

Summary
In response to oxidative stress, Nrf2 and p21 Cip1/WAF1 are both upregulated to protect cells from oxidative damage. Nrf2 is constantly ubiquitinated by a Keap1 dimer that interacts with a weak-binding 29DLG motif and a strong-binding 79ETGE motif in Nrf2, resulting in degradation of Nrf2. Modification of the redox-sensitive cysteine residues on Keap1 disrupts the Keap1-29DLG binding, leading to diminished Nrf2 ubiquitination and activation of the antioxidant response. However, the underlying mechanism by which p21 protects cells from oxidative damage remains unclear. Here, we present molecular and genetic evidence suggesting that the antioxidant function of p21 is mediated through activation of Nrf2 by stabilizing the Nrf2 protein. The 154KRR motif in p21 directly interacts with the 29DLG and 79ETGE motifs in Nrf2, and thus, competes with Keap1 for Nrf2 binding, compromising ubiquitination of Nrf2. Furthermore, the physiological significance of our findings was demonstrated in vivo using p21-deficient mice.
Introduction
The transcription factor Nrf2 has emerged as a master regulator of an intracellular antioxidant response through transcriptional activation of an array of genes, including phase II detoxifying enzymes, antioxidants, and transporters that protect cells from toxic and carcinogenic chemicals (Kensler et al., 2007; Kobayashi and Yamamoto, 2005; Zhang, 2006). Nrf2-deficient mice are prone to chemical-induced toxicity and tumorigenesis (Aoki et al., 2001; Cho et al., 2004; Lau et al., 2008; Ramos-Gomez et al., 2001). The Nrf2 signaling pathway is negatively controlled by Keap1. Keap1 contains several domains that are essential for its functions: the BTB domain is required for homodimerization; the linker domain contains several cysteine residues that sense intracellular redox imbalance; and the Kelch domain binds the N-terminal Neh2 domain of Nrf2 (Itoh et al., 1999; Wakabayashi et al., 2004; Yamamoto et al., 2008; Zhang and Hannink, 2003; Zipper and Mulcahy, 2002). Under basal conditions, Keap1, functioning as a substrate adaptor for a Cul3-based E3 ubiquitin ligase, constantly targets Nrf2 for ubiquitin-dependent degradation to maintain low levels of Nrf2 (Itoh et al., 1999; Kobayashi et al., 2004; Kobayashi et al., 2006; McMahon et al., 2003; Zhang and Hannink, 2003; Zhang et al., 2004). Very recently, a two-site substrate recognition model, also called the hinge and latch model, was presented to explain how Keap1 recruits Nrf2 and assists in ubiquitination of Nrf2 (McMahon et al., 2006; Tong et al., 2006; Tong et al., 2007). In this model, each Kelch domain from a Keap1 homodimer binds one Nrf2 protein through a weak-binding 29DLG motif (latch) or a strong-binding 79ETGE motif (hinge), located in the N-terminal Neh2 domain of Nrf2. The binding affinity of Kelch to the 79ETGE motif is approximately 100-fold higher than that of Kelch to the 29DLG motif (Tong et al., 2006). It is believed that the Keap1 homodimer binds both the 29DLG and 79ETGE motifs in Nrf2 to align the seven ubiquitin accepting lysine residues between these motifs into a conformation suitable for ubiquitin conjugation (McMahon et al., 2006; Tong et al., 2006; Tong et al., 2007; Zhang et al., 2004). Upon oxidative stress, modification of the cysteine residues on Keap1, such as C151, C273 or C288 in the BTB or linker domain, imposes a conformational change that disrupts the weak Kelch-29DLG binding, resulting in diminished Nrf2 ubiquitination without dissociation of Nrf2 from Keap1 (McMahon et al., 2006; Tong et al., 2006; Tong et al., 2007). As a consequence, Nrf2 protein levels are increased and the Nrf2 signaling pathway is activated (Itoh et al., 1999; Kobayashi et al., 2004; Kobayashi et al., 2006; McMahon et al., 2003; Zhang and Hannink, 2003; Zhang et al., 2004).
p21Cip1/WAF1 regulates many cellular processes such as cell cycle arrest, DNA replication and repair, cell differentiation, senescence, and apoptosis (Dotto, 2000; Gartel and Tyner, 2002; Li et al., 1994; O'Reilly, 2005). In response to oxidative stress, p21 is upregulated to promote cell survival (Esposito et al., 1998; Gartel and Tyner, 2002; O'Reilly et al., 2001; Poon and Hunter, 1998). However, the underlying mechanism by which p21 protects cells from oxidative damage remains unclear. In this report, we present the first molecular and genetic evidence suggesting that p21-dependent cell survival under oxidative stress is mediated through activation of the Nrf2 signaling pathway. The protein level of Nrf2 is upregulated by p21 through direct interaction between p21 and Nrf2. The interacting motifs have been mapped to the 29DLG and 79ETGE motifs in Nrf2 and the 154KRR in p21. p21 activates the Nrf2 pathway by competing with Keap1 for Nrf2 binding, particularly through the 29DLG motif, compromising the Keap1-dependent ubiquitination of Nrf2. Furthermore, p21-mediated upregulation of Nrf2 under both basal and induced conditions was confirmed using p21-deficient mice, demonstrating the physiological significance of our findings.
Results
Nrf2 was Required for the p21-Dependent Cellular Protection in Response to Oxidative Stress
To determine a possible functional link between p21 and Nrf2, a series of experiments were performed in mouse embryonic fibroblasts (MEFs) from Keap1- or Nrf2- deficient mice, or in HCT116 cells with or without a somatic p21 deletion. First, the protective role of p21 against the cellular stress response was verified by measuring intracellular reactive oxygen species (ROS) in HCT116-p21+/+ and HCT116-p21-/- cells following H2O2 challenge. While H2O2 did not induce a measurable amount of ROS in HCT116-p21+/+ cells, it increased the level of ROS in HCT116-p21-/- cells in a dose-dependent manner (Figure 1A). To exclude the possibility that the difference in their response to H2O2 is due to clonal artifacts, p21-cDNA was re-introduced into HCT116-p21-/- cells by transient transfection. Ectopic expression of p21 rescued HCT116-p21-/- cells from H2O2-induced ROS production (Figure 1A). The residual dose-dependent increase of ROS is likely due to the fact that 100% transfection efficiency could not be achieved. Next, the antioxidant function of Nrf2 was demonstrated in HCT116-p21+/+ cells using a siRNA approach. Nrf2-siRNA significantly reduced Nrf2 protein level, compared with the control siRNA (Figure 1B). Consequently, Nrf2-siRNA transfected cells had an elevated basal ROS level compared to the control-siRNA transfected cells (Figure 1B). In addition, the antioxidant response mediated by p21 and by Nrf2 was further evaluated by comparing the survival curves in response to H2O2 treatment between the pair of HCT116-p21+/+/p21-/- cells and the pair of MEF-Keap1+/+/Keap1-/-cells. The HCT116-p21+/+ cells had a higher basal level of Nrf2 compared to the HCT116-p21-/- cells, and the basal level of Nrf2 was higher in MEF-Keap1-/- cells than in MEF-Keap1+/+ cells (data not shown). Interestingly, similar toxicity curves were obtained between these two groups (Figure S1), indicating that p21 and Nrf2 render cells more resistant to H2O2. Next, the requirement of Nrf2 in p21-mediated cell survival in response to H2O2 was tested in both MEF-Nrf2+/+ and MEF-Nrf2-/- cells. Although overexpression of p21 protected cells from oxidative cell death in MEF-Nrf2+/+ cells, it had no effect on cell survival in MEF-Nrf2-/- cells (Figure 1C). In contrast, ectopic expression of Nrf2 had an effect on both cell types. In order to eliminate the possibility that the protection observed is due to cell cycle arrest under conditions of p21 overexpression, a similar experiment in which p21 expression was inhibited by p21-siRNA was performed in both MEF-Nrf2+/+ and MEF-Nrf2-/- cells. Reduced p21 expression sensitized MEF-Nrf2+/+ cells to H2O2 insult, but not MEF-Nrf2-/- cells (Figure 1D). Collectively, these results demonstrate that both Nrf2 and p21 have antioxidant functions that promote survival of cells under oxidative stress. More importantly, p21-mediated protection requires Nrf2, suggesting crosstalk between p21 and the Nrf2 signaling pathway.
Figure 1. Nrf2 was Required for the p21-Dependent Cellular Protection in Response to Oxidative Stress.
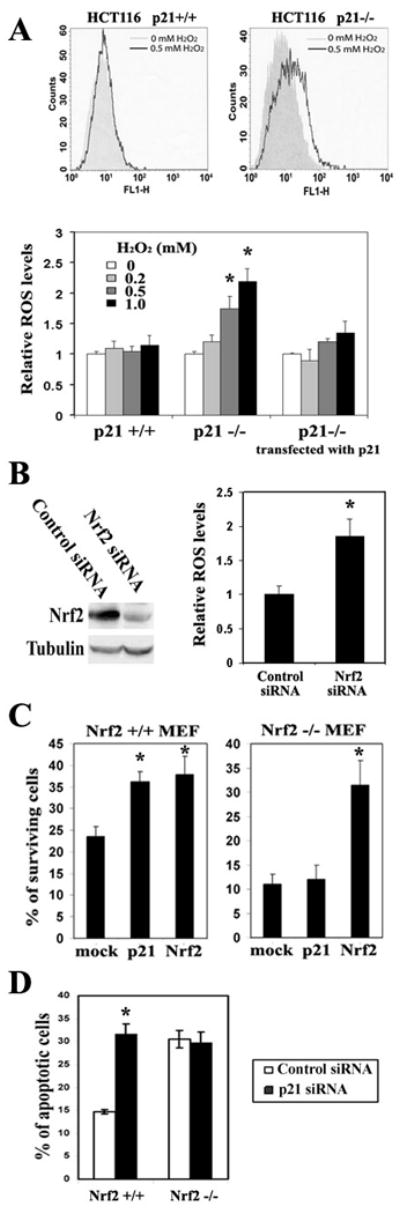
(A) p21 exhibited antioxidant functions. HCT116-p21+/+, HCT116-p21-/-, and HCT116-p21-/- cells transfected with an expression vector for p21, were treated with the indicated doses of H2O2 for 12 hrs. Intracellular ROS levels were measured by the dichlorofluorescein/flow cytometry method. The representative histograms of ROS analysis in HCT116-p21+/+cells and HCT116-p21-/- cells are shown in the upper two panels. (B) HCT116-p21+/+ cells with knockdown of Nrf2 had higher intracellular levels of ROS. HCT116-p21+/+ cells were transfected with either control siRNA or Nrf2-siRNA. At 72 hrs post-transfection, levels of Nrf2 and intracellular ROS were measured. (C) Ectopic expression of p21 enhanced cell survival in response to H2O2 in MEF-Nrf2+/+, but not in MEF-Nrf2-/- cells. MEF-Nrf2+/+ or MEF-Nrf2-/- cells were cotransfected with the indicated expression vectors for 24 hrs. GFP was used to mark the transfected cells. Following treatment with H2O2, over 150 GFP positive cells were counted for both surviving and apoptotic cells by fluorescence microscopy. The percentage of surviving cells was plotted. (D) Knockdown of p21 sensitized cells to H2O2-induced cell death in MEF-Nrf2+/+, but not in MEF-Nrf2-/- cells. Cells were transfected with p21-siRNA or control siRNA for 72 hrs before treatment with H2O2. Apoptotic cells were detected using the Annexin V-FITC/flow cytometry method. All error bars indicate standard deviations calculated from three independent experiments. *p<0.05 compared with its control.
p21 Upregulated the Protein Level of Nrf2 and its Downstream Genes
To further verify our hypothesis that the antioxidant function of p21 is mediated through upregulation of the Nrf2 signaling pathway, effects of p21 on the transcription of Nrf2 and Nrf2-target genes were measured by qRT-PCR in MEF-Nrf2+/+ cells. p21-siRNA reduced the expression of endogenous p21 protein approximately 60% with a concomitant decrease of the Nrf2 protein level in MEF-Nrf2+/+ cells (Figure 2A). p21-siRNA had no effect on the mRNA expression of Nrf2 or Keap1, while knockdown of p21 expression resulted in a decrease of the Nrf2-dependent transcription of NAD(P)H quinone oxidoreductase 1 (NQO1) and heme oxygenase-1 (HO-1) in MEF-Nrf2+/+ cells (Figure 2A). In contrast, p21 siRNA had no effect on transcription of NQO1 or HO-1 in MEF-Nrf2-/- cells (Figure 2B). Together, these data indicate that p21-mediated upregulation of the Nrf2 signaling pathway requires Nrf2. This notion was further verified by overexpression of p21 in HCT116-p21-/-cells. Transient expression of p21 enhanced the levels of Nrf2 protein in a p21-dose-dependent manner (Figure 2C). Consistent with the increased protein level of Nrf2, the transcriptional activity of Nrf2 was also enhanced by overexpression of p21 when a NQO1-ARE firefly luciferase reporter was used (Figure 2C). Therefore, p21 is able to activate the Nrf2 signaling pathway by enhancing Nrf2 protein levels. A similar experiment was also performed in COS-1 cells, coexpression of p21 with Nrf2 enhanced the protein level of Nrf2 in the absence or presence of exogenous Keap1 and overexpression of Keap1 decreased the protein level of Nrf2 as expected (Figure S2).
Figure 2. p21 Upregulated the Protein Level of Nrf2 and its Downstream Genes.

(A) Knockdown of p21 diminished expression of the Nrf2-target genes in MEF-Nrf2+/+ cells. MEF-Nrf2+/+ cells were transfected with either control siRNA or p21-siRNA. The endogenous levels of p21 and Nrf2 proteins were detected by immunoblot analysis at 48 hrs post-transfection, while the mRNA expression of Nrf2, Keap1, NQO1 and HO-1 was measured 72 hrs after transfection. The error bars indicate standard deviations calculated from triplicate samples. *p<0.05 compared with its control. (B) Knockdown of p21 had no effects in MEF-Nrf2-/- cells. The same experiment was performed in MEF-Nrf2-/- cells as described in 2A. (C) p21 upregulated the ARE-reporter gene activity. HCT116-p21-/- cells were transfected with different amounts of p21 expression vectors, along with expression vectors for the NQO1-ARE firefly luciferase and TK-renilla luciferase. The protein levels of p21 and Nrf2 were detected with anti-p21 and anti-Nrf2 antibodies (lower panels). The firefly luciferase activities were normalized to renilla luciferase activities and the standard deviations were calculated from three independent experiments, each with duplicate samples. *p<0.05 compared with its control.
HCT116-p21-/- Cells had Reduced Basal and Induced Levels of the Nrf2-Dependent Antioxidant Response
It has been well documented that p21 is induced in both a p53-dependent and – independent manner at the transcriptional and post-transcriptional level in response to oxidative stress. The endogenous p21 level was measured in HCT116-p21+/+ cells treated with Nrf2 activators, tert-butyl hydroquinone (tBHQ) and sulforaphane (SF). Both tBHQ and SF are able to induce p21 protein levels in a dose-dependent manner (Figure 3A). Next, the Nrf2-dependent response under basal and induced conditions in HCT116-p21+/+ and HCT116-p21-/- cells was compared. First, the basal level of Nrf2 is slightly lower in HCT116-p21-/- cells (Fig3B). tBHQ and SF enhanced the protein level of Nrf2 in both cell lines, although the Nrf2 level was consistently lower in HCT116-p21-/-cells compared to that in HCT116-p21+/+ (Figure 3B). Second, the Nrf2 transcriptional activity is consistently lower under both basal and induced conditions in HCT116-p21-/-cells, compared to HCT116-p21+/+ cells, when the NQO1-ARE-dependent firefly luciferase reporter was used (Figure 3C). In addition, tBHQ or SF markedly enhanced the ARE-luciferase activity in HCT116-p21+/+ cells; however, only moderate inductions were observed upon treatment in HCT116-p21-/- cells, indicating an additive effect of p21 and oxidative stress in activating Nrf2 (Figure 3C). A similar result was obtained using a glutathione S-transferase (GST)-ARE firefly luciferase reporter (Figure S3). Third, the mRNA expression of Nrf2 and several Nrf2 target genes was measured by qRT-PCR. No significant difference was detected in Nrf2 mRNA expression in any of the conditions between these two cell types, which is consistent with the result demonstrating that p21 siRNA had no effect on Nrf2 mRNA expression in MEF-Nrf2+/+ cells (Figures S4 and 2A). However, mRNA expression of NQO1, glutathione peroxidase 2 (Gpx2), and multidrug resistance protein 2 (Mrp2) was increased by the Nrf2 activators in both cell types (Figure 3D). Furthermore, the basal level and the induction fold of these genes were lower in HCT116-p21-/- cells compared to HCT116-p21+/+ cells (Figure 3D). Lastly, in correlation with the lower level of NQO1 mRNA in HCT116-p21-/- cells, the enzymatic activity of NQO1 was also reduced in HCT116-p21-/-cells, compared to HCT116-p21+/+ cells (Figure 3E). These results clearly demonstrate that p21 is able to activate the Nrf2 signaling pathway by enhancing Nrf2 protein levels.
Figure 3. HCT116-p21-/- Cells had Reduced Basal and Induced Levels of the Nrf2-Dependent Antioxidant Response.

(A) p21 was upregulated in response to oxidative stress. HCT116-p21+/+ cells were either untreated or treated with the indicated compounds for 16 hrs. Endogenous p21 and β-actin were detected with anti-p21 and anti-β-actin antibodies (left panel). The band intensity of p21 and β-actin was quantified and the p21 protein level was normalized to the β-actin protein and plotted (right panel). (B) The basal and induced Nrf2 protein levels were lower in HCT116-p21-/- cells. HCT116-p21+/+ or HCT116-p21-/-cells were either untreated or treated with different doses of tBHQ or SF for 16 hrs. Endogenous Nrf2 was detected with an anti-Nrf2 antibody. Nrf2 appears as a double band because a lower percentage gel (7.5%) was used. (C) Knockdown of p21 diminished the transcriptional activity of Nrf2. HCT116-p21+/+ and HCT116-p21-/- cells were transfected with expression vectors for NQO1-ARE firefly luciferase and TK-renilla luciferase. The transfected cells were treated with the indicated concentration of tBHQ or SF for 16 hrs prior to the measurement of luciferase activities. The standard deviations were calculated from three independent experiments, each with duplicate samples. *p<0.05 compared with its control. (D) mRNA expression of the Nrf2-downstream genes were reduced in HCT116-p21-/- cells. HCT116-p21+/+ and HCT116-p21-/- cells were either left untreated, or treated with 50 μM tBHQ or 10 μM SF for 16 hrs. Relative amounts of NQO1, Gpx2, and Mrp2 mRNAs were measured by qRT-PCR. The standard deviations were calculated from triplicate samples. *p<0.05 compared with its control. (E) HCT116-p21-/- cells had lower NQO1 activity. NQO1 enzyme activity in HCT116-p21+/+ and HCT116-p21-/- cells was measured as the dicoumarol-inhibitable fraction of DCPIP reduction. NQO1 activity was normalized to the total protein level and the standard deviations were calculated from triplicate samples. *p<0.05 compared with its control.
p21 Directly Interacted with Nrf2
The interaction of these two proteins was explored in vivo and in vitro. p21-Myc and HA-Nrf2 were transiently expressed in COS-1 cells. p21-Myc was detected in the HA-immunoprecipitates (Fig 4a, left panel). In a reciprocal immunoprecipitation assay, HA-Nrf2 was present in the Myc-immunoprecipitates (Fig 4a, right panel), indicating the presence of both p21 and Nrf2 in the same complex in vivo. To test direct binding and to identify the Nrf2-binding domain in p21, a series of GST-tagged p21 deletion mutants were purified and incubated with the 35S-labeled Nrf2 protein. Direct interaction of these two proteins was detected (Figure 4B, lane 1-164). Based on the fact that p21 70-140 lost interaction with Nrf2, whereas p21 70-164 did not, it was deduced that the C-terminal domain of p21 containing 24 amino acids (from 140-164) is likely to be the interaction site for Nrf2 binding (Figure 4B). Reciprocally, GST-tagged Nrf2 was also able to pull-down 35S-labeled p21 protein, but not the negative control luciferase (Figure 4C). To identify the p21-binding domain in Nrf2, an immunoprecipitation analysis was performed with cell lysates coexpressing p21-Myc and each of the several HA-Nrf2 deletion mutants. All mutants retained their ability to bind p21, including the smallest one (the N-terminal 1-115), indicating that this N-terminal domain (1-115) contains a binding site for p21 (Figure 4D). More importantly, the interaction of endogenous Nrf2 with endogenous p21 was assessed in HCT116-p21+/+and MEF-Nrf2+/+ cells. The anti-p21 antibody, but not IgG or HA, immunoprecipitated Nrf2 in both HCT116-p21+/+ and MEF-Nrf2+/+ cells (Figures 4E and S5). Furthermore, there was an increased association of p21 with Nrf2 in response to oxidative stress, likely due to increased expression of Nrf2 and p21 in response to tBHQ treatment (Figure 4E). These results suggest that p21 upregulates the Nrf2 signaling pathway mainly through direct interaction with Nrf2 to enhance the protein level of Nrf2.
Figure 4. p21 Directly Interacted with Nrf2.

(A) p21 interacted with Nrf2. COS-1 cell lysates coexpressing p21-Myc and HA-Nrf2 were immunoprecipitated with anti-HA (left panel) or anti-Myc (right panel) antibodies. The total lysates (lower two panels) and the immunoprecipitates (upper two panels) were subjected to immunoblot analysis with both anti-HA and anti-Myc antibodies for detection of Nrf2 and p21. (B) The C-terminal domain of p21 was required for direct interaction with Nrf2. GST pull-down analysis were performed with GST-p21 fusion proteins containing different regions of p21 and the 35S-labeled Nrf2 protein. (C) p21 directly interacted with Nrf2. The GST-Nrf2 fusion protein was used to pulldown 35S-labeled p21. Luciferase was used as a negative control. (D) The N-terminal domain of Nrf2 interacted with p21. p21 and each of the Nrf2 C-terminal deletion mutants were coexpressed in COS-1 cells. The Nrf2-containing complexes were immunoprecipitated with HA-beads and blotted with anti-HA and anti-Myc antibodies for detection of Nrf2 and p21. Bands labeled with asterisks are Nrf2 and its deletion mutants. (E) Binding of endogenous p21 to Nrf2 was enhanced in response to oxidative stress. Cell lysates from HCT116-p21+/+cells untreated or treated with tBHQ were subjected to immunoprecipitation analysis with an anti-p21 antibody and then blotted with an anti-Nrf2 antibody (upper panel). An aliquot of total lysate was subjected to immunoblot analysis with anti-Nrf2, anti-p21 and anti-tubulin antibodies (lower three panels).
p21 Interfered with the Keap1-Dependent Ubiquitination of Nrf2 by Competing with Keap1 for Nrf2 Binding Mainly through the 29DLG Motif
The immunoprecipitation result indicates that the N-terminal domain (1-115) of Nrf2 interacts with p21 (Figure 4D). The N-terminal Neh2 domain of Nrf2 contains many functional motifs or residues that have been reported to be important for the function of Nrf2. In particular, it contains seven lysine residues that are required for ubiquitination and degradation of Nrf2. It also contains two Keap1 binding motifs, 29DLG and 79ETGE, that are crucial for Keap1-dependent ubiquitination and degradation of Nrf2. To further narrow down the precise amino acid residues in the Neh2 domain of Nrf2 that interact with p21, two Nrf2 mutants were generated: mDLG and mETGE in which the 29DLG or 79ETGE motif was replaced with alanine residues. Immunoprecipitation analyses were performed in COS-1 cells cotransfected with p21-Myc and Nrf2 wild-type or each of its mutants. In order to compare the binding affinity of Nrf2 to p21, with that of Nrf2 to Keap1, Nrf2 wild-type or each of its mutants was also coexpressed with the Kelch domain of Keap1 in another set of experiments. Both sets of samples were prepared and analyzed together using the immunoprecipitation/immunoblot method. Since mutation of 29DLG or 79ETGE affects Keap1 binding to Nrf2 and therefore, the stability of the Nrf2 proteins, the expression levels of Nrf2, mDLG, and mETGE differed significantly when equal amounts of Nrf2 were transfected. For that reason, the amounts of Nrf2 were adjusted to give rise to similar levels of Nrf2 protein in the total lysates (Figure 5A, lower panels). p21-Myc and Kelch-Myc were expressed at comparable levels when the total lysates were analyzed with an anti-Myc antibody (Figure 5A, lower anti-myc panel). All the lysates were immunoprecipitated with an anti-Nrf2 antibody and the immunoprecipitated proteins were subjected to immunoblot analysis with anti-myc for detection of p21-Myc and Kelch-Myc, and anti-HA to detect HA-Nrf2 (Figure 5A, upper panels). In agreement with the two-site model reported previously, mutation of 79ETGE significantly diminished the Nrf2-Kelch interaction while replacement of 29DLG with AAA weakened Nrf2 binding slightly (Figure 5A, compare lanes 8 and 7 with lane 6). Interestingly, the association of p21 to either of the Nrf2 mutants was weaker compared to Nrf2 wild-type, indicating that both 29DLG and 79ETGE are the interacting motifs for p21 (Figure 5A, compare lanes 3 and 4 with lane 2). It seems that the binding affinities of Nrf2- p21 and Nrf2-Kelch were comparable since similar amounts of p21-Myc and Kelch-Myc were immunoprecipitated by an anti-Nrf2 antibody. In contrast, the binding was weaker for mETGE to Kelch than mETGE to p21 (Figure 5A, compare lane 8 with lane 4), indicating that p21 may readily compete with Keap1 for the 29DLG site (latch). Next, a competition assay was carried out to examine if p21 is able to displace Keap1. Since ectopic coexpression of p21 changes Nrf2 levels, it is impossible to express equal amounts of Nrf2 among samples when different amounts of p21 are expressed. Therefore, the competition assay between p21 and Keap1 for Nrf2 binding was performed in vitro. HA-Nrf2, chitin binding domain (CBD) tagged Keap1, or p21-Myc was individually transfected into COS-1 cells. The same amount of lysate containing HA-Nrf2 and CBD-Keap1 was mixed with an increasing amount of p21-Myc lysate. The untransfected COS-1 lysate was used to match the total volume of the mixture. Keap1-containing complexes were pulled down by chitin beads and subjected to immunoblot analysis with anti-HA and anti-CBD antibodies. Our results show that p21 is inefficient in dissociating the Nrf2-Keap1 complex (Figure S6). We reasoned that p21 is probably able to compete for 29DLG binding, but not for 79ETGE due to the strong 79ETGE-Kelch interaction. In the hinge and latch model, it was proposed that disruption of the latch (29DLG-Kelch interaction) is sufficient to compromise the Keap1-dependent ubiquitination of Nrf2. It is conceivable that p21 interferes with the Keap1-dependent ubiquitination of Nrf2 by competing with Keap1 for Nrf2 binding through the 29DLG motif. Thus, endogenous Nrf2 ubiquitination in HCT116-p21+/+ and HCT116-p21-/- cells was measured under basal and stressed conditions. Cells were treated with a proteasome inhibitor, MG132, for 4 hours to block ubiquitinated Nrf2 from degradation. The results show a reduced basal level of ubiquitin-conjugated Nrf2 in HCT116-p21+/+ cells, compared to that in HCT116-p21-/- cells (Figure 5B). As expected, tBHQ was able to reduce ubiquitination of Nrf2 in both cell lines (Figure 5B). Next, the half-life of Nrf2 in HCT116-p21+/+ cells was compared to that in HCT116-p21-/- cells using cycloheximide (CHX) and immunoblot analysis. Nrf2 had a longer half-life in HCT116-p21+/+ cells than in HCT116-p21-/- cells (27.9 min. vs. 14.4 min in the untreated condition; 45.3 min. vs. 23.9 min in the tBHQ-treated condition) (Figure 5C). A similar result was obtained when a pulse-chase 35S-labeling method was utilized for measurement of the half-life of Nrf2 (Figure S7). Taken together, our data indicate that p21 is able to prevent ubiquitination of Nrf2 by disrupting the latch site (29DLG-Kelch) of the Nrf2-Keap1 interaction without displacement of Keap1 from the complex.
Figure 5. p21 Interfered with the Keap1-Dependent Ubiquitination of Nrf2 by Competing with Keap1 for Nrf2 Binding Mainly through the DLG Motif.

(A) p21 interacted with the DLG and ETGE motifs of Nrf2. Two Nrf2 mutants were generated: mDLG and mETGE in which the DLG or ETGE motif was replaced with alanine residues respectively. COS-1 cells were either cotransfected with expression vectors for the indicated Nrf2 protein and p21-Myc; or expression vectors for the indicated Nrf2 protein and Kelch-Myc. Transfected cells were immunoprecipitated with an anti-Nrf2 antibody and the immunoprecipitated proteins were subjected to immunoblot analysis with anti-HA and anti-Myc antibodies (upper panels). An aliquot of total lysate was analyzed with anti-HA, anti-Myc, and anti-tubulin antibodies (lower panels). (B) p21 reduced ubiquitination of Nrf2. HCT116-p21+/+ and HCT116-p21-/- cells were either untreated or treated with 100 μM tBHQ for 4 hrs in the presence of 10 μM of MG132, which blocks degradation of the ubiquitinated proteins. Cell lysates were subjected to an in vivo ubiquitination assay for detection of the ubiquitin-conjugated endogenous Nrf2 protein. Lysates were denatured and immunoprecipitated with an anti-Nrf2 or an anti-HA antibody (negative control), and blotted with an anti-ubiquitin antibody (upper panel). An aliquot of total lysate was analyzed for Nrf2 and GAPDH expression (lower panels). (C) p21 stabilized Nrf2 in basal and stressed conditions. HCT116-p21+/+ or HCT116-p21-/- cells were left untreated or pre-treated with 100 μm tBHQ for 4 h, followed by addition of 25 μM CHX and incubated for the time periods indicated. Endogenous Nrf2 was detected and the intensity of the Nrf2 bands was quantified and plotted on a semi-log graph. The amount of Nrf2 before addition of CHX was set as 1.
The C-terminal 154KRR in p21 was Essential for Binding and Upregulating Nrf2
29DLG and 79ETGE motifs bind to each of the Kelch domains from the Keap1 homodimer. The Kelch domain contains six conserved Kelch repeats that form a 6-bladed beta-propeller structure (Li et al., 2004; Padmanabhan et al., 2006). The arginine triad (R380, R415, and R483) located in the central channel of Keap1 was found to be important for interaction with either the 29DLG motif or the 79ETGE motif (McMahon et al., 2006; Tong et al., 2006; Tong et al., 2007). Since p21 does not have a rigid structure, but it binds to the same 29DLG or 79ETGE motif as Kelch does, it is possible that basic amino acid clusters consisting of arginine/lysine residues may be the Nrf2 interaction site in p21. In addition, the result from the GST pull-down experiment demonstrates that the 24 amino acids (140-164) in the C-terminus of p21 contained the Nrf2 binding site(s) (Figure 4B). Hence, we examined the arginine/lysine clusters within the 24 amino acids. Three clusters, 140RKRR, 154KRR, and 161KRK, were found. Accordingly, three p21 mutants (mRKRR, mKRR, and mKRK) were constructed in which arginine or lysine residues were replaced with methionine residues. Next, p21 wild-type or each of its mutants was coexpressed with HA-Nrf2 in COS-1 cells for immunoprecipitation analysis. Expression of p21 wild-type or its mutants was roughly equal (Figure 6A, lower panel). Remarkably, the Nrf2 level in the presence of mKRR was significantly lower and was comparable to the Nrf2 level in the absence of exogenous p21, while p21-WT or the other p21 mutants markedly enhanced the protein levels of Nrf2 (Figure 6A, lower panel, compare lanes 4 and 6, with other lanes). The result of the immunoprecipitation analysis indicates that mKRR lost interaction with Nrf2 (Figure 6A, upper panel, lane 4). Next, modulation of the Nrf2 transcriptional activity by p21 or its mutants was assessed in COS-1 cells. In concordance with the protein level of Nrf2, cotransfection of p21-WT, mRKRR, or mKRK significantly increased the luciferase activity (Figure 6B). In contrast, cotransfection of mKRR had no effect on the luciferase activity that was similar to the vector-cotransfected sample (Figure 6B). Combined with the data shown in Figure 5, these results demonstrate that direct interaction between Nrf2 and p21 requires the 29DLG and 79ETGE motifs in Nrf2 and the 154KRR cluster in p21. Next, cellular localization of Nrf2 and p21 was examined using indirect immunofluorescence staining. Nrf2 was primarily localized in the cytosol under basal conditions (Figure 6C, panel A). tBHQ and SF enhanced nuclear translocation of Nrf2 (Figure 6C, panels E and I). Heterogeneous localization of p21 was observed and the majority of cells had both cytoplasmic and nuclear localization (Fig 6C, panels B, F and J). In untreated cells, p21 and Nrf2 colocalized primarily in the cytoplasm, whereas in treated cells, there was more overlap staining in the nucleus, primarily due to enhanced nuclear translocation of Nrf2 (Fig 6C, panels D, H, and L). Localization of ectopically expressed p21 and Nrf2 were also determined. Ectopically expressed p21-WT had a similar staining pattern as endogenous p21, i.e. whole cell or whole cell with more nuclear staining (Fig 6D, panel B). mRKRR shifted staining more to the cytoplasm, resulting in whole cell staining (Fig 6D, panel F). Since 140RKRR has been identified as a classical nuclear localization signal (NLS) of p21(Child and Mann, 2006; Dotto, 2000), and also the size of p21 allows it to pass through the nuclear pore without the assistance of nuclear transporters, the slight shift to more cytoplasmic localization of mRKRR is expected. Interestingly, mKRR had a similar localization pattern as mRKRR (Figure 6D, panels F and J). At this point, the reason for this is still unclear. It may represent another part of the NLS since most classical NLSs are bipartite, meaning that two clusters of the basic amino acids are separated by several amino acids. mKRK had a cytoplasmic localization with a punctate staining pattern (Figure 6D, panel N). These data suggest that p21 mutants with more cytoplasmic localization are still capable of stabilizing Nrf2, except mKRR, which lost interaction with Nrf2. Therefore, it is most likely that competing with Keap1 for 29DLG binding occurs in the cytosol. This fits well with the established notion that Keap1-mediated Nrf2 ubiquitination and degradation occurs in the cytosol.
Figure 6. The C-terminal 154KRR in p21 was Essential for the Binding and Upregulation of Nrf2.

(A) mKRR in p21 interacted with Nrf2 and was required for upregulation of Nrf2. Three p21 mutants were made. mRKRR, mKRR, and mKRK are mutants with RKRR, KRR, or KRK replaced with methionine residues, respectively. COS-1 cells were cotransfected with expression vectors for Nrf2 and the indicated p21 protein. Transfected cells were subjected to immunoprecipitation with an anti-HA antibody. The immunoprecipitated proteins and an aliquot of total lysate were electrophoresed and analyzed with anti-HA and anti-Myc antibodies. (B) mKRR was unable to upregulate the transcriptional activity of Nrf2. COS-1 cells were transfected with the expression vector for the indicated p21 protein, along with expression vectors for the NQO1-ARE firefly luciferase and TK-renilla luciferase. The reporter gene analysis was performed as described in Fig 2C. All error bars indicate standard deviations calculated from three independent experiments. *p<0.05 compared with wild-type. (C) Localization of endogenous Nrf2 and p21. Nrf2 and p21 in MDA-MB-231 cells were determined by double-label indirect immunofluorescence with anti-Nrf2 and anti-p21 antibodies. Colocalization of Nrf2 and p21 is indicated by the presence of yellow in the merge images. (D) Localization of p21 and its mutants. NIH 3T3 cells were cotransfected with HA-Nrf2 and each of the Myc-tagged p21 proteins. Double-label indirect immunofluorescence was performed with anti-HA and anti-Myc antibodies for detection of Nrf2 and p21, respectively.
p21-Deficient Mice had Reduced Basal and Induced Levels of Nrf2 and Nrf2 Target Genes
p21-dependent Nrf2 upregulation under physiological conditions was confirmed using liver tissue from p21-deficient and p21 wild-type control mice that were either untreated or treated with tert-butyl-hydroxyanisole (BHA). BHA is metabolized into tBHQ following absorption and is a known chemical inducer of oxidative stress in animals (Hayes et al., 2000). As shown in Figure 7A, the basal Nrf2 level was slightly lower in p21-deficient mice than in wild-type mice. Strikingly, the protein level of Nrf2 enhanced by BHA was substantial in wild-type mice whereas BHA enhanced Nrf2 to a lesser extent in p21-deficient mice (Figure 7A). Consistent with the Nrf2 protein level, both NQO1 and HO-1 had lower basal and induced levels in p21-deficient mice, compared to p21 wild-type mice (Figure 7A). The induction of the Nrf2-dependent antioxidant response appears to be more substantial in animal models than in cultured cells. Collectively, these results provide solid evidence that upregulation of the Nrf2 antioxidant response by p21 under both basal and induced conditions does not only occur in cell models, but also under physiological conditions in animals.
Figure 7. p21-Deficient Mice had Reduced Basal and Induced Levels of Nrf2 and Nrf2 Target Genes.

(A) p21-deficient mice had reduced basal and induced levels of Nrf2 and Nrf2 target genes. Wild-type or p21-deficient mice (n=3) were treated with 350 mg/kg BHA for 12 hrs through intraperitoneal injection. Liver tissues were subjected to immunoblot analysis with anti-Nrf2, anti-NQO1, anti-HO-1, and anti-GAPDH antibodies. (B) A model by which p21 upregulates the Nrf2-dependent antioxidant response at both basal and induced conditions. Keap1, an E3 ubiquitin ligase containing the BTB and Kelch domains, constantly targets Nrf2 for ubiquitination and subsequent degradation under basal conditions. The binding of Keap1 to Nrf2 is through a hinge and latch mechanism, in which each Kelch domain from a Keap1 homodimer binds Nrf2 through two binding sites: a weak-binding DLG motif and a strong-binding ETGE motif. Binding of both sites are essential to present the seven ubiquitin-accepting lysine residues of Nrf2 in the correct orientation to accept ubiquitin, which targets Nrf2 for degradation. In response to oxidative stress, Keap1 is able to detect an imbalance in intracellular redox homeostasis through modification of its cysteine residues. The three important cysteine residues, C151, C273, and C288 are labeled. These modifications alter conformation of Keap1 and loosen the latch (DLG-Kelch), which puts p21 in a better position to compete with Keap1 for binding to the DLG motif. Once the latch is open, the Keap1-dependent ubiquitination of Nrf2 is compromised even though Keap1 still associates with Nrf2 through the hinge (ETGE). Thus, Nrf2 is stabilized and Nrf2-dependent cytoprotective genes are expressed under oxidative stress.
Discussion
In this report, we present our finding that p21 is able to upregulate the Nrf2 signaling pathway under both basal and induced conditions through direct interaction with Nrf2. Based on the genetic and molecular data obtained, we present a model that best explains how p21 is able to upregulate the Nrf2-dependent antioxidant response under both basal and induced conditions (Figure 7B). Keap1, an E3 ubiquitin ligase, constantly targets Nrf2 for ubiquitination and subsequent degradation under unstressed conditions. The binding of Keap1 to Nrf2 is through a hinge and latch mechanism, in which each Kelch domain from a Keap1 homodimer binds Nrf2 through two binding sites: a weak-binding 29DLG motif and a strong-binding 79ETGE motif. Binding of both sites are essential to present the seven ubiquitin-accepting lysine residues of Nrf2 in the correct orientation to accept ubiquitin, and thus targets Nrf2 for degradation. In response to oxidative stress, Keap1 is able to detect an imbalance in intracellular redox homeostasis through modification of its cysteine residues. These modifications alter its conformation and loosen the latch (29DLG-Kelch), which puts p21 in a better position to compete with Keap1 for binding to the 29DLG motif. Once the latch is open, the Keap1-dependent ubiquitination of Nrf2 is compromised even though Keap1 still associates with Nrf2 through the hinge (79ETGE). Thus, Nrf2 is stabilized and the Nrf2-dependent cytoprotective genes are expressed under oxidative stress (Fig 7B). It is noteworthy that our finding of p21-mediated upregulation of the Nrf2 signaling pathway should not devalue the importance of Keap1 in sensing and regulating Nrf2. Rather, this p21-Keap1 competition model requires the presence of Keap1 and provides solid support of the hinge and latch model. In general, our data indicate that p21 is able to upregulate the Nrf2-dependent antioxidant response more substantially under induced conditions compared to basal conditions. This is most likely due to increased p21 protein levels under induced conditions leading to additive effects.
p21 is the founding member of the cyclin-dependent kinase inhibitor (CKI) family, which also include p27 and p57. The N-terminal regions are well conserved among the three members, including the cyclin/CDK binding motifs (Gartel and Tyner, 2002). In our study, the Nrf2-interacting motif is mapped to the C-terminal 154KRR. The C-terminal sequences of the CKIs are poorly conserved among the family members, implying that upregulation of the Nrf2-dependent antioxidant response is likely specific to p21. A large body of literature shows that this motif may overlap with many other p21 interacting proteins identified so far, including the PCNA-interacting region (143-160), a second cyclin binding region (155-157), C8 α–subunit (140-164), Calmodulin (145-164), c-Myc (139-164), GADD45 (139-164), MDM2 (87-164), SET (145-164), TOK1 (149-164), HPV-16E7 (139-164), and C/EBP-α (84-164) (Child and Mann, 2006; Dotto, 2000; Gartel and Tyner, 2002). The physiological roles of this pleiotropic protein mainly rely on its cellular localization and its interacting proteins, which determine the ultimate fate of cells, such as proliferation, differentiation, cell-cycle arrest, and apoptosis. Although we did not address the interplay of these putative p21-interacting proteins in this study, the physiological significance of p21 in upregulating the Nrf2-dependent antioxidant response was confirmed in animals. Both the basal and induced Nrf2-antioxidant response was reduced substantially in p21-deficient mice, compared to wild-type. Thus, our finding that p21 upregulates the Nrf2-dependent antioxidant response pathway adds another dimension to the complexity of the p21-mediated signaling network.
Experimental Procedures
Antibodies, Transfection, Animal Treatment, Immunoblot Analysis, and Reporter Gene Assay
Anti-Nrf2, anti-Keap1, and anti-p21 (Santa Cruz); anti-HA beads (Sigma); anti-HA (Covance); Chitin beads and the anti-CBD (New England Biolab) were purchased from commercial sources. Transfection of cDNA was performed with Lipofectamine Plus (Invitrogen). Nrf2-siRNA and p21-siRNA were purchased from Qiagen. HiPerfect transfection reagent (Qiagen) was used to deliver siRNA. Male wild-type and p21-deficient mice were purchased from the Jackson Laboratory. Mice at 6 weeks of age were randomly assigned to BHA (350 mg/Kg in corn oil, i.p.) or control group (corn oil, i.p.), and euthanized at 12 hrs following administration. Livers were excised and subjected to immunoblot analysis. The detailed procedures are described in the supplement. Reporter assays were performed using the Promega dual-luciferase reporter gene assay system according to the manufacturer's instructions.
In vivo ubiquitination of Nrf2, GST pull-down, and Immunofluorescence assays
To detect endogenous Nrf2 that is ubiquitin-conjugated, cells were exposed to 10 μM MG132 (Sigma) for 4 prior to lysis. Cell lysates were subjected to immunoprecipitation with an anti-Nrf2 antibody and precipitated proteins were immunoblotted with an anti-Ub antibody (Sun et al., 2007). GST fusion proteins were expressed in E. coli and purified using GST beads (Amersham Biosciences). The 35S-labeled Nrf2 was generated by an in vitro transcription and translation kit (Promega). Colocalization of Nrf2 with p21 was detected using double-label indirect immunofluorescence with anti-p21 and anti-Nrf2 antibodies for detection of endogenous p21 and Nrf2; with anti-Myc and anti-HA antibodies to detecting ectopically expressed p21-Myc and HA-Nrf2 (Sun et al., 2007).
Cell Viability (MTT) and Cell-death assays
Cell viability was performed using MTT assay (Wang et al., 2007). For detection of H2O2-induced apoptotic cells, two methods were used: (i) transfected cells were treated with H2O2 and cells with the condensed chromatin were detected according to the reported method (Gong et al., 1999); (ii) apoptotic cells were detected using Annexin V-FITC apoptosis detection kit (Sigma) in combination with flow cytometry.
ROS detection, NQO1 activity, and qRT-PCR
To detecte ROS, cells were treated with H2O2 for 12 hrs and incubated with dichlorofluorescein (DCF) (Sigma) before measuring ROS by flow cytometry. NQO1 activity was measured as the dicoumarol-inhibitable fraction of DCPIP reduction described previously (Wang et al., 2007). Total mRNA extraction and qRT-PCR analysis were performed as described in the supplement and reported previously (Wang et al., 2008).
Protein half-life analysis
The half-life of Nrf2 was measured using both cycloheximide/immunoblot and pulse-chase 35S-labeling methods as described in the supplement.
Statistical Analysis
Experiments were conducted in triplicate and data are shown as mean ± SD. Statistical analysis was performed using two-tailed Student's t-tests to compare means. Significance was set at the p value <0.05.
Supplementary Material
Acknowledgments
We thank Dr. Masayuki Yamamoto for the MEF Keap1+/+ and Keap1-/- cells; Dr. Jeff Chan for the MEF Nrf2+/+ and Nrf2-/- cells; Dr. Bert Vogelstein and Dr. Kenneth W. Kinzler for HCT116-p21+/+ and p21-/- cells. We also thank Dr. Stuart A. Aaronson and Dr. Salvador Macip for helpful discussion, and Drs. John Regan and Cathy Smith for thoughtful comments. We thank Nicole Villeneuve and Alexandria Lau for critical reading of the manuscript. This work was supported by research grants from NIEHS (ES015010) and American Cancer Society (RSG-07-154) awarded to D. D. Zhang, and ES006694.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aoki Y, Sato H, Nishimura N, Takahashi S, Itoh K, Yamamoto M. Accelerated DNA adduct formation in the lung of the Nrf2 knockout mouse exposed to diesel exhaust. Toxicol Appl Pharmacol. 2001;173:154–160. doi: 10.1006/taap.2001.9176. [DOI] [PubMed] [Google Scholar]
- Child ES, Mann DJ. The intricacies of p21 phosphorylation: protein/protein interactions, subcellular localization and stability. Cell Cycle. 2006;5:1313–1319. doi: 10.4161/cc.5.12.2863. [DOI] [PubMed] [Google Scholar]
- Cho HY, Reddy SP, Yamamoto M, Kleeberger SR. The transcription factor NRF2 protects against pulmonary fibrosis. Faseb J. 2004;18:1258–1260. doi: 10.1096/fj.03-1127fje. [DOI] [PubMed] [Google Scholar]
- Dotto GP. p21(WAF1/Cip1): more than a break to the cell cycle? Biochim Biophys Acta. 2000;1471:M43–56. doi: 10.1016/s0304-419x(00)00019-6. [DOI] [PubMed] [Google Scholar]
- Esposito F, Cuccovillo F, Russo L, Casella F, Russo T, Cimino F. A new p21waf1/cip1 isoform is an early event of cell response to oxidative stress. Cell Death Differ. 1998;5:940–945. doi: 10.1038/sj.cdd.4400427. [DOI] [PubMed] [Google Scholar]
- Gartel AL, Tyner AL. The role of the cyclin-dependent kinase inhibitor p21 in apoptosis. Mol Cancer Ther. 2002;1:639–649. [PubMed] [Google Scholar]
- Gong JG, Costanzo A, Yang HQ, Melino G, Kaelin WG, Jr, Levrero M, Wang JY. The tyrosine kinase c-Abl regulates p73 in apoptotic response to cisplatin-induced DNA damage. Nature. 1999;399:806–809. doi: 10.1038/21690. [DOI] [PubMed] [Google Scholar]
- Hayes JD, Chanas SA, Henderson CJ, McMahon M, Sun C, Moffat GJ, Wolf CR, Yamamoto M. The Nrf2 transcription factor contributes both to the basal expression of glutathione S-transferases in mouse liver and to their induction by the chemopreventive synthetic antioxidants, butylated hydroxyanisole and ethoxyquin. Biochem Soc Trans. 2000;28:33–41. doi: 10.1042/bst0280033. [DOI] [PubMed] [Google Scholar]
- Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, Yamamoto M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999;13:76–86. doi: 10.1101/gad.13.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. 2007;47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- Kobayashi A, Kang MI, Okawa H, Ohtsuji M, Zenke Y, Chiba T, Igarashi K, Yamamoto M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol. 2004;24:7130–7139. doi: 10.1128/MCB.24.16.7130-7139.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi A, Kang MI, Watai Y, Tong KI, Shibata T, Uchida K, Yamamoto M. Oxidative and electrophilic stresses activate Nrf2 through inhibition of ubiquitination activity of Keap1. Mol Cell Biol. 2006;26:221–229. doi: 10.1128/MCB.26.1.221-229.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi M, Yamamoto M. Molecular mechanisms activating the Nrf2-Keap1 pathway of antioxidant gene regulation. Antioxid Redox Signal. 2005;7:385–394. doi: 10.1089/ars.2005.7.385. [DOI] [PubMed] [Google Scholar]
- Lau A, Villeneuve NF, Sun Z, Wong PK, Zhang DD. Dual roles of Nrf2 in cancer. Pharmacol Res. 2008;58:262–270. doi: 10.1016/j.phrs.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Waga S, Hannon GJ, Beach D, Stillman B. Differential effects by the p21 CDK inhibitor on PCNA-dependent DNA replication and repair. Nature. 1994;371:534–537. doi: 10.1038/371534a0. [DOI] [PubMed] [Google Scholar]
- Li X, Zhang DD, Hannink M, Beamer LJ. Crystal structure of the Kelch domain of human Keap1. J Biol Chem. 2004;279:54750–54758. doi: 10.1074/jbc.M410073200. [DOI] [PubMed] [Google Scholar]
- McMahon M, Itoh K, Yamamoto M, Hayes JD. Keap1-dependent proteasomal degradation of transcription factor Nrf2 contributes to the negative regulation of antioxidant response element-driven gene expression. J Biol Chem. 2003;278:21592–21600. doi: 10.1074/jbc.M300931200. [DOI] [PubMed] [Google Scholar]
- McMahon M, Thomas N, Itoh K, Yamamoto M, Hayes JD. Dimerization of substrate adaptors can facilitate cullin-mediated ubiquitylation of proteins by a “tethering” mechanism: a two-site interaction model for the Nrf2-Keap1 complex. J Biol Chem. 2006;281:24756–24768. doi: 10.1074/jbc.M601119200. [DOI] [PubMed] [Google Scholar]
- O'Reilly MA. Redox activation of p21Cip1/WAF1/Sdi1: a multifunctional regulator of cell survival and death. Antioxid Redox Signal. 2005;7:108–118. doi: 10.1089/ars.2005.7.108. [DOI] [PubMed] [Google Scholar]
- O'Reilly MA, Staversky RJ, Watkins RH, Reed CK, de Mesy Jensen KL, Finkelstein JN, Keng PC. The cyclin-dependent kinase inhibitor p21 protects the lung from oxidative stress. Am J Respir Cell Mol Biol. 2001;24:703–710. doi: 10.1165/ajrcmb.24.6.4355. [DOI] [PubMed] [Google Scholar]
- Padmanabhan B, Tong KI, Ohta T, Nakamura Y, Scharlock M, Ohtsuji M, Kang MI, Kobayashi A, Yokoyama S, Yamamoto M. Structural basis for defects of Keap1 activity provoked by its point mutations in lung cancer. Mol Cell. 2006;21:689–700. doi: 10.1016/j.molcel.2006.01.013. [DOI] [PubMed] [Google Scholar]
- Poon RY, Hunter T. Expression of a novel form of p21Cip1/Waf1 in UV-irradiated and transformed cells. Oncogene. 1998;16:1333–1343. doi: 10.1038/sj.onc.1201897. [DOI] [PubMed] [Google Scholar]
- Ramos-Gomez M, Kwak MK, Dolan PM, Itoh K, Yamamoto M, Talalay P, Kensler TW. Sensitivity to carcinogenesis is increased and chemoprotective efficacy of enzyme inducers is lost in nrf2 transcription factor-deficient mice. Proc Natl Acad Sci U S A. 2001;98:3410–3415. doi: 10.1073/pnas.051618798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z, Zhang S, Chan JY, Zhang DD. Keap1 controls postinduction repression of the Nrf2-mediated antioxidant response by escorting nuclear export of Nrf2. Mol Cell Biol. 2007;27:6334–6349. doi: 10.1128/MCB.00630-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong KI, Katoh Y, Kusunoki H, Itoh K, Tanaka T, Yamamoto M. Keap1 recruits Neh2 through binding to ETGE and DLG motifs: characterization of the two-site molecular recognition model. Mol Cell Biol. 2006;26:2887–2900. doi: 10.1128/MCB.26.8.2887-2900.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong KI, Padmanabhan B, Kobayashi A, Shang C, Hirotsu Y, Yokoyama S, Yamamoto M. Different electrostatic potentials define ETGE and DLG motifs as hinge and latch in oxidative stress response. Mol Cell Biol. 2007;27:7511–7521. doi: 10.1128/MCB.00753-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakabayashi N, Dinkova-Kostova AT, Holtzclaw WD, Kang MI, Kobayashi A, Yamamoto M, Kensler TW, Talalay P. Protection against electrophile and oxidant stress by induction of the phase 2 response: fate of cysteines of the Keap1 sensor modified by inducers. Proc Natl Acad Sci U S A. 2004;101:2040–2045. doi: 10.1073/pnas.0307301101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XJ, Sun Z, Chen W, Eblin KE, Gandolfi JA, Zhang DD. Nrf2 protects human bladder urothelial cells from arsenite and monomethylarsonous acid toxicity. Toxicol Appl Pharmacol. 2007;225:206–213. doi: 10.1016/j.taap.2007.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XJ, Sun Z, Villeneuve NF, Zhang S, Zhao F, Li Y, Chen W, Yi X, Zheng W, Wondrak GT, et al. Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs, the dark side of Nrf2. Carcinogenesis. 2008;29:1235–1243. doi: 10.1093/carcin/bgn095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto T, Suzuki T, Kobayashi A, Wakabayashi J, Maher J, Motohashi H, Yamamoto M. Physiological significance of reactive cysteine residues of Keap1 in determining Nrf2 activity. Mol Cell Biol. 2008;28:2758–2770. doi: 10.1128/MCB.01704-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang DD. Mechanistic studies of the Nrf2-Keap1 signaling pathway. Drug Metab Rev. 2006;38:769–789. doi: 10.1080/03602530600971974. [DOI] [PubMed] [Google Scholar]
- Zhang DD, Hannink M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol Cell Biol. 2003;23:8137–8151. doi: 10.1128/MCB.23.22.8137-8151.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang DD, Lo SC, Cross JV, Templeton DJ, Hannink M. Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol Cell Biol. 2004;24:10941–10953. doi: 10.1128/MCB.24.24.10941-10953.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zipper LM, Mulcahy RT. The Keap1 BTB/POZ dimerization function is required to sequester Nrf2 in cytoplasm. J Biol Chem. 2002;277:36544–36552. doi: 10.1074/jbc.M206530200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.