

Abstract
Modulation of the N-methyl-d-aspartate (NMDA)-selective glutamate receptors by extracellular protons and Zn2+ may play important roles during ischemia in the brain and during seizures. Recombinant NR1/NR2A receptors exhibit a much higher apparent affinity for voltage-independent Zn2+ inhibition than receptors with other subunit combinations. Here, we show that the mechanism of this apparent high-affinity, voltage-independent Zn2+ inhibition for NR2A-containing receptors results from the enhancement of proton inhibition. We also show that the N-terminal leucine/isoleucine/valine binding protein (LIVBP)-like domain of the NR2A subunit contains critical determinants of the apparent high-affinity, voltage-independent Zn2+ inhibition. Mutations H42A, H44G, or H128A greatly increase the Zn2+ IC50 (by up to ≈700-fold) with no effect on the potencies of glutamate and glycine or on voltage-dependent block by Mg2+. Furthermore, the amino acid residue substitution H128A, which mediates the largest effect on the apparent high-affinity Zn2+ inhibition among all histidine substitutions we tested, is also critical to the pH-dependency of Zn2+ inhibition. Our data revealed a unique interaction between two important extracellular modulators of NMDA receptors.
Glutamate receptors are ligand-gated ion channels that mediate excitatory synaptic transmission in the central nervous system. Although overactivation of the N-methyl-d-aspartate (NMDA)-selective glutamate receptors can trigger neurodegeneration in neuropathological conditions such as stroke, NMDA receptor function is regulated under normal conditions by several extracellular ions (Mg2+, H+, and Zn2+), which exert strong tonic inhibition in a subunit selective manner (1). Inhibition by extracellular protons is particularly relevant for the neuropathological consequences of occlusive stroke because acidification of the extracellular environment during ischemia has been hypothesized to inhibit NMDA receptor overactivation by extrasynaptic glutamate that accumulates following metabolic failure (2, 3). The proton inhibition of NMDA receptors could delay their contribution to subsequent neuronal death until the pH gradients are restored, and this delay may provide a therapeutic window for postinsult treatment with NMDA receptor antagonists. Acidification of the extracellular space during electrographic seizure may also contribute to seizure termination through inhibition of NMDA receptor function (4, 5). Because of these potentially important aspects of the pH sensitivity of the NMDA receptor, we have sought to understand the structural basis by which the receptor might control its regulation by extracellular protons.
In the central nervous system, the extracellular Zn2+ concentration has been shown to vary under normal brain function as well as neuropathological conditions (6, 7). In addition, there are large amounts of chelatable Zn2+ in the glutamatergic terminals of hippocampus (8–10), which are released in a Ca2+-dependent manner (for review, see ref. 6). It is well established that Zn2+ can inhibit neuronal NMDA receptors through a dual mechanism involving both voltage-dependent channel block and a voltage-independent inhibition (11–14). The voltage-independent Zn2+ inhibition appears to be strongly dependent on subunit composition, being influenced by the NR2 subunits as well as NR1 splice variants in recombinant NMDA receptors (15–18). Interestingly, recombinant receptors comprised of the NR2A subunit are much more sensitive to Zn2+ than NR2B-, NR2C-, or NR2D-containing receptors, being inhibited in the nanomolar range by as much as 70% (15–17). Although cultured or acutely dissociated hippocampal and spinal dorsal horn neurons show lower sensitivity than recombinant NR2A-containing receptors (19), NMDA receptors in some brain areas such as mossy fiber synapses in the hippocampus (20, 21) may possess a sensitivity for Zn2+ in the nanomolar range and be tonically regulated by endogenous extracellular Zn2+ in physiologically and pathologically relevant ways.
Our previous study (18) as well as more recent work (22) suggested that there may be structural and/or functional links between inhibition of the NMDA receptor by extracellular protons and Zn2+. Here, we have explored the nature of the coupling between proton and Zn2+ inhibition and show that there is a unique interaction between Zn2+ and proton inhibition for NR2A- but not NR2B-containing receptors. We have also used site-directed mutagenesis to systematically probe for coordinate structural determinants of proton and Zn2+ inhibition in the N-terminal leucine/isoleucine/valine binding protein (LIVBP)-like domain of NR2A subunit (22, 23). This region is critical for the rapidly reversible form of redox modulation (24), which has been suggested to reflect chelation of contaminant Zn2+ (17). Here, we identify several histidine residues within the NR2A LIVBP-like domain that are candidate electron donors to the Zn2+ coordination site. In addition, a single histidine residue (His128) that caused the largest shift in Zn2+ sensitivity is also critical to the pH-dependence of voltage-independent Zn2+ inhibition of NR2A-containing receptors.
Materials and Methods
Site-Directed Mutagenesis.
The mutants of NR2A were constructed by using the pCIneo template with Pfu DNA polymerase (Stratagene) to linearly replicate the parental strand with desired mismatch(es) incorporated into the primer. Methylated parental DNA template was then degraded with DpnI. The nicked double-stranded mutant DNA was transformed into MAX Efficiency DH5α (GIBCO/BRL) or XL-Blue (Stratagene). Colonies were screened for a silent mutation that introduces a new restriction site. The mutations were verified by dideoxy sequencing through the region of the mutations. For NR2A containing mutations H42A, H44G, and H128A, their full coding sequences were verified by sequencing. The starting wild-type NR2A template used for all NR2A mutants construction carries an 11-amino acid residues insert in the C terminus, which has no effect on the function of the receptor.
Expression of NMDA Receptors in Xenopus Oocytes.
cRNA was synthesized from linearized template of cDNA according to manufacturer's specification (Ambion, Austin, TX). The quality and quantity of cRNA were assessed by gel electrophoresis. Preparation of oocytes and injection of cRNAs coding for wild-type and mutant NMDA receptors were performed as described (18).
Buffered Zn2+ Solutions.
The tricine-buffered Zn2+ solutions used to obtain the Zn2+ dose-response curves were prepared as described by Zheng et al. (25) by using the empirically established binding constant of 10−5 M (17). The following nominal concentrations of Zn2+ (0.78, 2.6, 7.8, 26, 77.6, and 254 μM), were added into 10 mM (pKa 8.15) tricine-buffer. The corresponding estimated concentrations of free Zn2+ at pH 7.3 were calculated with winmac (26) and bad (27), and were (in nM): 7.71; 25.3; 77.1; 253; 777; and 2,590.
Voltage-Clamp Recordings from Xenopus Oocytes.
Two electrode voltage-clamp recordings were made 2–7 days postinjection as described (18). Briefly, oocytes were perfused with a solution comprised of (in mM) 90 NaCl, 1 KCl, 10 Hepes, and 0.5 BaCl2 at pH 7.3, and held under voltage clamp at −20 to −30 mV (or stated otherwise). Oocytes were prewashed with a given Zn2+ concentration before application of Zn2+ and agonist/coagonist. Similar prewash for pH changes was also performed. Electrodes were filled with 300 mM KCl and had resistance of 1–10 MΩ. Solution exchange was computer controlled through a 16-valve manifold. ZnCl2 solutions (10 mM) were prepared fresh every 10 h and added directly to the recording solution to obtain the desired Zn2+ concentrations for all experiments. EDTA (pH 7.3, 1–10 μM) was added to control solutions containing saturating concentrations of glutamate (20–50 μM) or NMDA (200 μM) and glycine (10–100 μM) to chelate the contaminant Zn2+. The ambient Zn2+ concentration (17) in our solution was ≈300 nM (25) and arised predominantly as a contaminant from the NaCl. Tricine was used to buffer Zn2+ concentrations 0.003–3 μM (as described above) and ADA [N-2-acetamidoiminodiacetic acid (Sigma); log stability constants K1 = 7.1 and K2 = 9.22] (1 mM) was used to buffer Zn2+ concentrations 0.1–100 nM (28) for experiments with NR2A-containing NMDA receptors. For Zn2+ inhibition experiments performed at pH 8.0 or higher, the nominal Zn2+ concentration was increased to compensate for the formation of zinc hydroxide complex as described previously (18).
Data Analysis.
The IC50 value for a single-binding-site isotherm was determined for each oocyte recording by fitting the data with the following equation I/Imax = (1 − minimum)/(1 + ([Zn2+]/IC50)n) + minimum, where n is the Hill slope and minimum is the residual response (18). If the fitting algorithm returned a value less than 0.05 for minimum (our estimated limit for detection), we fixed minimum to 0 and refit the Zn2+ and proton inhibition data. The overall mean IC50 was then determined by averaging all of the individual IC50 values for a given wild-type or mutant receptor. All pooled data were expressed as mean ± SEM. The number of oocytes recorded for each mutant is shown in parentheses.
Simulation of the Zn2+-Inhibition Curve.
If Zn2+ inhibits NR1/NR2A receptors by enhancing proton inhibition, Zn2+ should alter proton IC50 (IC50(H)) in a concentration-dependent manner. Proton IC50(H) as a function of free Zn2+ was calculated by the following equation:
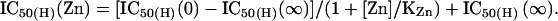 |
1 |
We set the IC50(H) (0) and IC50(H) (∞) to 125 and 27.5 nM, respectively. These values are a close approximation of measured proton IC50 values for NR1/NR2A receptors under nominally Zn2+-free condition (in the presence of 2–10 μM EDTA) and in the presence of nominally saturating Zn2+ (1 μM; see Fig. 2A). The KZn is set to 10 nM, an arbitrary value chosen based on the reported Zn2+ IC50 (15–18).
Figure 2.

The voltage-independent Zn2+ inhibition of NR1-1a/NR2A reflects the enhancement of proton inhibition. (A) Zn2+ causes an alkaline-shift of the proton inhibition curve of NR1-1a/NR2A receptors. Proton inhibition curve was measured both in the presence of 1 μM EDTA to remove the contaminant Zn2+ in the recording solution and in the presence of 1 μM Zn2+. All current responses were normalized to the current responses at the most alkaline pH 8.8. For pH 8.8 and pH 8.2, 6.64 μM and 1.48 μM Zn2+, respectively, were added to the recording solutions to achieve 1 μM free Zn2+. Error bars are SEM (whenever larger than the symbol). (B) Zn2+ inhibition curves at various pH values were predicted as described in Materials and Methods. The residual responses should be pH dependent when the apparent high affinity Zn2+ site is saturated. (C) Correlation between predicted residual currents (dotted line) and experimentally obtained residual currents (solid dot) of NR1/NR2A receptors at pHs 6.8, 7.3, and 8.0. (D–F) Zn2+ inhibition curves recorded at three different pH values. As predicted, the residual responses are pH dependent. The IC50 values of Zn2+ inhibition at pHs 6.8, 7.3, and 8.0 are 74.2 ± 9.0 nM, 23.9 ± 2.9 nM, and 4.4 ± 1.0 nM, respectively. For pH 8.0, Zn2+ inhibition curve was obtained with buffered Zn2+ by 1 mM ADA. All currents were measured in the presence of saturating concentrations of glutamate (20–50 μM) and glycine (10–30 μM).
The amount of Zn2+ inhibition as a function of free Zn2+ (I/Imax(Zn)) at any given pH could be predicted by the ratio of current responses in the presence of Zn2+ (IZn (H)) and absence of Zn2+ (I0 (H)). Assuming that enhancement of proton inhibition is the only mechanism by which Zn2+ inhibits NR1/NR2A receptors, then:
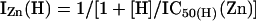 |
2 |
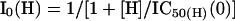 |
3 |
Therefore, at any given pH, the amount of Zn2+ inhibition can be determined by:
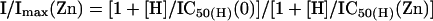 |
4 |
Results
Zn2+ Inhibition Is pH Dependent for NR2A- but Not for NR2B-Containing Receptors.
We have previously shown that alternative exon splicing and point mutations of NR1 that alter proton sensitivity also alter Zn2+ sensitivity of recombinant NMDA receptors. One straightforward explanation for this apparent correlation between proton sensitivity and Zn2+ sensitivity is that the voltage-independent Zn2+ inhibition results from enhancement of proton inhibition. If this were the case, one would predict that Zn2+ could not inhibit NMDA receptors at alkaline pHs. However, reduction of Zn2+ inhibition at alkaline pH cannot be viewed as strong evidence supporting an interaction between Zn2+ and proton inhibition because of the formation of zinc hydroxide and zinc carbonate complexes at alkaline pHs. If the total Zn2+ remains constant at 1 μM, the free Zn2+ will be reduced to 0.82 μM at pH 8.0, 0.68 μM at pH 8.2, and 0.39 μM at pH 8.5 (18, 28). These large reductions of free Zn2+, which have not always been compensated (22), are sufficient to produce the reduction of Zn2+ inhibition at alkaline pHs (see Fig. 6F of ref. 22). Quantitatively, the reduction of free Zn2+ because of the formation of zinc hydroxide complex could account for most of the reduction of Zn2+ inhibition that we observed for NR1/NR2B at alkaline pH, leading us previously to conclude that there is no interaction between Zn2+ and protons for NR1/NR2B receptors (18).
In the present study, we have addressed the link between pH and Zn2+ for both NR2B- and NR2A-containing receptors by using a different approach. We coexpressed NR2A or NR2B with NR1(D669N). Mutation at Asp669 to Asn, which does not affect the potencies of glutamate and glycine, reduces the pH sensitivity such that there is only negligible proton inhibition at pH 7.5–8.0 (29). The use of this mutant receptor, with its acid-shifted pH inhibition curve, removes the need to use solutions more alkaline than pH 8.0. Because zinc hydroxide formation below pH 8.0 is minimal, the change of free Zn2+ because of zinc hydroxide complexing will be negligible at the pH values used in these experiments. If Zn2+ inhibition is significantly reduced for NR1(D669N)-containing receptors by raising pH to 8.0, then the change cannot be accounted for by the formation of zinc hydroxide complex. Evaluation of the pH dependence of Zn2+ inhibition at pH values below 8.0 therefore can provide an opportunity to probe the link between Zn2+ and pH without having to deal with formation of zinc hydroxide complexes. To test the interaction between pH- and Zn2+-inhibition of recombinant NMDA receptors, Zn2+ concentrations that cause approximately 50% inhibition at the most acidic pH tested (pH 6.4) were chosen. As reported previously for NR2B-containing receptors (18), Zn2+ inhibition for NR1(D669N)/NR2B is unaltered at pHs 6.4–8.0 (Fig. 1 B and C). On the other hand, Zn2+ inhibition for NR1(D669N)/NR2A receptors are steeply pH-dependent (Fig. 1 A and C). It should be noted that the Zn2+ inhibition for NR1(D669N)/NR2A at pH 7.4 is only half of that at pH 6.4, whereas the free Zn2+ concentrations (considering the degree of zinc hydroxide complex formation) at these two pH values differ by less than 3%. Furthermore, there is no detectable Zn2+ inhibition at pH 8.2 for NR1(D669N)/NR2A. To qualitatively verify that the pH sensitivity of Zn2+ inhibition of NR2A-containing receptors is not influenced by mutation of Asp669 in NR1, we repeated these experiments with wild-type receptors. The free Zn2+ concentration for complex formation is corrected by increasing the total Zn2+ (18). Wild-type NR1-1a containing receptors possessed similar differences as NR1-1a(D669N)-containing receptors in the proton sensitivity of the voltage-independent Zn2+ inhibition when coexpressed with NR2A and NR2B (Fig. 1D). Because a Zn2+ concentration of 15 μM at a holding potential of −15 to −30 mV was used to determine NR1-1a/NR2B receptor's pH-dependent Zn2+ inhibition, it is possible that some voltage-dependent Zn2+ block occurs under our recording conditions. Therefore, we examined outward responses recorded at +20 mV at either extreme pH values and obtained identical results as those recorded at −20 mV (Fig. 1D). Our data confirmed that there is no voltage-dependent channel block caused by Zn2+ at the highest Zn2+ concentration used in this study. Taken together, these data strongly suggest that voltage-independent Zn2+ inhibition of NR1/NR2A receptors is distinct from that of NR1/NR2B receptors, and there is a potential interaction between proton and Zn2+ for NR2A-containing, but not NR2B-containing receptors.
Figure 1.

Zn2+ inhibition is pH dependent for NR2A but not NR2B-containing receptors. (A) Inhibition of NR1-1a(D669N)/NR2A receptors by 1 μM added Zn2+ at two indicated pHs from the same oocyte. (B) Inhibition of NR1-1a(D669N)/NR2B receptor by 15 μM added Zn2+ at two indicated pHs from the same oocyte. All control currents for NR2A-containing receptors were recorded in the presence of 10 μM EDTA. The NR1(D669N) mutant was used to shift the pH sensitivity to a range where formation of Zn2+ hydroxide complex is negligible at alkaline pHs. (C) Plot of the degree of Zn2+ inhibition at various pH values for NR1-1a(D669N) containing NR2A (−20 to −40 mV) or NR2B (−15 to −30 mV) receptors. (D) Wild-type NR1-1a receptors containing NR2A or NR2B subunit in the presence of 3 μM and 15 μM added Zn2+, respectively, confirm the observation in C. ○, Outward responses recorded at +20 mV for NR1-1a/NR2B. All error bars are SEM. Numbers in parentheses are the number of oocytes.
Mechanism of the Voltage-Independent Zn2+ Inhibition of NR1/NR2A Receptors.
To further test the extent to which Zn2+ inhibition reflected enhancement of tonic proton inhibition of NR2A-containing NMDA receptors, we performed the following two experiments. First, we measured the proton sensitivity of NR1/NR2A receptors in nominal absence of free Zn2+ (in the presence of 1–10 μM EDTA) and in the presence of 1 μM added Zn2+. We found that the receptors are differentially sensitive to protons in Zn2+-free solution (proton IC50, 120 nM) and in the presence of 1 μM Zn2+ (proton IC50, 29 nM) (Fig. 2A) (22).
The apparent high-affinity Zn2+ inhibition curve has a residual fractional response of about 0.2–0.4 (Fig. 3C, ref. 17, see also refs. 15 and 16). However, the nature of this residual response is not clear. If Zn2+ inhibition of NR2A-containing receptors reflected enhancement of proton sensitivity, as has been reported for phenolethanolamines (30), the maximal Zn2+ inhibition would depend on the concentration of protons. Based on the shift of proton curve in the presence of saturating Zn2+, the proton inhibition at neutral pH is still far from the maximal, resulting in a significant residual response. At more alkaline pH, the residual current in the presence of saturating Zn2+ would be large because the amount of proton inhibition is further reduced. At more acidic pH, the residual current would be smaller because of the larger degree of proton inhibition. We simulated the Zn2+ inhibition curves at various pH values (see Methods), and from these simulated results, we predict that the residual responses of Zn2+ inhibition curves will be pH dependent (Fig. 2B). Additional simulations as in Fig. 2B (data not shown) using a range of KZn values (1–200 nM) confirm that the pH dependence of the residual current is independent of the KZn for the Zn2+ site provided that the Zn2+ concentration tested is saturating (i.e., 10-fold of KZn). If the predicted residual responses agree with those determined experimentally, that would be consistent with our assumption that Zn2+ inhibition reflects enhancement of tonic proton inhibition.
Figure 3.

Histidines in the LIVBP-like domain of NR2A are critical for the apparent high-affinity Zn2+ inhibition. (A) Schematic of the NR2A subunit showing the positions of histidine in the LIVBP-like domain of NR2A that were investigated. The labels at the bottom of this figure indicate putative signal peptide (sp), the region with homology to the Leu/Ile/Val binding protein (LIVBP; ref. 31), the four membrane-associated domains (TM1–4; ref. 1), the agonist binding domains (S1, S2; ref. 32, 33), the region controlling the slow component of desensitization (preM1; refs. 34 and 35). (B) Mean-fitted IC50 values determined for voltage-independent Zn2+ inhibition of NR1-1a/NR2A receptors containing the histidine mutations. These histidines were categorized into three groups based on their effect on the IC50 values of the voltage-independent Zn2+ inhibition. Group I histidines, when mutated, have micromolar IC50 values (**). Group II histidines, when mutated, have moderate but significant effect on the IC50 values compared with wild-type (P < 0.01, ANOVA and Newman–Keul's post hoc test). Group III histidines do not have significantly different IC50 values compared with wild-type (P > 0.05, ANOVA and Newman–Keul's post hoc tests). (C) Composite Zn2+ inhibition curves are shown for NR2A mutants (H42A, H44G, and H128A) and wild-type NR1-1a/NR2A. Zn2+ inhibition of glutamate-evoked NR2A wild-type and mutant receptor-mediated currents were recorded at −20 to −30 mV. Zn2+ concentrations (0.007 to 2.6 μM) were achieved by using tricine buffer. Values are mean ± SEM. Numbers in parentheses are the number of oocytes.
In the second set of experiments, we measured the residual responses of the apparent high affinity Zn2+ inhibition at three different pH values (Fig. 2 D–F). Consistent with our simulated Zn2+ inhibition curves in Fig. 2B, we found that the residual response of the Zn2+ inhibition curve has shifted from 0.24 at pH 6.8 to 0.38 and 0.70 at pHs 7.3 and 8.0, respectively (Fig. 2 D–F, n = 6 at each pH). The residual responses obtained experimentally at these pH values correlate closely with the predicted values (Fig. 2C). These data support the conclusion that the nanomolar component of the voltage-independent Zn2+ inhibition of NR2A-containing receptors is pH-dependent and operates in a manner consistent with the enhancement of proton sensitivity. These data further suggest that the residual current during Zn2+ inhibition of NR2A-containing receptors reflects the fact that Zn2+ inhibits receptors by causing a modest shift in pH inhibition.
In addition to the shift in residual currents, we also observed an enhancement of apparent Zn2+ affinity at alkaline pH (Fig. 2 D and F). This measured shift in apparent affinity is consistent with the contribution of an uncharged histidine residue to the stabilization of Zn2+ binding. In this scenario, protonation of a histidine would increase the partial positive charge on the side chain thus diminishing its ability to contribute an electron pair to Zn2+ coordination. Alternatively, removal of a proton at alkaline pH values would enhance the ability of a histidine to coordinate Zn2+. To evaluate this idea, we sought to identify candidate histidine residues involved in Zn2+ coordination.
Histidine Residues in the Proximal LIVBP-like Domain of NR2A Subunit Contribute to the Voltage-Independent Zn2+ Inhibition.
Because recent data suggests that the LIVBP-like domain (N-terminal 370 residues) of NR2A harbors critical residues influencing the voltage-independent Zn2+ inhibition of NMDA receptors (24), we mutated each histidine residue in this region of NR2A subunit to alanine, glycine, or valine (Fig. 3A) and screened the mutant receptors for altered Zn2+ binding. The results from the site-directed mutagenesis experiments (Fig. 3B) show that histidine residues in the LIVBP-like domain of NR2A subunit can be stratified into three groups based on their effect on the voltage-independent Zn2+ inhibition. Group I is comprised of H42, H44, and H128, which exerted the largest shift (100-fold or greater) in the IC50 values of all mutant NR1/NR2A receptors. The Zn2+ inhibition curves for NR1-1a/NR2A(H42A), NR1-1a/NR2A(H44G), and NR1-1a/NR2A(H128A) current responses recorded at holding potentials −20 to −30 mV can be fit with a single binding site isotherm, yielding IC50 values of 2,651 (122-fold), 6,267 (289-fold), and 15,078 (695-fold) nM, respectively (Fig. 3C). Mutations of Group I histidine residues had little or no effect on the EC50 of the coagonists glutamate and glycine, and voltage-dependent Mg2+ block (Table 1). The Group II residues H85, H96, H332, and H335 exert a more modest shift on the IC50 values of recombinant NR1/NR2A receptors, between 4- and 8-fold. Group III is comprised of H168, H223, H358, and H387, which showed no significant change compared with the wild-type NR1-1a/NR2A receptors (IC50 21.7 nM) when mutated to alanine, glycine, or valine (P > 0.05, ANOVA and Newman–Keul's post hoc tests). Similar reduction in Zn2+ sensitivity was confirmed in HEK293 cells transiently expressing mutant NR1/NR2A(H44G) (n = 3) and NR1/NR2A(H128A) (n = 3) (data not shown).
Table 1.
Histidine residues that alter Zn2+ sensitivity do not alter basic receptor properties
| Constructs | Glutamate
|
Glycine
|
Mg2+
|
||
|---|---|---|---|---|---|
| EC50 μM (n) | NHill | EC50 μM (n) | NHill | I100μM/Icontrol | |
| NR1-1a/NR2A | 5.9 ± 0.2 (7) | 1.6 ± 0.1 | 3.3 ± 0.2 (14) | 1.5 ± 0.1 | 0.29 (8) |
| NR1-1a/NR2A(H42A) | 5.7 ± 0.2 (9) | 1.5 ± 0.1 | 2.4 ± 0.2 (7) | 1.6 ± 0.2 | 0.33 (10) |
| NR1-1a/NR2A(H44G) | 7.2 ± 0.2 (9) | 1.8 ± 0.1 | 4.1 ± 0.3 (10) | 1.8 ± 0.2 | 0.32 (10) |
| NR1-1a/NR2A(H128A) | 4.5 ± 0.2 (8) | 1.3 ± 0.1 | 2.1 ± 0.2 (8) | 1.7 ± 0.2 | 0.35 (9) |
The glutamate and glycine EC50 values of wild-type and NR2A Group I histidine mutants show no significant differences at −20 to −30 mV holding potentials (p > 0.05, ANOVA and Tukey-post-hoc tests). The Mg2+ inhibition (IMg/Icontrol at 100 μM Mg2+) of NR2A wild-type and Group I histidine mutants showed no significant differences (p > 0.05, ANOVA and Tukey-post-hoc tests) at −50 mV. There was no detectable differences between wild-type NR2A receptors and Group I histidine mutants at 300 μM Mg2+ (n = 37, data not shown).
Because there may be common structural determinants of proton and Zn2+ as suggested previously by the correlation between the proton sensitivity and the Zn2+ sensitivity for NR1 splice variants and point mutations of NR1 subunit (18), some mutations of NR2A that alter Zn2+ sensitivity may alter proton sensitivity. Therefore, we tested the effect of all histidine mutants in the LIVBP-like domain of NR2A on the proton sensitivity. The pH sensitivity was determined in the absence of contaminant Zn2+ by addition of 10 μM EDTA. Our results are summarized in Table 2. There was no correlation (correlation coefficient, r = 0.095, P > 0.5) between the effects of amino acid exchanges on Zn2+ and pH sensitivity.
Table 2.
Proton inhibition of NR-1-1a/NR2A receptors
| Constructs | pH IC50 | Free proton concentration, nM |
|---|---|---|
| NR1-1a/NR2A | 7.02 (7) | 120 |
| NR1-1a/NR2A(H42A) | 6.76 (6) | 215 |
| NR1-1a/NR2A(H44G) | 6.88 (10) | 166 |
| NR1-1a/NR2A(H85G) | 6.70 (4) | 251 |
| NR1-1a/NR2A(H96A) | 6.80 (7) | 196 |
| NR1-1a/NR2A(H128A) | 6.82 (5) | 189 |
| NR1-1a/NR2A(H168G) | 7.01 (5) | 123 |
| NR1-1a/NR2A(H223A) | 7.02 (5) | 120 |
| NR1-1a/NR2A(H332/5A) | 7.01 (5) | 121 |
| NR1-1a/NR2A(H358V) | 7.07 (5) | 106 |
| NR1-1a/NR2A(H387A) | 7.16 (4) | 86 |
Fitted pH IC50 values of NR2A histidine mutants screened when coexpressed with NR1-1a. IC50 values in free H+ concentration are given to the nearest 1 nM (activity coefficient of 0.8). Experiments were performed with saturating concentrations of NMDA (200 μM) and glycine (100 μM). All solutions contain 10 μM EDTA to remove contaminant Zn2+.
Histidine 128 Is Critical for the pH Dependency of Zn2+ Inhibition of NR1/NR2A Receptors.
Because the NR2A subunit controls the pH dependency of Zn2+ inhibition in the recombinant NMDA receptors (see above), we tested whether those histidines in NR2A that have the largest effect on the voltage-independent Zn2+ inhibition (H42, H44, and H128) might be the molecular determinant(s) for this phenomenon. By using nominal Zn2+ concentrations that caused approximately 50% inhibition of each mutant, we found that the Zn2+ inhibition for H42A and H44G mutations are pH-dependent in a similar fashion to the wild-type NR2A (Fig. 4 A, B, and D), even though the Zn2+ inhibition curves are shifted by as much as 289-fold. Similar pH-dependent Zn2+ inhibition was found for receptors containing the double mutations H42A/H44G (n = 6, data not shown). However, amino acid substitution at histidine 128 renders Zn2+ inhibition pH-insensitive (Fig. 4 C and E). These data suggest that H128 may be important for the pH-dependency of Zn2+ inhibition of NR1/NR2A receptors.
Figure 4.

Mutations of histidine 128 abolished pH-dependency of Zn2+ inhibition of NR1/NR2A receptors. (A) Zn2+ inhibition of glutamate-evoked NR1-1a/NR2A(H42A) receptor-mediated currents recorded in 3 μM added Zn2+ at −20 mV in pHs 6.8 and 7.8. (B) Zn2+ inhibition of glutamate-evoked NR1-1a/NR2A(H44G) receptor-mediated currents recorded in 10 μM added Zn2+ at −20 mV in pHs 6.8 and 7.8. (C) Zn2+ inhibition of glutamate-evoked NR1-1a/NR2A(H128A) receptor-mediated currents recorded in 16 μM added Zn2+ at −30 mV in pHs 6.8 and 7.8. All control solutions contain 10 μM EDTA to chelate any contaminant Zn2+. (D) The pH-dependency of voltage-independent Zn2+ inhibition of NR2A-containing receptors harboring H42A or H44G mutations is similar to that of wild-type NR1/NR2A receptors (dotted line, data from Fig. 1D). (E) Exchange of His128 to Ala is sufficient to abolish the pH-dependency of voltage-independent Zn2+ inhibition. Free Zn2+ concentration is compensated at pH 8.2 for the formation of zinc hydroxide complexes. Error bars are SEM. Numbers in parentheses are the number of oocytes.
Discussion
The three most important conclusions to emerge from this study are: (i) that the voltage-independent Zn2+ inhibition is pH-dependent for NR2A- but not NR2B-containing NMDA receptors, (ii) that the inhibition of NR2A-containing receptors by nanomolar concentrations of Zn2+ reflects enhancement of tonic proton inhibition, and (iii) that the LIVBP-like domain of NR2A contain residues that control the voltage-independent Zn2+ inhibition of NMDA receptors, including a single amino acid residue (His128) that is also critical for the pH-dependency of Zn2+ inhibition. These findings have important structural and functional implications about the manner by which protons and Zn2+ control NMDA receptor function.
Subunit Dependence of the pH-Dependent Zn2+ Inhibition of Recombinant NMDA Receptors.
From our previous studies of the interaction between protons and Zn2+, we concluded that Zn2+ did not inhibit NR1/NR2B receptor function by enhancement of pH inhibition (18). However, it has recently been suggested that the voltage-independent Zn2+ inhibition of recombinant NMDA receptors may involve the enhancement of proton inhibition (22). Given these apparently conflicting data, we have investigated the possible interaction between protons and Zn2+ in NR1/NR2A and NR1/NR2B receptors by using an experimental paradigm designed to avoid pH-dependent reduction of free Zn2+ concentration (18). Our results show pH-dependence of Zn2+ inhibition of NR2A but not NR2B receptors, and thus are consistent with our previous results as well as some of the results reported by Choi and Lipton (22). The difference in the pH sensitivity underscores the different structural determinants within the NR2A subunit that control Zn2+ inhibition, and suggests that the Zn-binding domains of the NR2A subunit may have functional or structural interactions with the proton sensor. Furthermore, the strong pH-dependence of Zn2+ inhibition of NR2A-containing receptors raises the idea that Zn2+ inhibition of these receptors may proceed by a fundamentally different mechanism than Zn2+ inhibition of NR2B receptors.
Mechanism of Zn2+ Inhibition of Recombinant NR1/NR2A Receptors.
Phenolethanolamines inhibit recombinant NR2B-containing NMDA receptors by enhancing proton inhibition such that, at physiological pH, receptor function is almost fully inhibited (30). Could a similar mechanism also account for Zn2+ inhibition of recombinant NMDA receptors? The pH independence of Zn2+ inhibition of NR2B-containing receptors argues against the idea that Zn2+ binding to the receptor enhances tonic proton inhibition (see also ref. 18). However, the pH-dependence of NR2A receptors is consistent with this possibility, as first suggested by Choi and Lipton (22). If enhancement of proton inhibition is the mechanism by which Zn2+ could cause voltage-independent inhibition of NR1/NR2A, then the residual current observed at physiological pH (7.3–7.4) could be explained by the submaximal proton inhibition. Furthermore, one would predict that the degree of the residual current would be pH dependent. Our data confirm this prediction (Fig. 2C), providing strong evidence that Zn2+-bound receptors have an enhanced proton sensitivity, and also provides a mechanistic explanation for the residual current that remains when the apparent high affinity Zn2+ binding site is saturated. In addition, this finding increases the list of substances (polyamines, ifenprodil, Zn2+) that exert their actions through modification of the pH sensitivity of NMDA receptors (30, 36). Thus, the control of channel gating by protonation of an ionizable group within the receptor appears to be a common denominator for several extracellular binding sites on NMDA receptors.
Structural Basis for Zn2+ Inhibition of Recombinant NR1/NR2A Receptors.
We have identified three histidine residues (Group I residues: H42, H44, H128) within the LIVBP-like domain that are critical for Zn2+ inhibition. Our data on the voltage-independent Zn2+ inhibition of H42A and H44G mutations are consistent with a previous report (ref. 22, see also ref. 23). Given the magnitude of the effect of amino acid substitutions at these sites and the propensity of histidine residues to participate in Zn2+ coordination in other metalloproteins, these residues are likely candidates for Zn2+ coordination. Given the flexible coordination chemistry of Zn2+, it is possible that additional amino acid residues may also participate in coordination of Zn2+ ions. Group II residues (H85, H96, H332, H335) should also be viewed as candidates for this effect. However, any Group II residue that participated in Zn2+ coordination would energetically contribute less to Zn2+ coordination given the data showing amino acid exchanges at these sites have smaller effects on Zn2+ sensitivity. If Group I and II residues contribute to the Zn2+ site, the differential contribution of Group I and II residues to Zn2+ coordination could reflect geometrical constraints of the protein that allow certain residues (H42, H44, H128) to form more stable molecular orbitals with Zn2+ than other residues. Although, these mutagenesis data cannot alone prove participation of these histidine residues in Zn2+ coordination or rule out the possibility that these mutations disrupt an allosteric transduction mechanism (37, 38), they strongly suggest that important structural determinants of Zn2+ actions reside within the LIVBP-like domain of NR2A subunit.
The potential for protons and Zn2+ to interact with each other's respective binding sites makes interpretation of experiments that co-vary their concentrations complex. However, we believe the most parsimonious explanation for the data reported in this study is a functional convergence for protons and Zn2+ inhibition on NR2A-containing receptors. This interaction follows a precedent for phenolethanolamine enhancement of tonic proton inhibition of NMDA receptors (30), and is consistent with all of our results (see also ref. 22). Thus, Zn2+ and ifenprodil may be functional analogous ligands for the LIVBP domains of NR2A- and NR2B-containing receptors. Substitution of H128 to alanine abolished the pH-dependence of apparent high-affinity Zn2+ inhibition. This observation suggests that the protonation of H128 may play an important role in the regulation of NR1/NR2A receptors. The protonation of H128, by decreasing the electronegativity of the side chain, reduces the ability of this residue to stabilize Zn2+ binding. This idea is consistent with the increase in apparent affinity of Zn2+ inhibition that occurs at alkaline pH values (compare Fig. 2 D and F) at which a larger fraction of H128 would be expected to be unprotonated. Although more work including structural data will be needed to ultimately understand Zn2+ inhibition, our experiments have emphasized interesting structural features of the link in the effects of extracellular proton and Zn2+ for NR2A-containing receptors.
Acknowledgments
We thank T. G. Smart and J. Neyton for critical comments on this manuscript; A. Fayyazuddin, A. Villarroel, A. Le Goff, J. Lerma, J. Neyton, and P. Paoletti for sharing data before publication; and R. Dingledine and D. Mott for their helpful discussion. We are grateful to S. F. Heinemann (NR1-1a and NR2B, accession nos. U08261 and U11419, respectively), S. Nakanishi (NR2A, accession no. D13211), and K. Williams for sharing cDNAs with us. This work was supported by National Institute of Neurological Diseases and Stroke (S.F.T.), National Alliance for Research on Schizophrenia and Depression (F.Z.), and the John Merck Fund (S.F.T.).
Abbreviations
- NMDA
N-methyl-d-aspartate
- LIVBP
leucine/isoleucine/valine binding protein
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.180307497.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.180307497
References
- 1.Dingledine R, Borges K, Bowie D, Traynelis S F. Pharmacol Rev. 1999;51:7–61. [PubMed] [Google Scholar]
- 2.Giffard R G, Monyer H, Christine C W, Choi D W. Brain Res. 1990;506:339–342. doi: 10.1016/0006-8993(90)91276-m. [DOI] [PubMed] [Google Scholar]
- 3.Traynelis S F, Cull-Candy S G. Nature (London) 1990;345:347–350. doi: 10.1038/345347a0. [DOI] [PubMed] [Google Scholar]
- 4.Balestrino M, Somjen G G. J Physiol (London) 1988;396:247–266. doi: 10.1113/jphysiol.1988.sp016961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Traynelis S F, Cull-Candy S G. J Physiol (London) 1991;433:727–763. doi: 10.1113/jphysiol.1991.sp018453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smart T G, Xie X, Krishek B J. Prog Neurobiol. 1994;42:393–441. doi: 10.1016/0301-0082(94)90082-5. [DOI] [PubMed] [Google Scholar]
- 7.Harrison N L, Gibbons S J. Neuropharmacology. 1994;33:935–952. doi: 10.1016/0028-3908(94)90152-x. [DOI] [PubMed] [Google Scholar]
- 8.Charlton G, Rovira C, Ben-Ari Y, Leviel V. Exp Brain Res. 1985;58:202–205. doi: 10.1007/BF00238969. [DOI] [PubMed] [Google Scholar]
- 9.Aniksztejn L, Charlton G, Ben-Ari Y. Brain Res. 1987;404:58–64. doi: 10.1016/0006-8993(87)91355-2. [DOI] [PubMed] [Google Scholar]
- 10.Slomianka L. Neuroscience. 1992;38:843–854. doi: 10.1016/0306-4522(90)90076-g. [DOI] [PubMed] [Google Scholar]
- 11.Westbrook G L, Mayer M L. Nature (London) 1987;328:640–643. doi: 10.1038/328640a0. [DOI] [PubMed] [Google Scholar]
- 12.Peters S, Koh J, Choi D W. Science. 1987;236:589–593. doi: 10.1126/science.2883728. [DOI] [PubMed] [Google Scholar]
- 13.Christine C W, Choi D W. J Neurosci. 1990;10:108–116. doi: 10.1523/JNEUROSCI.10-01-00108.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Legendre P, Westbrook G L. J Physiol (London) 1990;429:429–449. doi: 10.1113/jphysiol.1990.sp018266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Williams K. Neurosci Lett. 1996;215:9–12. doi: 10.1016/s0304-3940(96)12924-4. [DOI] [PubMed] [Google Scholar]
- 16.Chen N, Moshaver A, Raymond L A. Mol Pharmacol. 1997;51:1015–1023. doi: 10.1124/mol.51.6.1015. [DOI] [PubMed] [Google Scholar]
- 17.Paoletti P, Asher P, Neyton J. J Neurosci. 1997;17:5711–5725. doi: 10.1523/JNEUROSCI.17-15-05711.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Traynelis S F, Burgess M F, Zheng F, Lyuboslavsky P, Powers J L. J Neurosci. 1998;18:6163–6175. doi: 10.1523/JNEUROSCI.18-16-06163.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xiong Z-G, Pelkey K A, Lu W Y, Lu Y M, Roder J C, MacDonald J F, Salter M W. J Neurosci. 1999;19:1–6. doi: 10.1523/JNEUROSCI.19-21-j0003.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Monyer H, Burnashev N, Laurie D J, Sakmann B, Seeburg P H. Neuron. 1994;12:529–540. doi: 10.1016/0896-6273(94)90210-0. [DOI] [PubMed] [Google Scholar]
- 21.Vogt K, Mellor J, Tong G, Nicoll R. Neuron. 2000;26:187–196. doi: 10.1016/s0896-6273(00)81149-6. [DOI] [PubMed] [Google Scholar]
- 22.Choi Y-B, Lipton S A. Neuron. 1999;23:171–180. doi: 10.1016/s0896-6273(00)80763-1. [DOI] [PubMed] [Google Scholar]
- 23.Fayyazuddin A, Villarroel A, Le Goff A, Lerma J, Neyton J. Neuron. 2000;25:683–694. doi: 10.1016/s0896-6273(00)81070-3. [DOI] [PubMed] [Google Scholar]
- 24.Köhr G, Eckardt S, Lüddens H, Monyer H, Seeburg P H. Neuron. 1994;12:1031–1040. doi: 10.1016/0896-6273(94)90311-5. [DOI] [PubMed] [Google Scholar]
- 25.Zheng F, Gingrich M B, Traynelis S F, Conn P J. Nat Neurosci. 1998;1:185–191. doi: 10.1038/634. [DOI] [PubMed] [Google Scholar]
- 26.Bers D, Patton C, Nuccitelli R. Methods Cell Biol. 1994;40:3–29. doi: 10.1016/s0091-679x(08)61108-5. [DOI] [PubMed] [Google Scholar]
- 27.Brooks S P J, Storey K B. Anal Biochem. 1992;201:119–126. doi: 10.1016/0003-2697(92)90183-8. [DOI] [PubMed] [Google Scholar]
- 28.Martell A E, Smith R M. Critical Stability Constants. Vol. 1. New York: Plenum; 1989. –6. [Google Scholar]
- 29.Kashiwagi K, Fukuchi J-I, Chao J, Igarashi K, Williams K. Mol Pharmacol. 1996;49:1131–1141. [PubMed] [Google Scholar]
- 30.Mott D D, Doherty J J, Zhang S, Washburn M S, Fendley M J, Lyuboslavsky P, Traynelis S F, Dingledine R. Nat Neurosci. 1998;1:659–667. doi: 10.1038/3661. [DOI] [PubMed] [Google Scholar]
- 31.O'Hara P J, Sheppard P O, Thogersen H, Venezia D, Haldeman B A, McGrane V, Houamed, Thomsen C, Gilbert T L, Mulvihill E R. Neuron. 1993;11:41–52. doi: 10.1016/0896-6273(93)90269-w. [DOI] [PubMed] [Google Scholar]
- 32.Stern-Bach Y, Bettler B, Hartley M, Sheppard P O, O'Hara P J, Heinemann S F. Neuron. 1994;13:1345–1357. doi: 10.1016/0896-6273(94)90420-0. [DOI] [PubMed] [Google Scholar]
- 33.Armstrong N, Sun Y, Chen G-Q, Gouaux E. Nature (London) 1998;395:913–917. doi: 10.1038/27692. [DOI] [PubMed] [Google Scholar]
- 34.Krupp J, Vissel B, Heinemann S F, Westbrook G L. Neuron. 1998;20:317–327. doi: 10.1016/s0896-6273(00)80459-6. [DOI] [PubMed] [Google Scholar]
- 35.Villarroel A, Regalado M P, Lerma J. Neuron. 1998;20:329–339. doi: 10.1016/s0896-6273(00)80460-2. [DOI] [PubMed] [Google Scholar]
- 36.Traynelis S F, Hartley M, Heinemann S F. Science. 1995;268:873–876. doi: 10.1126/science.7754371. [DOI] [PubMed] [Google Scholar]
- 37.Lynch J W, Jacques P, Pierce K D, Schofield P. J Neurochem. 1998;71:2159–2168. doi: 10.1046/j.1471-4159.1998.71052159.x. [DOI] [PubMed] [Google Scholar]
- 38.Harvey R J, Thomas P, James C H, Wilderspin A, Smart T G. J Physiol. 1999;520:53–64. doi: 10.1111/j.1469-7793.1999.00053.x. [DOI] [PMC free article] [PubMed] [Google Scholar]