

Abstract
Background: Chondrocyte death has been linked to injury-induced oxidative damage, suggesting that antioxidants could substantially improve viability. However, since reactive oxygen species play roles in normal physiology, there are concerns that antioxidants may have deleterious side effects. To address these issues, we studied the effects of N-acetylcysteine, a potent free radical scavenger, on chondrocyte viability and cartilage proteoglycan content in an in vitro cartilage injury model. We hypothesized that treatment with N-acetylcysteine soon after an impact injury would have significant chondrocyte-sparing effects and would prevent injury-induced proteoglycan losses.
Methods: Bovine osteochondral explants were subjected to a single impact load with use of a drop-tower device. Chondrocyte viability was measured at multiple time points post-impact with use of fluorescent probes and confocal microscopy. Forty-eight hours after impact, the effects on viability of immediate post-impact treatment with N-acetylcysteine were compared with the effects of the caspase inhibitor N-CBZ-Val-Ala-Asp(O-Me) fluoromethyl ketone and those of the cell-membrane-stabilizing surfactant poloxamer 188. The effect of N-acetylcysteine on proteoglycan content was determined at seven and fourteen days post-impact.
Results: Chondrocyte viability declined sharply within an hour and reached a steady state within six to twelve hours after impact. Immediate treatment with N-acetylcysteine doubled the number of viable chondrocytes assayed forty-eight hours after impact, and this effect was significantly greater than that of N-CBZ-Val-Ala-Asp(O-Me) fluoromethyl ketone. Even when N-acetylcysteine treatment was delayed for up to four hours after injury, it still had significant positive effects on cell viability at forty-eight hours. Moreover, N-acetylcysteine treatment significantly improved proteoglycan content at the impact sites at both seven and fourteen days after injury.
Conclusions: Treatment with N-acetylcysteine soon after a blunt impact injury can reduce chondrocyte death and proteoglycan loss measured seven to fourteen days after injury.
Clinical Relevance: These findings suggest that antioxidant protection in the early aftermath of joint injury may help to reduce chondrocyte death and stabilize the articular cartilage.
Articular fractures necessarily subject articular cartilage to high-energy impact loading. Cartilage damage caused by a single impact is thought to be a major risk factor for posttraumatic osteoarthritis1. One pathoetiologic component of impact injury is chondrocyte death1. At the high loading rates characteristic of impact injuries, local strains in the superficial zone can exceed 40%. There, matrix fissuring and chondrocyte death are evident post-impact1-5. Several studies have shown that chondrocytes begin to die within moments after impact and continue to die for up to several days6-10. Studies involving use of the cell-membrane-stabilizing surfactant poloxamer 188, a necrosis inhibitor, have indicated that up to 15% of chondrocytes at impact sites succumb to necrosis soon after injury6,7,11. However, chondrocyte apoptosis is the predominant mode of death in the hours and days after impact injury12.
Chondrocyte death in injured cartilage can be dramatically reduced by apoptosis inhibitors9,13. Treatment of injured cartilage with N-CBZ-Val-Ala-Asp(O-Me) fluoromethyl ketone (z-VAD-fmk), a pan-caspase inhibitor, improves cell viability, inhibits proteoglycan loss, and ameliorates the progression of osteoarthritis in animal models14. These findings associating chondrocyte death with cartilage degeneration suggest that there is a window of opportunity to prevent chondrocyte depletion in the early aftermath of injury.
Previous work in our laboratory revealed that a variety of antioxidants including N-acetylcysteine, superoxide dismutase, and vitamin E can prevent up to 80% of chondrocyte death associated with injurious cyclic compression15. In addition, superoxide dismutase has been shown to block chondrocyte death induced by impact loading16. These studies indicate a role for reactive oxygen species in impact-associated chondrocyte apoptosis.
Previous studies have suggested that superoxide radicals released by mitochondria cause oxidative damage and apoptosis of cells exposed to a variety of stresses9,17-19. Superoxide may be released from the NADH (nicotinamide adenine dinucleotide)-coenzyme Q1 reductase in complex I of the mitochondrial electron transport chain20-23. This subunit is specifically inhibited by rotenone, which has been shown to reduce superoxide release after an insult such as serum deprivation20,24-26. Rotenone blocked mitogen-activated kinase activation and catabolic gene induction in chondrocytes stimulated with fibronectin fragments27. Moreover, production of oligomycin-sensitive reactive oxygen species in chondrocytes plated on polylysine is suppressed by intact fibronectin28. These two related studies suggest that disruption of integrin-matrix interactions induces production of mitochondrial reactive oxygen species.
N-acetylcysteine is a powerful scavenger of highly damaging hydroxyl and hypochlorous radicals, which are formed in cells following exposure to superoxide29. Its efficient free radical scavenging activity and well-characterized biocompatibility make N-acetylcysteine an attractive candidate for cytotherapy following joint injury. We hypothesized that treatment with N-acetylcysteine soon after an impact injury would have significant chondrocyte-sparing effects and would prevent injury-induced proteoglycan loss. To test this, we evaluated the effects of N-acetylcysteine on chondrocyte viability and proteoglycan content after a blunt impact injury in a bovine osteochondral explant model. The time course of impact-induced chondrocyte death and the effects of delaying the initiation of the N-acetylcysteine treatment were investigated to determine the window of opportunity for preserving chondrocyte viability.
Materials and Methods
Specimen Preparation
Mature bovine stifle joints were obtained immediately after slaughter from a local abattoir. Osteochondral explants were prepared by manually sawing a 25 by 25-mm square from the lateral tibial plateau, which included the central loaded area of the articular surface that was not covered by menisci. Subchondral bone was cut from the underside of each specimen to a thickness of 4 to 8 mm. One explant was cut from one stifle joint, and each joint was obtained from a different animal. The explants were placed immediately in culture medium containing 45% Dulbecco modified Eagle medium, 45% Ham F-12, and 10% fetal bovine serum (Invitrogen, Carlsbad, California) and incubated at 37°C in an atmosphere of 5% CO2 in air.
Impact Loading
The osteochondral explants were secured into custom testing fixtures for impact loading and were kept submerged in culture medium at all times. A drop tower was used to impart loads to an indenter resting on the explant surface. The indenter was a flat-faced 5.0-mm-diameter brass rod with rounded edges (radius = 1 mm). Impact energy was modulated by dropping a 2-kg mass from a height of 14 or 7 cm, which resulted in impact energy densities of 14 J/cm2 and 7 J/cm2. The mass was removed from the indenter immediately after impact.
Viability Analysis
Explants were stained in culture medium for one hour with calcein acetoxymethylester (calcein AM) and ethidium homodimer-2 (Invitrogen), at a concentration of 1.0 mM each. Calcein AM is metabolized in living cells to form a bright green fluorescent product that accumulates in the cytosol. Ethidium homodimer is a red fluorophore that stains the DNA of nonviable cells but cannot penetrate living cells with intact plasma membranes. The staining medium was aspirated, and new medium was added to wash off any residual stain on the explant surface before fluorescent illumination. Confocal microscopy was performed on a BioRad 1024 confocal microscope equipped with a krypton-argon laser (BioRad, Hercules, California). Explants were scanned at wavelengths of 568 and 488 nm with use of a 10× objective with a field size of ∼1.0 mm2. More than 2400 cells were counted in each explant. Z-axis projections were assembled from images made from the cartilage surface to a depth of 200 μm at 40-μm intervals. Three different projections were recorded within each impact or nonimpact control site. The three ∼1.0-mm2 sites imaged for each specimen represented 15% of the ∼20 mm2 area of the impact sites. ImageJ software (National Institutes of Health, Bethesda, Maryland) was used for automated counting of red and green-stained chondrocytes in each projection and the percentage of viable cells was calculated (green-stained chondrocytes/[red + green-stained chondrocytes] × 100). At least 800 cells were counted in each z-axis projection; thus, counts for each explant totaled more than 2400 cells. The percentage of viable cells for each specimen was calculated by averaging the values for the three different projections, each composed of six planes. Thus, each viability determination is based on eighteen images. The average group viabilities for the time-course studies are based on three explants multiplied by eighteen images per explant, for a total of fifty-four images, whereas the average group viabilities in the drug comparison studies are based on six explants multiplied by eighteen images, for a total of 108 images.
Proteoglycan Content
Cartilage proteoglycan content was measured by glycosaminoglycan assay with use of dimethyl methylene blue (Sigma-Aldrich, St. Louis, Missouri) essentially as previously described30. The assay was performed on papain digests of full-thickness cartilage samples with use of shark chondroitin sulfate (Sigma-Aldrich) as a standard. Samples were dissected from explant surfaces with use of a 4-mm-diameter dermal punch and an osteotome. On the basis of pilot studies indicating substantial interexplant variability in glycosaminoglycan content, glycosaminoglycan concentration (μg of glycosaminoglycan/mg wet weight) at impact sites was normalized to the concentration at nearby, nonimpacted (control) sites in the same explant and reported in terms of the percentage of the control value (glycosaminoglycanimpact/glycosaminoglycancontrol × 100). Six explants were assayed for each treatment group.
Testing Protocols
The details for each of the tests described below are presented in Table I.
TABLE I.
Experimental Design
| Time Post-Impact | Impact Energy (J/cm2) | Treatment | No. of Specimens |
|---|---|---|---|
| Test 1 | 48 | ||
| 1 hr | 7/14 | None | 3/3 |
| 2 hr | 7/14 | None | 3/3 |
| 4 hr | 7/14 | None | 3/3 |
| 6 hr | 7/14 | None | 3/3 |
| 12 hr | 7/14 | None | 3/3 |
| 24 hr | 7/14 | None | 3/3 |
| 48 hr | 7/14 | None | 3/3 |
| 72 hr | 7/14 | None | 3/3 |
| Test 2 | 24 | ||
| 48 hr | 7 | Control | 6 |
| 48 hr | 7 | N-acetylcysteine | 6 |
| 48 hr | 7 | Tocopherol | 6 |
| 48 hr | 7 | Poloxamer-188 | 6 |
| Test 3 | 30 | ||
| 48 hr | 7 | N-acetylcysteine, 1-hr delay | 6 |
| 48 hr | 7 | N-acetylcysteine, 2-hr delay | 6 |
| 48 hr | 7 | N-acetylcysteine, 3-hr delay | 6 |
| 48 hr | 7 | N-acetylcysteine, 4-hr delay | 6 |
| 48 hr | 7 | N-acetylcysteine, 12-hr delay | 6 |
| Test 4 | 24 | ||
| 7 days | 7 | N-acetylcysteine | 6 |
| 7 days | 7 | Control | 6 |
| 14 days | 7 | N-acetylcysteine | 6 |
| 14 days | 7 | Control | 6 |
Test 1: Effect of Impact Energy on Severity and Timing of Chondrocyte Death
The first test was designed to determine how the amount of impact energy affected the severity and time course of chondrocyte death following an impact injury. Specimens were impacted with 7 or 14 J/cm2, and chondrocyte viability was determined as detailed above. After being placed in medium, three nonimpacted specimens were analyzed at one, twelve, and seventy-two hours, for a total of nine nonimpacted specimens. Three impacted specimens were analyzed after impact at each impact energy level, for a total of forty-eight specimens.
Test 2: Effect of Treatment of Impacted Specimens with N-Acetylcysteine, Poloxamer 188, α and γ Tocopherol, and z-VAD-fmk
On the basis of the results from test 1, the 7-J/cm2 energy level was selected for subsequent testing. Both impact energy levels resulted in chondrocyte death, but the specimens impacted at the 14-J/cm2 level had greater surface damage and irregularity, making assessment with confocal microscopy more difficult.
In test 2, specimens were treated with drugs at concentrations previously shown to inhibit mechanically induced chondrocyte death. N-acetylcysteine (Sigma-Aldrich) was used at a concentration of 2.0 mM15, α and γ tocopherol (Sigma-Aldrich) were mixed for use at concentrations of 25 μM each31, poloxamer 188 (Sigma-Aldrich) was used at a concentration of 8 mg/mL7, and z-VAD-fmk (Calbiochem, San Diego, California) was used at a concentration of 100 μM32. Impacted specimens were treated within five minutes post-impact (immediate treatment) and incubated for forty-eight hours before the viability analysis. Six specimens were tested for each therapeutic intervention, for a total of twenty-four specimens.
Test 3: Effect of Timing of N-Acetylcysteine Treatment on Chondrocyte Viability
In this test, the effect of the timing of the treatment with N-acetylcysteine was determined in specimens subjected to an impact of 7 J/cm2. N-acetylcysteine (2.0 mM) was applied to specimens at one, two, three, four, and twelve hours after impact. Viability was determined forty-eight hours after impact. Six specimens were tested at each interval, for a total of thirty specimens.
Test 4: Effect of N-Acetylcysteine on Proteoglycan Content
Explants subjected to an impact of 7 J/cm2 were treated immediately after the impact with 2 mM N-acetylcysteine (six specimens) or were left untreated (six specimens). After twenty-four hours of incubation, the medium was replaced with fresh standard medium not containing N-acetylcysteine. Cartilage was harvested from the impact and control sites seven or fourteen days post-impact and was digested in papain. The glycosaminoglycan concentration in papain digests of each explant was assayed in triplicate.
Data Analysis
The significance of treatment-related differences in viability was assessed with one-way analysis of variance with the Tukey test for multiple comparisons. With the assumption of a minimum difference between group means of 0.15, a population standard deviation of 0.10, and alpha = 0.05, the power of the tests performed was >0.8.
Source of Funding
This work was supported by the National Institutes of Health/National Institute of Arthritis and Musculoskeletal and Skin Diseases Grant 1 P50 AR055533.
Results
Structural damage to the cartilage extracellular matrix was evident immediately after impact at both impact-energy levels (7 and 14-J/cm2) (Fig. 1). Disruption of the superficial zone in the form of delamination and cracks extending from the surface toward the transitional zone were observed in both groups. However, the damage from the 14-J/cm2 impacts tended to be more severe, with greater disruption of the superficial zone than was seen in the 7-J/cm2 group.
Fig. 1.
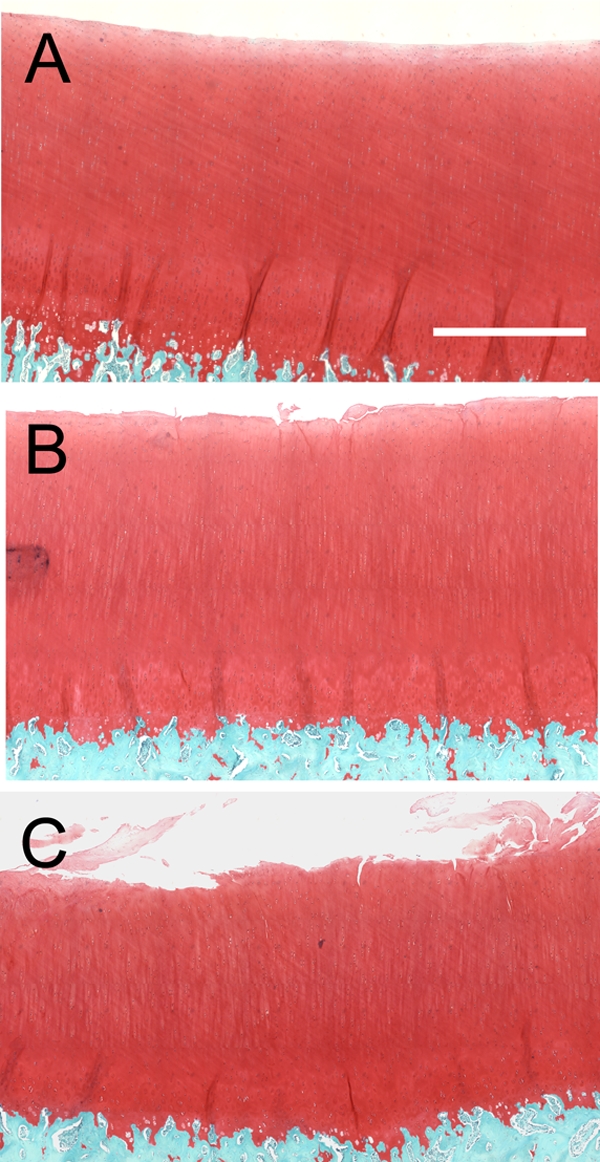
Structural damage from impact injuries. Micrographs show sections from a nonimpacted control cartilage explant (A), a 7-J/cm2-impact site (B), and a 14-J/cm2-impact site (C) (safranin-O and fast green). The bar in A is 0.5 mm long.
Figure 2 shows typical z-axis reconstructions of images made with confocal microscopic imaging at impacted and nonimpacted sites after use of calcein AM and ethidium homodimer stains as live and dead-cell probes, respectively. Although cell viability in nonimpacted controls remained stable, at nearly 90%, after seventy-two hours in culture, there were dramatic time-dependent increases in cell death in the impacted cartilage (Fig. 3). The viability in the 7 and 14-J/cm2 groups decreased to 45% and 35%, respectively. Greater than 85% of the chondrocyte death occurred within the first six to twelve hours after impact loading, with little additional accumulation of dead cells for up to seventy-two hours. In the 14-J/cm2 group, the number of dead chondrocytes was nearly maximal after only six hours. In contrast, the number of dead cells in the 7-J/cm2 group did not reach a maximum until twelve hours post-impact. Thus, both the magnitude and the kinetics of cell death varied with impact energy.
Fig. 2.

Laser confocal imaging of viability stains. A nonimpacted control cartilage explant (A), an explant subjected to a 7-J/cm2 impact (B), and an explant subjected to a 7-J/cm2 impact and treated immediately with N-acetylcysteine were stained with calcein AM (green) and ethidium homodimer (red) to label live and dead cells, respectively. The micrographs are z-axis reconstructions of images made at the surface and to 200 μm beneath the surface at intervals of 40 μm The bar in C represents 200 μm.
Fig. 3.

Time course of chondrocyte death after impact injury. Cell viability was evaluated in nonimpacted control explants (open triangles) and explants subjected to an impact of 7 J/cm2 (open circles) or 14 J/cm2 (filled squares). Each symbol represents the average of the results for three explants. The error bars indicate standard deviations.
Treatment with N-acetylcysteine immediately after impact improved viability at forty-eight hours, a time when death had reached a steady state. The improvement was from 35% (without treatment) to 64% (with N-acetylcysteine treatment) after a 14-J/cm2 injury and from 40% to 72% after a 7-J/cm2 injury (Fig. 4). These effects were significant (p = 0.001). Because similar N-acetylcysteine effects were observed in the 7 and 14-J/cm2 groups and because the highly irregular surfaces produced by the 14-J/cm2 injuries were difficult to image with confocal microscopy, we chose the 7-J/cm2 group for further study. Although significant numbers of chondrocytes were spared when N-acetylcysteine was used, treatment with tocopherol had no significant effect (p = 0.169). The anti-necrosis agent poloxamer 188 also did not result in sparing of significant numbers of chondrocytes (p = 0.9). The caspase inhibitor and anti-apoptosis agent z-VAD-fmk improved viability to 59%, which was a significant improvement compared with that in the untreated impacted controls (p < 0.001). However, viability in the z-VAD-fmk group was significantly lower than that in the N-acetylcysteine group (p < 0.001).
Fig. 4.

Effects of drug treatment on post-impact chondrocyte viability. Cell viability was evaluated in nonimpacted control explants (Con), explants subjected to an impact of 7 J/cm2 without drug treatment (No Tx), and explants subjected to an impact of 7 J/cm2 and treated immediately with N-acetylcysteine (NAC), tocopherol (Toc), poloxamer 188 (P188), or z-VAD-fmk (Z-Vad). Columns and error bars represent the means and standard deviations based on six explants.
Analyses performed forty-eight hours after a 7-J/cm2 impact revealed that 74% ± 7% of the cells (mean and standard deviation) were viable after N-acetylcysteine treatment that had been delayed for one hour after impact; 68% ± 5% of the cells remained viable with a two-hour delay; 67% ± 8%, with a three-hour delay; and 59% ± 6%, with a four-hour delay (Fig. 5). These values were all significantly greater than those for the untreated controls (p < 0.05). However, when the delay was increased to twelve hours, viability fell to 39% ± 6%, which was not significantly higher than the viability in the untreated group (p = 0.107).
Fig. 5.

Effect of delaying N-acetylcysteine treatment. Explants were subjected to an impact of 7 J/cm2 and were left untreated (No Tx) or were treated with N-acetylcysteine immediately (0) or after a delay of one, two, three, four, or twelve hours. Columns and error bars indicate means and standard deviations based on three explants.
At seven days after the impact injury, the glycosaminoglycan content at the impact site, calculated as the percentage of the value at the adjacent control site, averaged 81% ± 9% in the untreated explants and 103% ± 10% in the N-acetylcysteine-treated explants, a highly significant difference (p = 0.005) (Fig. 6). At fourteen days, the values at the impacted sites averaged 88% ± 10% in the untreated explants and 104% ± 10% in the N-acetylcysteine-treated explants; this difference was also significant (p = 0.001).
Fig. 6.

Effects of N-acetylcysteine on glycosaminoglycan (GAG) content seven and fourteen days after injury. Glycosaminoglycan content within each impact site was normalized to the glycosaminoglycan content of an adjacent, nonimpacted control site (percent of the control value). Results are shown for explants that were treated with N-acetylcysteine (NAC) and for untreated controls (Con). The columns and error bars indicate means and standard deviations based on six explants. The numbers above the bars are p values, which indicate significant differences between the N-acetylcysteine-treated and untreated specimens.
Discussion
Our results strongly support the hypothesis that N-acetylcysteine can help preserve chondrocytes following impact injury to articular cartilage. Moreover, we found that treatment with N-acetylcysteine significantly inhibited matrix glycosaminoglycan loss at impact sites for up to fourteen days following injury, an indication that this agent may have long-term beneficial effects beyond acute preservation of viability.
The effects of N-acetylcysteine were compared with those of z-VAD-fmk, which had been previously shown to decrease chondrocyte death induced by mechanical injury. We found a reduction in death from 60% (no treatment) to 41% in z-VAD-fmk-treated explants, which was a somewhat smaller effect than that shown by D'Lima et al., who observed that z-VAD-fmk reduced apoptosis from 65% to 31% in bovine explants at forty-eight hours post-impact (the same time point that we used)32. This difference might be attributable to the different methods used to measure cell death (superficial viability stains versus full-depth TUNEL [terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling] staining) or to differences in the mechanical injuries produced in the two studies. Drop towers such as the one used in our experiments load explants at rates of >1000 MPa/sec, whereas the injury described by D'Lima et al. was delivered at a much lower rate of 46 MPa/sec. These differences would be expected to lead to differences in the kinetics of accumulation or distribution of dead cells that could result in differences in the apparent magnitude of z-VAD-fmk effects.
The effects of N-acetylcysteine were also compared with those of poloxamer 188, which has been shown to reduce chondrocyte death by 10% to 15% in explants subjected to mechanical injury11. However, in that study, poloxamer 188 was effective as a post-impact treatment only if explants were manually “pumped” by gentle cyclic compression. We did not perform this step since it seemed impractical in a clinical context. Thus, the fact that we saw little effect of poloxamer 188 on death rates in our system was actually consistent with the previous study showing no effect without cyclic compression.
The N-acetylcysteine had to have been administered within four hours after the impact to significantly decrease impact-associated chondrocyte death. Our experiment showed that the impact energy had a modest effect on both the quantity and the timing of injury-related chondrocyte death. As expected, higher-energy impacts caused a greater quantity of chondrocyte death, with 35% viability in the 14-J/cm2-impact specimens compared with 45% viability in the 7-J/cm2-impact specimens. The results also showed that steady-state cell death occurred after six hours in the specimens subjected to the higher-energy impact compared with twelve hours in those subjected to the lower-energy impact.
Drop-tower devices reliably achieve stress rates (>1000 MPa/sec) comparable with the high-energy impacts sustained in automobile accidents that may result in joint fractures33,34. The injuries that we studied in this experiment were selected on the basis of pilot studies of matrix damage and chondrocyte death after impacts with energy densities ranging from 1.0 to 20 J/cm2. We found that 7 and 14-J/cm2 impacts caused superficial zone chondrocyte death rates and structural damage consistent with those thought to occur in severe joint injuries35.
Although bovine cartilage is used frequently as a surrogate for human cartilage in in vitro impact experiments, a direct comparison between bovine and human hip cartilage explants showed that impact-induced structural damage was more severe in the former than in the latter5. Thus, results from bovine models may overestimate damage from impact injury in humans and should be interpreted with caution.
Mechanical injury to cartilage may cause either acute chondrocyte death (within one hour after injury), due to necrosis, or subacute chondrocyte death (one to seventy-two hours after impact), presumably due to apoptosis. The balance of these modes of death may be determined by the mechanical characteristics of the injury, particularly with regard to the loading rate. Specimens subjected to injurious stresses at subimpact rates (<1000 MPa/sec) have consistently demonstrated cell death occurring over days by apoptosis. For example, human chondral explants subjected to 14-MPa loads at 140 MPa/sec demonstrated minimal increases in apoptotic chondrocytes within the first twenty-four hours after loading but had a 13% to 19% apoptosis rate forty-eight hours after loading. By ninety-six hours, approximately 25% of the cells were positive on TUNEL assay10. In another experiment, canine explants repeatedly loaded to 5 MPa at 60 MPa/sec for two hours had a 5.4% rate of apoptosis four hours after loading, which increased to 73% TUNEL-positive chondrocytes forty-eight hours after loading36. In bovine chondral explants subjected to a 25-MPa load at 25 MPa/sec, only 3% of cells were TUNEL positive one hour after loading6. This increased to 18% being TUNEL positive at ninety-six hours post-loading and 28% being TUNEL positive at 168 hours after loading.
In comparison with specimens subjected to subimpact loading, those subjected to a true impact injury (>1000 MPa/sec) show a much earlier wave of cell death, which occurs mainly in the superficial layer of cartilage or along impact-induced matrix cracks. Bush et al. subjected bovine chondral explants to a drop-tower impact and found that 28% of the cells were dead at three minutes post-impact; this increased to 40% at five minutes, 50% at twelve minutes, and 55% at twenty minutes8. In a study of a live rabbit model in which an impact was applied to the patellofemoral joint with a drop tower, there was a 45% rate of chondrocyte death in the superficial zone of cartilage and a 29% rate in the deeper cartilage on the day of impact7. The values were similar at four days post-impact (43% and 23%, respectively). In our study, we found that 28% to 36% of chondrocytes could not be rescued by immediate post-impact treatment with N-acetylcysteine. These data suggest that the experimental injuries caused by our drop-tower device caused acute necrosis at rates characteristic of true impact injury. However, these injuries also appeared to cause subacute apoptosis, indicating that both modes of death contributed to the overall depletion of viable chondrocytes. The pathophysiologic cascade(s) that lead to either of these forms of cell death are poorly understood. However, understanding the cellular events that lead to chondrocyte death, whether it is acute or subacute, will be paramount for the development of therapeutic interventions aimed to prevent impact-associated cell death.
Possible sources of excess impact-induced reactive oxygen species include mitochondria, which release superoxide radicals as a byproduct of respiration and in response to oxidative damage17. Chondrocytes also express inducible nitric oxide synthase (iNOS) and NADPH (nicotinamide adenine dinucleotide phosphate) oxidase (NOX), which generate nitric oxide and superoxide, respectively37,38. At low concentrations, reactive oxygen species downregulate chondrocyte responses to pro-inflammatory cytokines39. However, at higher concentrations, reactive oxygen species directly damage the cartilage extracellular matrix, activate matrix metalloproteinases, suppress extracellular matrix synthesis, and induce matrix metalloproteinase expression through activation of the transcription factors activator protein 1 and nuclear factor kappaB37,40,41. In addition, concomitant exposure to nitric oxide and to the superoxide radical contributes to chondrocyte apoptosis18,37. Reactive oxygen species have been shown to activate mitogen-activated protein kinases such as p38 and ERK, pathways known to be involved in apoptosis signaling13,37. Antioxidants have been shown to block activation of mitogen-activated protein kinases induced by reactive oxygen species and cytokines, which may explain their anti-apoptotic activities at impact sites.
Although group sizes were small in this experiment (three independent specimens were tested at each time point), eighteen separate samplings were taken from each specimen from the surface to a depth of 200 μm. The three ∼1.0-mm2 sites imaged for each specimen represented 15% of the impact site area. More than 2400 cells were counted in each explant. Thus, chondrocyte viabilities measured with this approach are likely to be representative of events throughout impact sites, at least to a depth of 200 μm. Pilot experiments indicated that >80% of the chondrocyte death associated with 7-J/cm2 impacts occurred within 200 μm of the cartilage surface. Therefore, our confocal analysis concentrating on the superficial zone of cartilage should have captured the majority of the viability changes induced by impact. While it is possible that there was significant death in the deeper zones, previous impact experiments have consistently shown that, at impact-loading levels, the majority of cell death is superficial or along matrix cracks3,4,10.
The data from our experiments demonstrate that chondrocyte death and subsequent proteoglycan loss can be significantly reduced by timely exposure of the impacted cartilage to N-acetylcysteine. Treatment with N-acetylcysteine within hours after injury essentially doubled chondrocyte viability compared with that in untreated specimens. This indicates that the primary pathophysiologic events attributable to reactive oxygen species occur very early after impact. It should be noted that N-acetylcysteine was withdrawn from the culture medium within twenty-four hours after the impact in our experiments. This short-term exposure was sufficient to improve viability for up to seventy-two hours post-impact and decrease proteoglycan loss for up to two weeks post-impact. However, considering that posttraumatic osteoarthritis may develop over months or years, two weeks in culture is a relatively brief interval. Thus, the efficacy of N-acetylcysteine in terms of halting the progression of posttraumatic osteoarthritis is still speculative. Testing in an animal model of joint injury will help to better elucidate the long-term effects of N-acetylcysteine on the stability of the cartilage extracellular matrix. 
Disclosure: In support of their research for or preparation of this work, one or more of the authors received, in any one year, outside funding or grants in excess of $10,000 from the National Institutes of Health/National Institute of Arthritis and Musculoskeletal and Skin Diseases (Grant 1 P50 AR055533). Neither they nor a member of their immediate families received payments or other benefits or a commitment or agreement to provide such benefits from a commercial entity. No commercial entity paid or directed, or agreed to pay or direct, any benefits to any research fund, foundation, division, center, clinical practice, or other charitable or nonprofit organization with which the authors, or a member of their immediate families, are affiliated or associated.
Investigation performed at the Orthopaedic Cell and Molecular Biology Laboratory, Department of Orthopaedics and Rehabilitation, The University of Iowa, Iowa City, Iowa
References
- 1.Borrelli J Jr. Chondrocyte apoptosis and posttraumatic arthrosis. J Orthop Trauma. 2006;20:726-31. [DOI] [PubMed] [Google Scholar]
- 2.Borrelli J Jr, Ricci WM. Acute effects of cartilage impact. Clin Orthop Relat Res. 2004;423:33-9. [DOI] [PubMed] [Google Scholar]
- 3.Torzilli PA, Grigiene R, Borrelli J Jr, Helfet DL. Effect of impact load on articular cartilage: cell metabolism and viability, and matrix water content. J Biomech Eng. 1999;121:433-41. [DOI] [PubMed] [Google Scholar]
- 4.Jeffrey J, Gregory D, Aspden R. Matrix damage and chondrocyte viability following a single impact load on articular cartilage. Arch Biochem Biophys. 1995;322:87-96. [DOI] [PubMed] [Google Scholar]
- 5.Jeffrey JE, Aspden RM. The biophysical effects of a single impact load on human and bovine articular cartilage. Proc Inst Mech Eng [H]. 2006;220:677-86. [DOI] [PubMed] [Google Scholar]
- 6.Baars DC, Rundell SA, Haut RC. Treatment with the non-ionic surfactant poloxamer P188 reduces DNA fragmentation in cells from bovine chondral explants exposed to injurious unconfined compression. Biomech Model Mechanobiol. 2006;5:133-9. [DOI] [PubMed] [Google Scholar]
- 7.Rundell SA, Baars DC, Phillips DM, Haut RC. The limitation of acute necrosis in retro-patellar cartilage after a severe blunt impact to the in vivo rabbit patello-femoral joint. J Orthop Res. 2005;23:1363-9. [DOI] [PubMed] [Google Scholar]
- 8.Bush PG, Hodkinson PD, Hamilton GL, Hall AC. Viability and volume of in situ bovine articular chondrocytes—changes following a single impact and effects of medium osmolarity. Osteoarthritis Cartilage. 2005;13:54-65. [DOI] [PubMed] [Google Scholar]
- 9.Kim HA, Blanco FJ. Cell death and apoptosis in osteoarthritic cartilage. Curr Drug Targets. 2007;8:333-45. [DOI] [PubMed] [Google Scholar]
- 10.D'Lima DD, Hashimoto S, Chen PC, Colwell CW Jr, Lotz MK. Impact of mechanical trauma on matrix and cells. Clin Orthop Relat Res. 2001;391 Suppl:S90-9. [DOI] [PubMed] [Google Scholar]
- 11.Phillips DM, Haut RC. The use of a non-ionic surfactant (P188) to save chondrocytes from necrosis following impact loading of chondral explants. J Orthop Res. 2004;22:1135-42. [DOI] [PubMed] [Google Scholar]
- 12.D'Lima DD, Hashimoto S, Chen PC, Lotz MK, Colwell CW Jr. Cartilage injury induces chondrocyte apoptosis. J Bone Joint Surg Am. 2001;83 Suppl 2(Pt 1):19-21. [DOI] [PubMed]
- 13.Malemud CJ, Islam N, Haqqi TM. Pathophysiological mechanisms in osteoarthritis lead to novel therapeutic strategies. Cells Tissues Organs. 2003;174:34-48. [DOI] [PubMed] [Google Scholar]
- 14.D'Lima D, Hermida J, Hashimoto S, Colwell C, Lotz M. Caspase inhibitors reduce severity of cartilage lesions in experimental osteoarthritis. Arthritis Rheum. 2006;54:1814-21. [DOI] [PubMed] [Google Scholar]
- 15.Beecher BR, Martin JA, Pedersen DR, Heiner AD, Buckwalter JA. Antioxidants block cyclic loading induced chondrocyte death. Iowa Orthop J. 2007;27:1-8. [PMC free article] [PubMed] [Google Scholar]
- 16.Kurz B, Lemke A, Kehn M, Domm C, Patwari P, Frank EH, Grodzinsky AJ, Schünke M. Influence of tissue maturation and antioxidants on the apoptotic response of articular cartilage after injurious compression. Arthritis Rheum. 2004;50:123-30. [DOI] [PubMed] [Google Scholar]
- 17.Terkeltaub R, Johnson K, Murphy A, Ghosh S. Invited review: the mitochondrion in osteoarthritis. Mitochondrion. 2002;1:301-19. [DOI] [PubMed] [Google Scholar]
- 18.Yasuhara R, Miyamoto Y, Akaike T, Akuta T, Nakamura M, Takami M, Morimura N, Yasu K, Kamijo R. Interleukin-1beta induces death in chondrocyte-like ATDC5 cells through mitochondrial dysfunction and energy depletion in a reactive nitrogen and oxygen species-dependent manner. Biochem J. 2005;389(Pt 2):315-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang H, Gajate C, Yu LP, Fang YX, Mollinedo F. Mitochondrial-derived ROS in edelfosine-induced apoptosis in yeasts and tumor cells. Acta Pharmacol Sin. 2007;28:888-94. [DOI] [PubMed] [Google Scholar]
- 20.Hoegger MJ, Lieven CJ, Levin LA. Differential production of superoxide by neuronal mitochondria. BMC Neurosci. 2008;9:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Adam-Vizi V. Production of reactive oxygen species in brain mitochondria: contribution by electron transport chain and non-electron transport chain sources. Antioxid Redox Signal. 2005;7:1140-9. [DOI] [PubMed] [Google Scholar]
- 22.Raha S, Robinson BH. Mitochondria, oxygen free radicals, disease and ageing. Trends Biochem Sci. 2000;25:502-8. [DOI] [PubMed] [Google Scholar]
- 23.Skulachev VP. Role of uncoupled and non-coupled oxidations in maintenance of safely low levels of oxygen and its one-electron reductants. Q Rev Biophys. 1996;29:169-202. [DOI] [PubMed] [Google Scholar]
- 24.Okun JG, Lümmen P, Brandt U. Three classes of inhibitors share a common binding domain in mitochondrial complex I (NADH:ubiquinone oxidoreductase). J Biol Chem. 1999;274:2625-30. [DOI] [PubMed] [Google Scholar]
- 25.Lee SB, Bae IH, Bae YS, Um HD. Link between mitochondria and NADPH oxidase 1 isozyme for the sustained production of reactive oxygen species and cell death. J Biol Chem. 2006;281:36228-35. [DOI] [PubMed] [Google Scholar]
- 26.O'Malley Y, Fink BD, Ross NC, Prisinzano TE, Sivitz WI. Reactive oxygen and targeted antioxidant administration in endothelial cell mitochondria. J Biol Chem. 2006;281:39766-75. [DOI] [PubMed] [Google Scholar]
- 27.Del Carlo M, Schwartz D, Erickson EA, Loeser RF. Endogenous production of reactive oxygen species is required for stimulation of human articular chondrocyte matrix metalloproteinase production by fibronectin fragments. Free Radic Biol Med. 2007;42:1350-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.DelCarlo M, Loeser RF. Chondrocyte cell death mediated by reactive oxygen species-dependent activation of PKC-betal. Am J Physiol Cell Physiol. 2006;290:C802-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aruoma OI, Halliwell B, Hoey BM, Butler J. The antioxidant action of N-acetylcysteine: its reaction with hydrogen peroxide, hydroxyl radical, superoxide, and hypochlorous acid. Free Radic Biol Med. 1989;6:593-7. [DOI] [PubMed] [Google Scholar]
- 30.Farndale RW, Buttle DJ, Barrett AJ. Improved quantitation and discrimination of sulphated glycosaminoglycans by use of dimethylmethylene blue. Biochim Biophys Acta. 1986;883:173-7. [DOI] [PubMed] [Google Scholar]
- 31.Huebbe P, Jofre-Monseny L, Boesch-Saadatmandi C, Minihane AM, Rimbach G. Effect of apoE genotype and vitamin E on biomarkers of oxidative stress in cultured neuronal cells and the brain of targeted replacement mice. J Physiol Pharmacol. 2007;58:683-98. [PubMed] [Google Scholar]
- 32.D'Lima DD, Hashimoto S, Chen PC, Lotz MK, Colwell CW Jr. Prevention of chondrocyte apoptosis. J Bone Joint Surg Am. 2001;83 Suppl 2(Pt 1):25-6. [DOI] [PubMed] [Google Scholar]
- 33.Aspden RM, Jeffrey JE, Burgin LV. Impact loading of articular cartilage. Osteoarthritis Cartilage. 2002;10:588-90. [DOI] [PubMed] [Google Scholar]
- 34.Haut RC. Contact pressures in the patellofemoral joint during impact loading on the human flexed knee. J Orthop Res. 1989;7:272-80. [DOI] [PubMed] [Google Scholar]
- 35.Repo RU, Finlay JB. Survival of articular cartilage after controlled impact. J Bone Joint Surg Am. 1977;59:1068-76. [PubMed] [Google Scholar]
- 36.Chen CT, Burton-Wurster N, Borden C, Hueffer K, Bloom SE, Lust G. Chondrocyte necrosis and apoptosis in impact damaged articular cartilage. J Orthop Res. 2001;19:703-11. [DOI] [PubMed] [Google Scholar]
- 37.Henrotin Y, Kurz B, Aigner T. Oxygen and reactive oxygen species in cartilage degradation: friends or foes? Osteoarthritis Cartilage. 2005;13:643-54. [DOI] [PubMed] [Google Scholar]
- 38.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245-313. [DOI] [PubMed] [Google Scholar]
- 39.Mathy-Hartert M, Martin G, Devel P, Deby-Dupont G, Pujol JP, Reginster JY, Henrotin Y. Reactive oxygen species downregulate the expression of pro-inflammatory genes by human chondrocytes. Inflamm Res. 2003;52:111-8. [DOI] [PubMed] [Google Scholar]
- 40.Tiku ML, Gupta S, Deshmukh DR. Aggrecan degradation in chondrocytes is mediated by reactive oxygen species and protected by antioxidants. Free Radic Res. 1999;30:395-405. [DOI] [PubMed] [Google Scholar]
- 41.Martin G, Andriamanalijaona R, Mathy-Hartert M, Henrotin Y, Pujol JP. Comparative effects of IL-1beta and hydrogen peroxide (H2O2) on catabolic and anabolic gene expression in juvenile bovine chondrocytes. Osteoarthritis Cartilage. 2005;13:915-24. [DOI] [PubMed] [Google Scholar]