.9, 27.0, 27.1, 27.6, 28.8, 29.1, 29.3, 29.4, 29.6, 29.7, 31.9, 39.6, 42.1, 42.8, 43.0, 43.5, 46.3, 46.9, 61.3, 69.3, 71.3, 168.3, 168.9, 169.3, 170.1, 170.3, 170.6, 170.7, 171.0, 173.2. IR (KBr): cm-1 3306, 3080, 2923, 2853, 1651, 1541, 1466, 1377, 1336, 1247, 1130, 1029, 721. Anal. Calcd for C67H125N9O10·H2O: C 65.17, H 10.37, N 10.21. Found: C 65.16, H 10.29, N 10.16.
CH3(CH2)17]2NCOCH2OCH2CO-Gly-Gly-Gly-Pro-Gly-Gly-Gly-N [(CH2)9CH3]2, “C18-didecylamide,” 16
182-[DGA]-GGGPGGG-N(C10H21)2
General procedure 2. Quantities used: 182-[DGA]-GGGPGGG-OH (0.25 g, 0.2 mmol), and didecylamine (0.07 g, 0.2 mmol). The crude product was chromatographed (silica gel, CHCl3:CH3OH 95:5 9:1) to give a white solid (0.12 g, 39%), mp 100-103 °C. 1H-NMR: 0.87 (12H, t, J = 7.2 Hz, CH3), 1.25 (84H, m, CH3(CH2)15CH2CH2N, CH3(CH2)6CH2CH2N), 1.50 (8H, bs, CH3(CH2)15CH2CH2N, CH3(CH2)6CH2CH2N), 1.90-2.25 (4H, m, Pro NCH2CH2CH2), 3.07 (2H, t, J = 7.8 Hz, CH3(CH2)16CH2N), 3.15-3.40(6H, CH3(CH2)8CH2NH, CH3(CH2)16CH2NH), 3.50-3.60 (1H, m, Pro NCH2CH2CH2), 3.65-4.20 (15H, m, Gly CH2, Pro NCH2CH2CH2, COCH2O), 4.29 (2H, s, COCH2O), 4.38 (1H, t, J = 7.5 Hz, Pro NCH), 7.36 (1H, bs, NH), 7.70 (2H, m, NH), 7.76 (1H, bs, NH), 7.93 (1H, bs, NH), 8.40 (1H, bs, NH). 13C-NMR: 14.0, 14.1, 22.7, 25.1, 26.9, 27.1, 27.6, 27.7, 28.7, 28.9, 29.3, 29.4, 29.5, 29.6, 29.7, 31.9, 41.1, 42.0, 42.9, 43.0, 43.1, 43.2, 46.3, 46.9, 47.0, 47.2, 61.3, 67.1, 69.6, 71.6, 167.4, 168.4, 169.1, 170.0, 170.1, 170.3, 171.2, 172.5. IR (KBr): cm-1 3304, 2925, 2852, 1651, 1541, 1465, 1376, 1334, 1245, 1129, 1030, 721 cm-1. Anal. Calcd. for C77H145N9O10·H2O: C 67.26, H 10.78, N 9.17. Found: C 67.25, H 10.71, N 9.18.
[CH3(CH2)17]2NCOCH2OCH2CO-Gly-Gly-Gly-Pro-Gly-Gly-Gly-NHC18H37, “C18-octadecylamide,” 17
182-[DGA]-GGGPGGG-NHC18H37
General procedure 2. Quantities used: 182-[DGA]-GGGPGGG-OH (0.25 g, 0.2 mmol), and octadecylamine (0.06 g, 0.2 mmol). The crude product was chromatographed (silica gel, CHCl3:CH3OH 95:5 9:1) to give a white solid (0.13 g, 42%), mp 168-170 °C. 1H-NMR: 0.87 (9H, t, J = 7.2 Hz, CH3), 1.24 (90H, m, CH3(CH2)15CH2CH2N, CH3(CH2)15CH2CH2NH), 1.46 (6H, bs, CH3(CH2)15CH2CH2N, CH3(CH2)7CH2CH2NH), 1.90-2.30 (4H, m, Pro NCH2CH2CH2), 3.07 (2H, t, J = 7.8 Hz, CH3(CH2)16CH2N), 3.15 (2H, t, J = 6.9 Hz, CH3(CH2)16CH2NH), 3.26 (2H, t, J = 7.5 Hz, CH3(CH2)16CH2N), 3.50-3.60 (1H, m, Pro NCH2CH2CH2), 3.65-4.20 (15H, m, Gly CH2, Pro NCH2CH2CH2, COCH2O), 4.29 (2H, s, COCH2O), 4.36 (1H, t, J = 7.5 Hz, Pro NCH), 7.18 (1H, J = 6.0 Hz, CONHC18H37), 7.61 (1H, J = 6.0 Hz, NH), 7.85 (1H, J = 6.0 Hz, NH), 7.94 (2H, m, NH), 8.18 (1H, t, J = 6.0 Hz, NH), 8.29 (1H, bs, NH). 13C-NMR: 14.0, 22.6, 25.0, 26.7, 26.8, 26.9, 27.4, 28.7, 29.0, 29.2, 29.6, 31.8, 39.5, 42.0, 42.6, 42.7, 42.8, 43.0, 46.2, 46.8, 46.9, 61.5, 68.9, 70.8, 168.3, 168.9, 169.2, 170.0, 170.5, 170.7, 171.3, 173.1. IR (CHCl3): cm-1 3305, 3082, 2924, 2853, 1653, 1540, 1467, 1377, 1335, 1247, 1131, 1029, 720. Anal. Calcd. for C75H141N9O10·H2O: C 66.88, H 10.70, N 9.36. Found: C 67.07, H 10.71, N 9.33.
[CH3(CH2)17]2NCOCH2OCH2CO-Gly-Gly-Gly-Pro-Gly-Gly-Gly-N [(CH2)6CH3]2, “C18-diheptylamide,” 18
Diheptylamine
A solution of n-heptylamine (5.0 g, 43.4 mmol), DMAP (0.4 g, 3.3 mmol), and NEt3 (8.8 mL) in CH2Cl2 (60 mL) was cooled to 0 °C and a solution of heptanoyl chloride (6.5 g, 43.4 mmol) in CH2Cl2 (50 mL) was added dropwise. After stirring at rt for 24 h, the solvent was evaporated, the residue was dissolved in EtOAc, washed with 5% citric acid (2 50 mL), 5% NaHCO3 dried over MgSO4 and evaporated to give a white solid (9.55 g, 97%) mp 44-46 °C. 1H-NMR: 0.85 (6H, t, J = 6.6 Hz, CH3-), 1.26 (14H, pseudo-s, CH3(CH2)4CH2CH2N, and CH3(CH2)3CH2CH2CO), 1.46 (2H, m, CH3(CH2)4CH2CH2N), 1.59 (2H, m, CH3(CH2)3CH2CH2CO), 2.13 (2H, t, J = 7.7 Hz, CH3(CH2)3CH2CH2CO), 3.20 (2H, m, CH3(CH2)4CH2CH2N), 5.66 (1H, bs, NH). 13C-NMR: 14.0, 22.4, 22.5, 25.8, 26.8, 28.9, 29.6, 31.5, 31.7, 36.8, 39.4, 173.1. IR (CHCl3): 3290, 3089, 2955, 2922, 2853, 1641, 1558, 1467, 1378, 1310, 1243, 1159, 947, 723 cm-1.
C6H13CONHC7H15 (5.0 g, 22.0 mmol) dissolved in THF (15 mL) was added dropwise to LiAlH4 (1.1 g, 29.0 mmol) suspended in dry THF (20 mL). The mixture was heated to reflux for 24 h, cooled, carefully diluted with water (1.1 mL) to decompose excess of LiAlH4, followed by 15% NaOH (1.1 mL), and water (3.6 mL). The solid obtained after filtration was washed with Et2O, and the solvent was evaporated. The residue was redissolved in Et2O (50 mL), washed with brine (2 30 mL), dried over MgSO4, and evaporated to give white solid (4.63 g, 99%) mp 30-1 °C. 1H-NMR: 0.87 (6H, t, J = 6.6 Hz, CH3(CH2)4CH2CH2NH), 1.27 (16H, pseudo-s, CH3(CH2)4CH2CH2NH), 1.47 (4H, m, CH3(CH2)4CH2CH2NH), 2.57 (4H, t, J = 7.2 Hz, CH3(CH2)4CH2CH2NH). 13C-NMR: 14.0, 22.6, 27.4, 29.2, 30.2, 31.8, 50.1. IR (CHCl3): cm-1 3400, 2957, 2926, 2855, 1466, 1378, 1130, 724 cm-1.
182-[DGA]-GGGPGGG-N(C18H37)2
General procedure 2. Quantities used: 182[DGA]-GGGPGGG-OH (0.24 g, 0.22 mmol), and diheptylamine (0.05 g, 0.23 mmol). The crude product was chromatographed (silica gel, CHCl3:CH3OH 95:5) to give a colorless oil (0.21 g, 75%). 1H-NMR: 0.80-0.94 (12H, m, CH3), 1.25 (68H, pseudo-s, CH3(CH2)15CH2CH2N and CH3(CH2)4CH2CH2NH), 1.47 (8H, bs, CH3(CH2)15CH2CH2N and CH3(CH2)7CH2CH2NH), 1.90-2.30 (4H, m, Pro NCH2CH2CH2), 3.07 (2H, t, J = 7.5 Hz, CH3(CH2)16CH2N), 3.17 (4H, t, J = 7.0 Hz, CH3(CH2)5CH2NH), 3.27 (2H, t, J = 7.5 Hz, CH3(CH2)16CH2N), 3.50-3.60 (1H, m, Pro NCH2CH2CH2), 3.65-3.73 (1H, m, Pro NCH2CH2CH2), 3.75-4.20 (14H, m, Gly NCH2, and COCH2O), 4.28 (2H, s, COCH2O), 4.39 (1H, bs, Pro NCH), 7.39 (1H, bs, Gly CONH), 7.72 (1H, bs, Gly CONH), 7.77 (1H, bs, Gly CONH), 7.89 (1H, bs, Gly CONH), 7.96 (1H, bs, Gly CONH) 8.37 (1H, bs, Gly CONH). 13C-NMR: 14.0, 14.1, 22.5, 22.6, 22.7, 25.1, 26.8, 26.9, 27.0, 27.1, 27.6, 28.7, 28.8, 28.9, 29.1, 29.3, 29.4, 29.5, 29.6, 29.7, 31.7, 31.9, 41.1, 41.9, 42.9, 43.2, 46.3, 47.1, 61.3, 69.5, 71.5, 167.5, 168.4, 169.0, 170.1, 170.4, 171.1, 172.7. IR (CHCl3): cm-1 3307, 2919, 2851, 1651, 1544, 1467, 1378, 1246, 1129, 1028, 721. Anal. Calcd for C71H133N9O10: C, 67.00; H, 10.53; N, 9.90 %. Found: C, 67.00; H, 10.53; N, 9.90 %.
[CH3(CH2)17]2NCOCH2OCH2CO-Gly-Gly-Gly-Pro-Gly-Gly-Gly-N [(CH2)17CH3]2, “C18-dioctadecylamide,” 19
182-[DGA]-GGGPGGG-N(C18H37)2
General procedure 2. Quantities used: 182-[DGA]-GGGPGGG-OH (0.21 g, 0.2 mmol), and dioctadecylamine (0.10 g, 0.2 mmol). The crude product was chromatographed (silica gel, CHCl3:CH3OH 95:5 85:15) and crystallized from CH3OH to give a white solid (0.10 g, 32%), mp 152-3 °C. 1H-NMR: 0.87 (12H, t, J = 6.6 Hz, CH3), 1.25 (120H, m, CH3(CH2)15CH2CH2N), 1.51 (4H, bs, CH3(CH2)15CH2CH2NCOCH2O), 1.61 (4H, bt, CH3(CH2)15CH2CH2NCOCH2N), 1.90-2.25 (4H, m, Pro NCH2CH2CH2), 3.07 (4H, t, J = 7.5 Hz, CH3(CH2)15CH2CH2N), 3.27 (4H, t, J = 7.5 Hz, CH3(CH2)15CH2CH2N), 3.47-3.77 (2H, m, Pro NCH2CH2CH2), 3.80-4.15 (16H, m, Gly CH2, COCH2O), 4.29 (2H, s, COCH2O), 4.37 (1H, t, J = 7.5 Hz, Pro NCH), 7.40 (1H, bt, NH), 7.54 (1H, bt, NH), 7.90 (3H, m, NH), 8.41 (1H, bs, NH). 13C-NMR: 14.3, 22.9, 25.4, 26.1, 27.1, 27.3, 27.8, 28.7, 29.1, 29.3, 29.6, 29.7, 29.9, 32.1, 41.5, 42.2, 43.2, 43.7, 46.6, 47.2, 61.6, 65.9, 69.7, 71.7, 168.7, 169.2, 170.32, 170.35, 170.5, 170.7, 171.0, 171.5, 173.5. Anal. Calcd. for C93H177N9O10: C 70.63, H 11.28, N 7.97. Found: C 70.63, H 11.28, N 7.97.
Formation of vesicles
1,2-Dioleoyl-sn-glycero-3-phosphocholine (DOPC) and 1,2-dioleoyl-sn-glycero-3-phosphate (DOPA) were obtained from Avanti Polar Lipids as CHCl3 solutions (vials with 25 mg in 2.5 mL). Dry lipid films of DOPC-DOPA (20 mg, 7:3 w/w) were dissolved in Et2O (0.5 mL) and then 0.5 mL of internal buffer (600 mM KCl:10 mM N-2-hydroxyethylpiperazine-N’-2-ethanesulfonic acid (HEPES), pH adjusted to 7.0) were added. The mixture was sonicated for approximately 10 s to give an opalescent dispersion. The organic solvent was then removed by evaporation under mild vacuum at 30 °C. The suspension was filtered through a 200 nm filter membrane (5 times) using a mini extruder and passed through a Sephadex G25 column, which had been equilibrated with external buffer (400 mM K2SO4:10 mM HEPES, pH adjusted to 7.0). The vesicles collected were subsequently characterized using laser light scattering, and their diameter was consistently ∼200 nm, as measured by a Coulter N4MD submicron particle analyzer. The final lipids concentration was assessed through colorimetric determination of the phospholipid-ammonium ferrothiocyanate complex.46
Studies of chloride release from liposomes
Chloride release was assayed directly on 2 00 nm, 0.23 mM phospholipid vesicles by using a chloride selective electrode (Accumet Chloride Combination Electrode. The electrode was introduced in a 2 mL vesicles solution and allowed to equilibrate. The voltage output was recorded, and after five minutes, aliquots of the solution of compound at study (∼9 mM in 2-propanol) were added. No more than 20 μL of 2-propanol were added in any experiment in order to avoid any effect of the solvent on the liposomes. Complete lysis of the vesicles was induced by addition of a 2 % aqueous solution of Triton X100 (100 μL) and the data collected were normalized to this value. The data were collected using a DigiData 1322A series interface and Axoscope 9.0 software.
Carboxyfluorescein Dequenching from Unilamellar Liposomes
Liposomes were prepared using the reverse phase procedure of Szoka and Papahadjopoulos.47 A lipid mixture (10 mg composed of 30% 1,2-dioleoyl-sn-glycero-3-phosphate and 70% 1,2-dioleoyl-sn-glycero-3-phosphocholine, Avanti Polar Lipids, AL) was dissolved in 0.5 mL of Et2O. To this was added 0.5 mL of 20 mM carboxyfluorescein in 100 mM KCl:10 mM HEPES (pH=7.0) and the final pH was adjusted to 7.0 with KOH. This mixture was sonicated at 1200 W for 3 20 seconds at 20 °C to produce a stable emulsion. The Et2O was removed (water aspirator, 15 °C) in a round bottom flask rotating at 40 rpm. The 0.5 mL suspension was supplemented with an additional 0.5 mL of 20 mM carboxyfluorescein solution. This mixture was passed (five times) through a 200 nm nucleopore filter. The extra-vesicular carboxyfluorescein was removed by passing the liposome-dye mixture over a 1 20 cm Sephacryl G-25 column (Sigma) in 100 mM KCl: 10 mM HEPES (pH=7.0). The liposome peak was collected and analyzed by dynamic light scattering, diameter = 182 ± 12 nm. Phospholipid concentration in this fraction was determined to be 3.3 mg/mL.45
Prior to use, carboxyfluorescein-containing vesicles were diluted to 0.66 μg/mL in a reaction volume of 500 μL. The fluorescence (excitation 497 nm: emission 520 nm at 2 nm bandpass) was monitored at 25 °C. Compounds were added as a 5 mM solution in 2-propanol with mixing to the indicated concentrations. Data were digitized at 10 points per second and subsequently reduced to one point per second by decimation. Dequenching, F520, was computed as the fraction of total release upon addition of 1% Triton X100:
| (1) |
with F0 and FTriton being the zero time and the fluorescence produced in the presence of Triton X-100. Dequenching data were fit using a nonlinear least squares method based upon the Levenberg-Marquardt algorithm, which generated 2 values indicating the goodness of fit, for each group of averaged data sets. The number of individual trials generating the data set (degrees of freedom) was used to obtain the p value for the individual fits and kinetic constants. In all cases, the 2 values for individual fits were less than 0.05 and the resulting p values varied from 0.05 to 0.001 depending upon the number of trials. Data were fit to the equation,
| (2) |
as described previously.39,48 In this equation, F0 is the fluorescence at time zero, A1 is the size of the exponential component, is the time constant for pore activation, and m is the slope of the linear dequenching resulting from multiple pores per vesicle.
Notes and References
- 1(a).Aidley DJ, Stanfield PR. Ion Channels: Molecules in Action. Cambridge University Press; Cambridge: 1996. [Google Scholar]; (b) Hille B. Ionic Channels of Excitable Membranes (Third Edition) 3rd Edn. Sinauer Associates; Sunderland, MA: 2001. [Google Scholar]
- 2(a).Doyle DA, Cabral JM, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, Chait BT, MacKinnon R. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]; (b) MacKinnon R, Cohen SL, Kuo A, Lee A, Chait BT. Science. 1998;280:106–109. doi: 10.1126/science.280.5360.106. [DOI] [PubMed] [Google Scholar]
- 3.Sato C, Ueno Y, Asai K, Takahashi K, Sato M, Engel A, Fujiyoshi Y. Nature. 2001;409:1047–1051. doi: 10.1038/35059098. [DOI] [PubMed] [Google Scholar]
- 4(a).Dutzler R, Campbell EB, Cadene M, Chait BT, MacKinnon R. Nature. 2002;415:287–294. doi: 10.1038/415287a. [DOI] [PubMed] [Google Scholar]; (b) Dutzler R, Campbell EB, MacKinnon R. Science. 2003;300:108–112. doi: 10.1126/science.1082708. [DOI] [PubMed] [Google Scholar]
- 5(a).Preston GM, Carroll TP, Guggino WB, Agre P. Science. 1992;256:385–387. doi: 10.1126/science.256.5055.385. [DOI] [PubMed] [Google Scholar]; (b) Borgnia M, Nielsen S, Engel A, Agre P. Annu Rev Biochem. 1999;68:425–458. doi: 10.1146/annurev.biochem.68.1.425. [DOI] [PubMed] [Google Scholar]; (c) Hummer G, Rasalah JC, Noworyta JP. Nature. 2001;414:188–190. doi: 10.1038/35102535. [DOI] [PubMed] [Google Scholar]
- 6(a).Chang G, Spencer RH, Lee AT, Barclay MT, Rees DC. Science. 1998;282:2220–2226. doi: 10.1126/science.282.5397.2220. [DOI] [PubMed] [Google Scholar]; (b) Spencer RH, Chang G, Rees DC. Curr. Opin. Struct. Biol. 1999;9:448–454. doi: 10.1016/S0959-440X(99)80063-3. [DOI] [PubMed] [Google Scholar]; (c) Scarsdale JN, Kazanina G, He X, Reynolds KA, Wright HT. J. Biol. Chem. 2001;276:20516–20522. doi: 10.1074/jbc.M010762200. [DOI] [PubMed] [Google Scholar]
- 7.Agre P. Angew. Chem. Int. Ed. Engl. 2004;43:4278–90. doi: 10.1002/anie.200460804. [DOI] [PubMed] [Google Scholar]
- 8.MacKinnon R. Angew Chem Int Ed Engl. 2004;43:4265–77. doi: 10.1002/anie.200400662. [DOI] [PubMed] [Google Scholar]
- 9(a).Reusch RN, Sadoff HL. Proc. Natl. Acad. Sci. 1988;85:4176–4180. doi: 10.1073/pnas.85.12.4176. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Reusch RN, Huang R, Bramble LL. Biophys. J. 1995;69:754–766. doi: 10.1016/S0006-3495(95)79958-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10(a).Seebach D, Brunner A, Buerger HM, Reusch RN, Bramble LL. Helv. Chim. Acta. 1996;79:507–517. [Google Scholar]; (b) Das S, Lengweiler UD, Seebach D, Reusch RN. Proc. Nat. Acad. Sci. (USA) 1997;94:9075–9079. doi: 10.1073/pnas.94.17.9075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11(a).Oiki S, Danho W, Montal M. Proc. Natl. Acad. Sci.(USA) 1988;85:2393–2397. doi: 10.1073/pnas.85.7.2393. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Montal M, Montal MS, Tomich JM. Proc. Nat. Acad. Sci. (USA) 1990;87:6929–6933. doi: 10.1073/pnas.87.18.6929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12(a).Iwamoto T, Grove A, Montal MO, Montal M, Tomich JM. nt. J. Pept. Protein. Res. 1994;43:597–607. doi: 10.1111/j.1399-3011.1994.tb00562.x. [DOI] [PubMed] [Google Scholar]; (b) Montal M. Annu. Rev. Biophys. Biomol. Struct. 1995;24:31–57. doi: 10.1146/annurev.bb.24.060195.000335. [DOI] [PubMed] [Google Scholar]
- 13(a).Lear JD, Wasserman ZR, DeGrado WF. Science. 1988;240:1177–1181. doi: 10.1126/science.2453923. [DOI] [PubMed] [Google Scholar]; (b) DeGrado WF, Wasserman ZR, Lear JD. Science. 1989;243:622–628. doi: 10.1126/science.2464850. [DOI] [PubMed] [Google Scholar]
- 14(a).Åkerfeldt KS, Lear JD, Wasserman ZR, Chung LA, DeGrado WF. Acc. Chem. Res. 1993;26:191–197. [Google Scholar]; (b) Dieckmann GR, DeGrado WF. Curr. Opin. Struct. Biol. 1997;7:486–494. doi: 10.1016/s0959-440x(97)80111-x. [DOI] [PubMed] [Google Scholar]
- 15.Tabushi I, Kuroda Y, Yokota K. Tetrahedron Lett. 1982:4601–4604. [Google Scholar]
- 16.Carmichael VE, Dutton PJ, Fyles TM, James TD, Swan JA, Zojaji M. J. Am. Chem. Soc. 1989;111:767–769. [Google Scholar]
- 17.Jullien L, Lehn J-M. Tetrahedron Lett. 1988:3803–3806. [Google Scholar]
- 18.Menger FM, Davis DS, Persichetti RA, Lee J-J. J. Am. Chem. Soc. 1990;112:2451–2452. [Google Scholar]
- 19(a).Beijnen A. J. M. v., Nolte RJM, Zwikker JW, Drenth W. Recl. Trav. Pays-Bas. 1982;101:409–410. [Google Scholar]; (b) Roks MFM, Nolte RJM. Macromolecules. 1992;25:5398–5407. [Google Scholar]
- 20.Kobuke Y, Ueda K, Sokabe M. J. Am. Chem. Soc. 1992;114:7618–7822. [Google Scholar]
- 21.Nakano A, Xie Q, Mallen JV, Echegoyen L, Gokel GW. J. Am. Chem. Soc. 1990;112:1287–1289. [Google Scholar]
- 22.Meillon J-C, Voyer N. Angew. Chem. Int. Ed. Engl. 1997;36:967–969. [Google Scholar]
- 23.Sakai N, Ni C, Bezrukov SM, Matile S. Bioorg. Med. Chem. Lett. 1998;8:2743–2746. doi: 10.1016/s0960-894x(98)00481-8. [DOI] [PubMed] [Google Scholar]
- 24.Schrey A, Vescovi A, Knoll A, Rickert C, Koert U. Angew. Chem. Int. Ed. 2000;39:900–902. [PubMed] [Google Scholar]
- 25(a).Gokel GW, Murillo O. Acc. Chem. Res. 1996;29:425–432. [Google Scholar]; (b) Gokel GW, Mukhopadhyay A. Chem. Soc. Reviews. 2001;30:274–286. [Google Scholar]
- 26.Boon JM, Smith BD. Curr Opin Chem Biol. 2002;6:749–756. doi: 10.1016/s1367-5931(02)00399-x. [DOI] [PubMed] [Google Scholar]
- 27.Bell TW. Curr. Opin. Chem. Biol. 1998;2:711–716. doi: 10.1016/s1367-5931(98)80108-7. [DOI] [PubMed] [Google Scholar]
- 28.Sakai N, Matile S. Chem. Commun. 2003:2514–2523. doi: 10.1039/b303649a. [DOI] [PubMed] [Google Scholar]
- 29.Bianchi A, Bowman-James K, Garcia-España E. Supramolecular Chemistry of Anions. Wiley-VCH; New York: 1997. [Google Scholar]
- 30(a).Wallace DP, Tomich JM, Eppler JW, Iwamoto T, Grantham JJ, Sullivan LP. Biochim. Biophys. Acta. 2000;1464:69–82. doi: 10.1016/s0005-2736(99)00248-5. [DOI] [PubMed] [Google Scholar]; (b) Broughman JR, Mitchell KE, Sedlacek RL, Iwamoto T, Tomich JM, Schultz BD. Am. J. Physiol. 2001;280:C451–C458. doi: 10.1152/ajpcell.2001.280.3.C451. [DOI] [PubMed] [Google Scholar]; (c) Gao L, Broughman JR, Iwamoto T, Tomich JM, Venglarik CJ, Forman HJ. Am. J. Physiol. 2001;281:L24–L30. doi: 10.1152/ajplung.2001.281.1.L24. [DOI] [PubMed] [Google Scholar]
- 31.Sidorov V, Kotch FW, Kuebler JL, Lam Y-F, Davis JT. J. Am. Chem. Soc. 2003;125:2840–2841. doi: 10.1021/ja029372t. [DOI] [PubMed] [Google Scholar]
- 32(a).Schlesinger PH, Ferdani R, Liu J, Pajewska J, Pajewski R, Saito M, Shabany H, Gokel GW. J. Am. Chem. Soc. 2002;124:1848–1849. doi: 10.1021/ja016784d. [DOI] [PubMed] [Google Scholar]; (b) Schlesinger PH, Ferdani R, Pajewski R, Pajewska J, Gokel GW. Chem. Comm. 2002:840–841. doi: 10.1039/b200126h. [DOI] [PubMed] [Google Scholar]; (c) Schlesinger PH, Djedovic NK, Ferdani R, Pajewska J, Pajewski R, Gokel GW. Chem. Comm. 2003:308–309. doi: 10.1039/b211629b. [DOI] [PubMed] [Google Scholar]; (d) Schlesinger PH, Ferdani R, Pajewska J, Pajewski R, Gokel GW. New J. Chem. 2003;27:60–67. [Google Scholar]
- 33.Sakai N, Houdebert D, Matile S. Chemistry. 2003;9:223–232. doi: 10.1002/chem.200390016. [DOI] [PubMed] [Google Scholar]
- 34.Gokel GW. Chem. Comm. 2000:1–9. [Google Scholar]
- 35.Bolard J. Biochim. Biophys. Acta. 1986;864:257–304. doi: 10.1016/0304-4157(86)90002-x. [DOI] [PubMed] [Google Scholar]
- 36.Zhou J, Lu X, Wang Y, Shi J. 2002;194-197:257–270. [Google Scholar]
- 37.Hervé M, Cybulska B, Gary-Bobo CM. Eur. Biophys. J. 1985;12:121–128. [Google Scholar]
- 38(a).Carmichael VE, Dutton PJ, Fyles TM, James TD, Swan JA, Zojaji M. J. Am. Chem. Soc. 1989;111:767–769. [Google Scholar]; (b) Fyles TM, James TD, Kaye KC. J. Am. Chem. Soc. 1993;115:12315–12321. [Google Scholar]
- 39.Riddell FG, Tompsett SJ. Biochim. Biophys. Acta. 1990;990:193–197. doi: 10.1016/0005-2736(90)90225-d. [DOI] [PubMed] [Google Scholar]
- 40.Murillo O, Watanabe S, Nakano A, Gokel GW. J. Am. Chem. Soc. 1995;117:7665–7679. [Google Scholar]
- 41.Saito M, Korsmeyer SJ, Schlesinger PH. Nature Cell Biology. 2000:553–555. doi: 10.1038/35019596. [DOI] [PubMed] [Google Scholar]
- 42.Koulov AV, Lambert TN, Shukla R, Jain M, Boon JM, Smith BD, Li H, Sheppard DN, Joos JB, Clare JP, Davis AP. Angew. Chem. Int. Ed. Engl. 2003;42:4931–4933. doi: 10.1002/anie.200351957. [DOI] [PubMed] [Google Scholar]
- 43.Ladokhin AS, Selsted ME, White SH. Biophys. J. 1997;72:1762–1766. doi: 10.1016/S0006-3495(97)78822-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hernandez JC, Trafton JE, Gokel GW. Tetrahedron Lett. 1991:6269–6272. [Google Scholar]
- 45.Huang HW, Chen F-Y, Lee M-T. Phys. Rev. Lett. 2004;92:198304/1–198304/4. doi: 10.1103/PhysRevLett.92.198304. [DOI] [PubMed] [Google Scholar]
- 46.Stewart JCM. Anal. Chem. 1980;104:10–14. [Google Scholar]
- 47.Szoka F, Papahadjopoulos D. Proc. Natl. Acad. Sci. (USA) 1978;75:4194–4198. doi: 10.1073/pnas.75.9.4194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rex S, Schwarz G. Biochem. 1998;37:2336–2345. doi: 10.1021/bi971009p. [DOI] [PubMed] [Google Scholar]