

Abstract
As the number of cancer survivors grows, prediction of radiotherapy-induced second cancer risks becomes increasingly important. Because the latency period for solid tumors is long, the risks of recently introduced radiotherapy protocols are not yet directly measurable. In the accompanying article, we presented a new biologically based mathematical model, which, in principle, can estimate second cancer risks for any protocol. The novelty of the model is that it integrates, into a single formalism, mechanistic analyses of pre-malignant cell dynamics on two different time scales: short-term during radiotherapy and recovery; long-term during the entire life span. Here, we apply the model to nine solid cancer types (stomach, lung, colon, rectal, pancreatic, bladder, breast, central nervous system, and thyroid) using data on radiotherapy-induced second malignancies, on Japanese atomic bomb survivors, and on background US cancer incidence. Potentially, the model can be incorporated into radiotherapy treatment planning algorithms, adding second cancer risk as an optimization criterion.
Introduction
Ionizing radiation is a well-known cytotoxic and carcinogenic agent. As such, it is effective as a treatment for cancer, but can also induce secondary malignancies (Travis et al. 1996, 1997, 2002, 2003, 2005; van Leeuwen et al. 2003). As more patients undergo cancer radiotherapy and live longer after treatment, the number of cancer survivors has tripled over the past three decades and continues to increase, reaching more than 10 million in the US (Editorial 2004). The lifetime risk of radiation-induced second cancers in these individuals is not negligible (e.g. Brenner et al. 2007), and these second cancers can result in high mortality and morbidity—for example, breast cancer radiotherapy can cause lung cancer (see below), and lung cancer has a poor prognosis. Consequently, second malignancies induced by radiotherapy are becoming a growing concern (Brenner et al. 2000; Ron 2006). This is the case particularly for patients irradiated in childhood, who have a long life expectancy during which second cancers can develop, and in whom the relative risk of some radiogenic second cancers is on the order of 10–100 (Ron 2006; Ronckers et al. 2006; Neglia et al. 2006).
It has only recently become apparent (Lindsay et al. 2001; Sachs and Brenner 2005) that even at high radiation doses—tens of Gy—radiation-induced cancer risk remains substantial, presumably due to cellular repopulation, instead of dropping essentially to zero due to cell inactivation (killing), as was previously thought (Bennett et al. 2004; Dasu et al. 2005). Thus, tissues surrounding the tumor, which unavoidably receive doses not much smaller than the prescribed treatment dose, may be a source of much of the second cancer risk attributable to radiotherapy (Hodgson et al. 2007; Koh et al. 2007).
A reasonable approach to minimizing radiation-induced second cancers would be to compare radiotherapy protocols of equal efficacy against the primary tumor, and identify the ones with the lowest second cancer risk. However, because the latency period for radiation-induced solid tumors is long (e.g. Tokunaga et al. 1979; Brenner et al. 2000; Ivanov et al. 2004, 2009), the carcinogenic effects of radiotherapy have been directly measured only for regimens used several decades ago. The risks that may be associated with newer treatment methods are for the most part not yet observable. Biologically motivated mathematical/computational models, calibrated using data from older protocols, can address this problem by predicting risks of current/prospective treatment regimens.
In the accompanying article (Shuryak et al. 2009), such a model is presented. To our knowledge, the model is the first comprehensive attempt to integrate a detailed analysis of pre-malignant cell dynamics during the comparatively short period of radiotherapy with a long-term model of carcinogenesis over the entire human life span. By comparison, existing models either emphasize the long-term processes and make simplistic assumptions about short-term ones (e.g. Armitage and Doll 1954; Moolgavkar 1978, 1980; Armitage 1985; Moolgavkar and Luebeck 2003), or vice versa (e.g. Upton 2003; Sachs and Brenner 2005; Shuryak et al. 2006; Sachs et al. 2007). A formalism that integrates both time scales is advantageous in terms of biological realism and should increase the accuracy of predictions.
In the accompanying article, the focus was on biological assumptions and mathematical implementation. Here, we apply our model specifically to the task of predicting radiotherapy-induced cancers, using second cancer data for nine solid cancer types—stomach, lung, colon, rectal, pancreatic, bladder, breast, CNS (central nervous system), and thyroid—in patients treated by radiotherapy for various primary cancers. Some of the model parameters are obtained from fitting radiogenic risks at comparatively low doses for Japanese atomic bomb survivors, or from background US cancer incidence data.
Materials and methods
Model
The model used emphasizes initiation, either by radiation or by spontaneous processes, of normal stem cells to produce pre-malignant stem cells, the cell kinetics of pre-malignant stem cells, and their transformation into malignant cells; it assumes the pre-malignant (i.e. initiated) cells to reside in stem cell niches or compartments, referred to generically as “niches”; and it emphasizes those niches that are “fully pre-malignant”, i.e. containing as many pre-malignant stem cells as the niche carrying capacity allows. The model uses a total of 11 parameters, which were introduced in Table 1 of the accompanying article (Shuryak et al. 2009). Three parameters, which characterize spontaneous stem cell initiation and subsequent malignant transformation (a, units = time−2), pre-malignant niche replication (b, units = time−1), and age-dependent stem cell senescence (c, units = time−2), can be determined with relatively high precision from background cancer incidence. Seven other parameters describe radiation-related effects: constants X (units = time × dose−1) and Y (units = dose−1) characterizing the dose-dependences of initiation and promotion, respectively; a parameter for homeostatic regulation of the number of pre-malignant stem cells per niche δ (units = time−1); the carrying capacity, Z (units = cells × niche−1), for the number of pre-malignant stem cells in a niche; the stem cell radiation-inactivation constants α (units = dose−1) and β (units = dose−2); and the maximum net stem cell repopulation rate λ (units = time−1). The last parameter—the lag period from the appearance of the first fully malignant cell until development of cancer, L (units = time)—can be estimated from the literature.
Table 1.
Best-fit parameter values for background incidence of all analyzed cancer types
| Cancer | Sex | a × 10−8 (years−2) | b (years−1) | c × 10−3 (years−2) |
|---|---|---|---|---|
| Stomach | M | 0.402 (0.380, 0.425) | 0.222 (0.219, 0.226) | 1.10 (1.08, 1.14) |
| Lung | M | 0.00291 (0.0028, 0.0030) | 0.460 (0.459, 0.462) | 2.91 (2.89, 2.92) |
| Colon | M | 1.78 (1.57, 2.10) | 0.204 (0.197, 0.208) | 0.944 (0.892, 0.985) |
| Rectum | M | 1.01 (0.852, 1.49) | 0.217 (0.201, 0.224) | 1.23 (1.09, 1.28) |
| Pancreas | M | 0.0732 (0.0397, 0.255) | 0.293 (0.156, 0.315) | 1.67 (0.174, 1.85) |
| Bladder | M | 0.0600 (0.0600, 0.0601) | 0.282 (0.279, 0.283) | 1.54 (1.51, 1.56) |
| Breast | F | 58.1 (55.6, 60.2) | 0.199 (0.197, 0.201) | 1.35 (1.34, 1.37) |
| CNS | Both | 0.243 (0.230, 0.259) | 0.257 (0.254, 0.260) | 1.66 (1.61, 1.68) |
| Thyroid | Both | 120 (105, 124) | 0.0768 (0.0740, 0.0872) | 0.669 (0.646, 0.770) |
Parameter interpretations are: a proportionality constant for spontaneous stem cell initiation, b pre-malignant niche replication rate, c age-dependent pre-malignant stem cell senescence constant
The numbers in parentheses represent the 95% confidence intervals for the corresponding parameter
The equation for the mean expected number of new fully malignant cells per individual per year under background conditions (A bac, units = time−1), which is an approximation for the cancer incidence hazard function L years later, was derived in the previous article. It is repeated below, using the notation where age is defined as the sum of age at exposure (T x) and the time after exposure (T y):
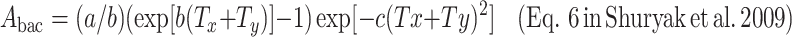 |
For data fitting, the exact hazard defined as H = A/(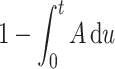 ) is used, but we use the simpler expression for A in the equations below, keeping in mind its interpretation and limitations.
) is used, but we use the simpler expression for A in the equations below, keeping in mind its interpretation and limitations.
The expression for the radiogenic excess relative risk (ERR) was also derived previously and is repeated below, where D is the total radiation dose, Sf(Z, D) is the probability that a pre-existing fully pre-malignant niche survives radiation, i.e. that not all the pre-malignant stem cells in the niche are inactivated, and ISf(D) (units = dose) represents a net outcome of initiation, inactivation, and cell repopulation (iir) during radiation exposure:
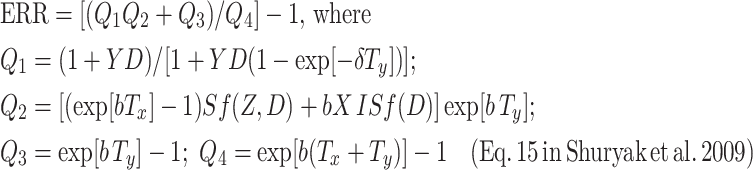 |
For a single-dose acute exposure, where there is no cell repopulation during irradiation, the functions ISf(D) and Sf(Z, D) simplify to the following expressions: Sf(Z, D) = 1 − (1 − exp[–αD − βD 2])Z and ISf(D) = D exp[–αD − βD 2]. For multi-fraction radiotherapy protocols, where substantial repopulation occurs between dose fractions, these functions are evaluated by the stochastic process formalism described in detail in the previous article.
As noted in the previous article, the term Q 2 in Eq. 15 of (Shuryak et al. 2009) is proportional to the number of fully pre-malignant stem cell niches shortly after exposure; Q 1 is the normalized size of such a niche, which can differ from the background carrying capacity Z due to radiation-promotion effects; Q 3 is proportional to the number of new niches spontaneously initiated, after exposure/recovery, by endogenous processes independent of radiation; and Q 4 is proportional to the total number of fully pre-malignant niches under background conditions. The parameters a and c cancel out of the ERR expression.
Data used
Here the model is applied by fitting it to data on nine cancer types: stomach, lung, colon, rectal, pancreatic, bladder, breast, CNS, and thyroid. These particular types were selected because for them radiation-induced ERR data are available both at comparatively low doses (ERR/Gy estimates based on Japanese atomic bomb survivors) and at high doses—second cancer ERRs estimated for patients treated with radiotherapy for existing malignancies, with at least two different high-dose points. There is a large body of second cancer data, and detailed comparisons to data on the atomic bomb survivors have been made previously (e.g. Little 2001). For our present purposes, the multiple dose points criterion for second cancer data was important because the analysis presented here was intentionally focused on modeling the shape of the radiation dose response at high doses.
The low-dose ERR/Gy estimates at attained age 70 and various ages at exposure were taken from an analysis of atomic bomb survivors by Preston et al. (2007), Tables 11 and 12. Gender-averaged ERR/Gy numbers were adjusted by sex-specific incidence ratios provided by the same authors. We used these particular ERR estimates because they approximate lifetime risk, were adjusted for several potential confounding variables, and were based on the most recent version of individual radiation dose calculations.
An important goal of our model development is second cancer risk estimation in Western populations. Consequently, we fitted the model to background incidence data for the selected cancers in the US, using the Surveillance Epidemiology and End Results (SEER) database (http://seer.cancer.gov), instead of using the incidence data from Japan provided by the Radiation Effects Research Foundation (RERF) (http://www.rerf.or.jp/). This raised the issue of risk transfer between the Japanese and Western populations. For simplicity, we used the fully multiplicative approach by directly transferring the ERR. Adequate fits were obtained (see below), which suggests the direct ERR transfer to be adequate for the present purpose and for the cancer types chosen, at least as a first approximation.
The high-dose radiotherapy-induced ERRs were taken from the following data sets which are, we believe, representative of currently available comprehensive second cancer epidemiologic studies that considered more than one dose:
Separate ERRs for bladder, colon, rectal, pancreatic, and stomach cancers reported from a case–control study embedded in a cohort of 28,843 patients, who were treated with radiotherapy for testicular cancer and survived for at least 1 year (Travis et al. 1997, 2005). The patients were gathered from 16 population-based tumor registries in North America and Europe. The mean age at radiotherapy was approximately 35 years, and the mean latency time before second cancer diagnosis was 18 years. Over 3,300 patients survived for more than 20 years. The particular five cancer types listed above were selected because they showed the most substantial radiation-induced ERRs, whereas the risk patterns for other cancer types, e.g. prostate, analyzed by the same studies were less clear. The data were transcribed from Table 3 in (Travis et al. 1997). A dose response could be constructed because radiotherapy regimens for non-seminoma testicular cancer typically involved a twofold higher dose, given in twice as many fractions, compared with regimens for seminoma testicular cancer [see Appendix Table 1 in (Travis et al. 1997)]. Error bars (95% CI) were estimated using Poisson assumptions about the distribution of cancer cases.
Separate ERRs for breast and lung cancers reported in patients treated with radiotherapy for Hodgkin’s disease (Travis et al. 2002, 2003; van Leeuwen et al. 2003; Gilbert et al. 2003). The breast cancer data were obtained from a case–control study within a cohort of 3,817 female patients, who were diagnosed with Hodgkin’s disease at an age of <30 years (mean age at radiotherapy = 22 years) in North America and Europe, and survived for at least 1 year after radiotherapy (mean latency time before second cancer diagnosis = 18 years) (Travis et al. 2003); and from another study of 650 patients treated for Hodgkin’s disease at an age of <41 years (mean age at radiotherapy = 21 years) in the Netherlands, who survived for at least 5 years (mean latency time before second cancer diagnosis = 18 years) (van Leeuwen et al. 2003). The lung cancer data were obtained from a case–control study within a cohort of 19,046 Hodgkin’s disease patients diagnosed in North America and Europe, who survived for at least 1 year after radiotherapy (Travis et al. 2002). The mean age at radiotherapy was 48 years, and the mean latency time before second cancer diagnosis was 11 years. Additional lung cancer data were obtained from another case–control study within the same cohort (Gilbert et al. 2003). The mean age at radiotherapy was 46 years, and the mean latency time before second cancer diagnosis was 8 years. In both lung cancer studies, the subjects were predominantly males, current or former tobacco smokers.
ERR for thyroid cancer reported from a case-control study within a cohort of 14,054 patients of both sexes, treated for various cancer types in childhood in North America, who survived for at least 5 years (Ronckers et al. 2006). The mean age at radiotherapy was 10 years, and the mean latency time before second cancer diagnosis was 15 years.
ERRs for CNS cancers, also reported from a case–control study within a cohort of 14,361 patients of both sexes, treated for various cancer types in childhood in North America, who survived for at least 5 years (Neglia et al. 2006). The mean age at radiotherapy was 5 years, and the mean latency time before second cancer diagnosis was 10 years. The glioma and meningioma data were pooled in our analysis.
Parameter estimation and data fitting procedure
Fitting of the model to the data was carried out using a customized random-restart simulated annealing algorithm implemented in the FORTRAN language (Press 1989). Standard least-squares inverse variance techniques were used for weighting individual data points.
To reduce the number of adjustable parameters, the stem cell inactivation (α and β) and repopulation (λ) constants were restricted to biologically plausible values for each cancer type, based on the literature (Bentzen et al. 1996; Challeton et al. 1997; Thames et al. 1989; Thames et al. 1990; Shimomatsuya et al. 1991; Rew and Wilson 2000; Fowler 2001; Chen et al. 2006; Ogawa et al. 2006; Phillips et al. 2006; Rachidi et al. 2007; Williams et al. 2008). The lag period from the appearance of the first fully malignant cell until development of cancer, L, was set at 10 years, which is consistent with available data (e.g. Brenner et al. 2000; Tokunaga et al. 1979; Ivanov et al. 2004).
The fitting procedure consisted of two sequential steps:
First, the expression for the background cancer hazard H was fitted to age- and sex-dependent US background cancer incidence for each cancer type from the SEER database. The population gender for each cancer type was chosen to be the same as that reported in the corresponding epidemiological study of radiotherapy ERRs. For example, only male patients were studied to determine ERR for bladder cancer following radiotherapy for testicular cancer, so the male-specific background incidence for bladder cancer was fitted. Gender-averaged background incidence was used where both sexes were studied after radiotherapy, such as in the case of CNS tumors. The relevant age at exposure T x was defined as T x = max (0, T – L), where T is the age at cancer incidence reported by SEER. It was assumed that L = 10 years, but the results were not substantially sensitive to variations of L in the plausible range of 3–15 years, provided the other parameters were adjusted correspondingly. This fitting step generated values for parameters a, b, and c for each cancer type.
The best-fit value of b was used in a second round of fitting the model-generated ERRs to radiation-induced ERR data sets: both the comparatively low-dose ERR/Gy from Japanese atomic bomb survivors, and the high-dose radiotherapy-induced ERR data for each cancer type. Mean age at exposure and time since exposure values from the references cited above were used for each epidemiological high-dose radiotherapy data set. Moderate variations in these numbers produced some corresponding changes in the best-fit parameter values, but did not alter the main conclusions. This second round of fitting generated values for the remaining adjustable parameters X, Y, δ, and Z.
This two-step procedure was chosen because the background cancer incidence data have much greater statistical precision, due to larger sample sizes, compared with ERR estimates. Therefore, it was deemed advantageous to set the values for a, b, and c from background data, before fitting the radiation-induced risks. Only parameter b is shared by the equations for background and radiation-induced cancer risks.
Estimation of parameter uncertainties
For each data set (SEER, atomic bomb survivors, and second cancers), 100 synthetic data sets were generated by Monte-Carlo simulation. For the SEER data, the simulations were based on the Poisson distribution (which is well approximated by the Normal distribution because the observed number of cancer cases for each 5-year age category is typically >100). For the atomic bomb survivors data, the simulations were based on the Chi-squared distribution with one degree of freedom, because this distribution was used by (Preston et al. 2007) to generate confidence intervals for their summary ERR values, which are used here. For the second cancer data, the Poisson distribution was used.
We fitted the model (the hazard function for background cancer incidence) first to the simulated SEER data, generating a distribution of values for each background parameter (parameters a, b, and c). For each parameter, 95% CI were generated based on these distributions. Then, the model-predicted ERR expression was fitted simultaneously to the simulated atomic bomb and second cancer data sets, generating distributions and 95% CI for the remaining adjustable parameters.
Results
The best-fit model parameters and predictions are shown in Tables 1 and 2 and Figs. 1, 2, 3. In general, the fits were adequate, particularly considering the uncertainties in the data and the biologically motivated constraints placed on four parameters (α, β, λ, L). We next highlight some features of the results on background cancers (Fig. 1), cancers in atomic bomb survivors (Fig. 2) and second cancers after radiotherapy (Fig. 3).
Table 2.
Best-fit parameter values for radiation-induced excess relative risk (ERR) of all analyzed cancer types
| Cancer | X (years × Gy−1) | Y (Gy−1) | δ × 10−3 (years−1) | log10 Z | α (Gy−1), β (Gy−2) | λ (day−1) |
|---|---|---|---|---|---|---|
| Stomach | 6.53 (1.94, 13.5) | 0.120 (0.089, 0.145) | 0.206 (0.00, 0.364) | 3.58 (2.65, 3.66) | 0.25, 0.025 | 0.05 |
| Lung | 0.860 (0.276, 10.5) | 0.387 (0.279, 0.690) | 9.49 (1.38, 23.3) | 4.99 (3.80, 5.56) | 0.25, 0.025 | 0.05 |
| Colon | 1.71 (0.010, 9.63) | 0.872 (0.465, 1.39) | 0.572 (0.00, 3.01) | 0.301 (0.00, 0.628) | 0.25, 0.025 | 0.10 |
| Rectum | 2.40 (0.048, 7.20) | 0.0717 (0.041, 0.246) | 0.0193 (0.00, 1.54) | 2.09 (0.978, 3.05) | 0.25, 0.025 | 0.10 |
| Pancreas | 3.12 (0.069, 10.7) | 0.107 (0.073, 0.370) | 0.019 (0.00, 28.6) | 3.23 (2.73, 4.09) | 0.25, 0.025 | 0.10 |
| Bladder | 0.151 (0.102, 13.1) | 0.626 (0.439, 0.878) | 1.68 (0.00, 3.92) | 1.96 (1.72, 2.13) | 0.25, 0.025 | 0.05 |
| Breast | 4.75 (0.880, 10.1) | 0.938 (0.653, 1.11) | 3.05 (0.00, 3.08) | 6.65 (5.47, ∞) | 0.25, 0.050 | 0.05 |
| CNS | 1.45 (0.046, 13.5) | 0.620 (0.273, 1.15) | 0.0316 (0.00, 20.5) | 5.69 (4.27, ∞) | 0.25, 0.025 | 0.10 |
| Thyroid | 34.4 (5.40, 69.1) | 0.237 (0.097, 0.809) | 0.0156 (0.00, 0.369) | 4.03 (3.02, 6.15) | 0.30, 0.030 | 0.05 |
Parameter interpretations are: X radiation-induced initiation constant, Y radiation-induced promotion constant, δ parameter for homeostatic regulation of the number of pre-malignant stem cells per niche, Z carrying capacity for pre-malignant stem cells per niche, α, β stem cell inactivation constants by radiation, λ maximum net stem cell repopulation rate
The numbers in parentheses represent the 95% confidence intervals for the corresponding parameter
Fig. 1.

Best-fit model predictions for US background incidence of each cancer type, from the SEER database. The sex of each population and the model parameter values are listed in Table 1. Error bars represent 95% confidence intervals
Fig. 2.

Best-fit model predictions for sex-adjusted ERR/Gy estimates from Japanese atomic bomb survivors for each cancer type, from (Preston et al. 2007). The error bars represent 95% confidence intervals. For rectal, pancreatic and CNS cancers, the ERR/Gy estimates had no statistically-significant age-dependence, as shown by dashed horizontal lines in the corresponding figure panels. The sex of each population and the parameter values are listed in Table 1
Fig. 3.

Best-fit model predictions for high-dose fractionated radiotherapy ERRs for each cancer type. The error bars represent 95% confidence intervals. The corresponding references for each data set are listed in the “Methods” section. The sex of each population and the parameter values are listed in Table 1
Special features of the results
The cancer type-specific background incidence rates were usually fitted well, using the three relevant parameters a, b, and c (Fig. 1). At older ages some of the fits were qualitatively better than those produced by the commonly used two-stage clonal expansion (TSCE) model with the same number of parameters (e.g. Heidenreich et al. 2007). The reason for this difference is that the TSCE model generates an incidence hazard function that asymptotically levels off at old age at some high value (e.g. Denes and Krewski 1996), whereas the recent data from SEER suggest the incidence of many cancers to decrease at the oldest recorded ages. Our model is based on the assumption that this decrease is due to age-dependent loss of stem cell function (senescence), although other causes, e.g. quality of diagnosis, birth-date cohort effects, or selection due to populations with heterogeneous risks (“frailty”), cannot be excluded.
Our model did not fit the background incidence of CNS cancers as well as that of other cancer types, because it underestimated the data at young ages, below 40 years. The incidence of CNS cancers at young ages may be produced by a different biological mechanism than that at older ages, and/or occur in a sub-population of genetically predisposed individuals, and therefore may require a separate set of parameters.
As expected, the parameter proportional to the spontaneous initiation rate (a) varies by orders of magnitude for the different cancer types, since the observed incidence rates for these cancers are also very different. The rates (b) for replication of pre-malignant stem cell niches, however, are much more tightly dispersed; most fall in the range of 0.2–0.46 years−1. The values qualitatively agree with the best-fit clonal expansion rates (birth minus death) found using the TSCE model (e.g. Heidenreich et al. 2007), and support the intuitive idea that pre-malignant stem cells have only a small net growth advantage over normal ones, so that their number increases only on the scale of years to decades. Numerically, the estimates of b tend to be somewhat larger than the net clonal expansion rates in the TSCE model, because in our formalism clonal growth by niche replication is partially offset by stem cell senescence (parameter c), which reduces the number of potentially carcinogenic stem cells in each niche.
Altering the model assumptions by postulating that more than one mutation is necessary to initiate a stem cell results in some reduction in the best-fit values of b. This is expected because the number of mutational stages and the rate of clonal expansion have somewhat similar effects on the predicted age-dependent hazard function (e.g. Kopp-Schneider and Portier 1991; Little and Wright 2003; Little and Li 2007), so that increasing the first results in a corresponding decrease in the second. However, the model variant with more than two mutational stages did not fit the data substantially better than the default two-stage formalism (not shown), and was not used because it requires an extra adjustable parameter (i.e. the number of mutations necessary for stem cell initiation).
The stem cell senescence parameter (c) is also relatively tightly distributed for most cancers, such that c/b = 6 − 7 × 10−3 years−1. These findings support an intuitive picture that the senescence rate is roughly proportional to the cell division rate—hence, the constancy of the c/b ratio.
The ERR/Gy estimates from Japanese atomic bomb survivors at age 70, as function of age at exposure, were also fitted quite well for all cancer types (Fig. 2). In the context of our model, the shape of the dependence of radiation-induced ERR on age at exposure provides insight into whether this ERR is dominated by initiation or promotion. As noted in the accompanying article (Shuryak et al. 2009), initiation-driven ERR should decrease markedly with age at exposure. In contrast, promotion-driven ERR should be relatively constant as function of age.
The atomic bomb survivor data (Preston et al. 2007) suggest that a substantial decrease in ERR/Gy with age at exposure occurs only for stomach and thyroid cancers, among those analyzed here. Consistently with the above arguments, the lower bounds of the 95% CI for the radiation-induced initiation parameter X for these particular cancers were substantially >0 (Table 2). For the seven other cancer types, ERR/Gy appears to be independent of age at exposure, or even to increase at older ages. For these cancers, the lower bounds of the 95% CI for parameter X were approaching zero. Indeed, restricting X to zero worsened the model fit only marginally in many of these cases.
The ERR/Gy data for lung cancer stand out from the others by apparently increasing with age at exposure (Fig. 2). This may be due to residual confounding by the rapid changes in smoking rates in Japan (Preston et al. 2007). However, since our model does not include the effects of smoking, the trend in the data points was attributed by the model to effects of both age at exposure and time since exposure: because the ERR/Gy was measured at a fixed age of 70, age at exposure and time since exposure were not independent—their sum had to equal 70. In the context of our model, an apparent increase in ERR with age at exposure was interpreted as a decrease in ERR with time since exposure, which occurs if radiation-induced promotion is reversible due to homeostatic regulation of the number of pre-malignant cells per niche, i.e. if parameter δ is >0. This explains why the best-fit value for δ was >0 for lung cancer, and the lower bound for its 95% CI was also >0 (Table 2). In contrast, the lower bounds for the 95% CI for δ were zero for the other cancers, and setting δ to zero did not alter the fits substantially in these cases.
Recent analyses of atomic bomb survivor data (Little 2009; Walsh 2009) suggest that an apparent increase in ERR/Gy for the oldest ages at exposure may occur for several other cancer types in addition to lung cancer. This phenomenon can be due to multiple factors, e.g. activation of microscopic dormant tumors by radiation. The explanation given by the current model for lung cancer is also a plausible hypothesis for explaining these new data for other cancers.
The best fits to the high-dose radiotherapy-induced ERRs were also generally adequate (Fig. 3). The fits to colon and bladder cancer data were the poorest. This was due to an inherent inconsistency in the data between ERR/Gy from the Japanese atomic bomb survivors, and ERR for multiple-Gy radiotherapy: at 1 Gy, the mean ERR/Gy for colon cancer is 0.61–0.75 (atomic bomb survivor data, Fig. 2), whereas at 6.9 Gy it is 0.03 and at 13.9 Gy it is 0.082 (second cancer data, Fig. 3). The same pattern can be seen by comparing other recent second cancer data sets (e.g. Chaturvedi et al. 2007) with atomic bomb survivor data. In general, the risk estimates of radiotherapy-induced colon cancer from different sources are highly variable (e.g. Inskip et al. 1990; Lundell and Holm 1995; Carr et al. 2002). Because of the more than tenfold inconsistency in the ERR/Gy slopes at different doses using the data sets we selected, our model overestimates the ERR at radiotherapeutic doses. A better fit to the high-dose data could be produced only by allowing unrealistically high cell inactivation, e.g. α > 1.0 Gy−1. A qualitatively similar picture was found for bladder cancer, where the ERR observed after >20 Gy of fractionated radiotherapy is roughly comparable to the ERR seen at 1 Gy in the atomic bomb survivors.
For the other seven cancers analyzed here, the atomic bomb survivors data at 1 Gy and the high-dose radiotherapy data produced relatively similar ERR/Gy slopes, at least at doses <20 Gy, so the model fits are substantially better (Fig. 3). There is a tendency to underestimate the risk at very high doses for some cancers, but this is probably due to the default values of α and β being too high for stem cells—better fits were produced by lowering α to say 0.15 Gy−1.
Discussion
We have presented a biologically motivated mathematical model of background and radiation-induced cancer risk, and applied it to data on nine solid adult-onset second cancer types. The model takes into account initiation, inactivation, and repopulation of target stem cells (iir processes) throughout the comparatively short-term period of radiotherapy and recovery by a stochastic formalism. This formalism is integrated with a deterministic long-term two-stage carcinogenesis model, which follows the kinetics of pre-malignant stem cells throughout the entire human lifetime, before and after radiation exposure.
Such an approach, unifying short- and long-term models, has some advantages over currently existing methods, as discussed in the previous article. Briefly, our model allows mechanistic risk predictions to be made at high radiotherapeutic doses as well as at low doses, can track the age and time dependencies of risk mechanistically, and is qualitatively better at describing background cancer incidence at old ages than the commonly used two-stage clonal expansion (TSCE) model, with the same number of adjustable parameters. Radiation-induced risks are calculated analytically, using plausible assumptions about underlying biology.
The current model is an improvement over our previous models. For example, in our deterministic solid tumor model (Sachs and Brenner 2005), the shape of the ERR dose response was determined by the relative proliferation rate of pre-malignant stem cells compared with normal ones—parameter r. To describe the data, r had to be smaller than 1, e.g. 0.8, implying that pre-malignant cells proliferate more slowly than normal ones, at least over the short term, i.e. during radiotherapy and for some weeks afterwards. Such an interpretation is at odds with the long-term tendency of pre-malignant cells to clonally expand. In our stochastic analysis of the same problem (Sachs et al. 2007), this inconsistency was removed, and a fit to the data was possible even if pre-malignant cells were assumed to proliferate as fast as, or faster than, their normal counterparts, i.e. r ≥ 1. However, the stochastic algorithm involved additional adjustable parameters. In the current model, repopulation of pre-malignant cells during radiotherapy is assumed to occur at the same rate as that of normal ones, i.e. r = 1 implicitly. The reasoning is that any proliferative advantage that pre-malignant cells have manifests itself only on the scale of multiple years in humans. The model describes the data adequately, so there is no need for r to be smaller than 1, and no need for additional parameters.
Our model applied to second cancer data can also provide some insight into the underlying biological mechanisms of carcinogenesis. An important outcome of our analysis is the finding that for many cancer types, radiation-induced risk, especially at ages >20, may be dominated by promotion, rather than initiation. Such an important role of promotion in low-LET radiation-induced risk for many cancers is consistent with the findings of other authors using the TSCE model on Japanese atomic bomb survivor data (e.g. Heidenreich et al. 2007). Our results indicate that promotion is the dominant mechanism of radiogenic risk for lung, colon, rectal, pancreatic, bladder, and CNS tumors; initiation and promotion both contribute to stomach and breast cancer risk; and thyroid cancer risk is dominated by initiation (see best-fit parameter values in Table 1). For example, we estimated that a 1 Gy dose leads to almost a doubling in the number of existing pre-malignant cells in the breast. Of course, this conclusion about the importance of radiogenic promotion is dependent on model assumptions, and needs to be tested experimentally.
For most cancers analyzed here, promotion appears to be permanent, i.e. pre-malignant niches are not reduced in size or number over time after irradiation. However, a gradual reduction in niche size after exposure, at a rate of ~1% per year, is suggested for lung cancer, and at a slower rate for some other cancers. These findings indicate that the radiation-induced hyper-proliferation of pre-malignant cells may be either permanent, or transient, depending on the organ.
The iir-based models in general, including the one described here, produce a dose response that has the same basic shape, with a maximum ERR at some intermediate dose, as the shape generated by older linear–quadratic–exponential (LQE) models, which neglected repopulation by cell proliferation (Little 2001; Bennett et al. 2004; Dasu et al. 2005). The main difference is in the dose range at which the maximum occurs. Models without repopulation predict that maximum ERR would occur at relatively low doses (below 5 Gy), and at typical clinically used doses (above 30 Gy) ERR would be essentially zero, due to the inactivation of nearly all target stem cells. In iir models, repopulation of both normal and pre-malignant stem cells between dose fractions compensates for much of the inactivation. Consequently, ERR peaks at much higher doses, e.g. 20–60 Gy. This prediction is much more consistent with the recent epidemiological data, which indicates that ERR can remain substantial even at doses as high as 40–50 Gy (e.g. Travis et al. 1996, 1997, 2002, 2003, 2005).
In the context of the model presented here, the radiation dose at which ERR peaks is determined by the carrying capacity for pre-malignant cells per niche, Z, in addition to the cell killing parameters α and β and the repopulation rate λ. Physiologically, Z can be interpreted as an estimate for the number of stem cells that contribute to repopulation within a given location of the target organ. For most cancers analyzed here, the best-fit values of Z (Table 2) suggested that up to several thousand stem cells may cooperate for local repopulation. The exceptions were breast and CNS cancers, where the upper bound of the 95% CI for the best-fit value of Z is infinity, suggesting that all the stem cells in the entire organ may participate in repopulation after irradiation.
The ability to reasonably predict cancer ERR at both low and high radiation doses using a biologically based mathematical model, incorporating formalisms for both short-term (iir) and long-term processes, should enable this model to be used for optimization of radiotherapy protocols, by introducing second cancer risk as an additional criterion. This can be done if a dose-volume histogram for the protocol is available, as is usually the case (e.g. Hodgson et al. 2007; Koh et al. 2007).
Almost by definition, all mathematical models are greatly simplified representations of complex and incompletely understood biological processes. Our model has the main inherent drawbacks of other two-stage and iir carcinogenesis formalisms; extension to more stages and processes may improve biological realism, but at the cost of extra parameters. For example, the model can be extended by treating long-term clonal expansion stochastically instead of deterministically, by incorporating more detailed cell–cell interactions other than just a slowdown in net cell proliferation after filling of a pre-malignant stem cell niche, by allowing the number of pre-malignant cells at birth to be greater than 0, by accounting for variability in the lag period for the development of clinical cancer after the appearance of the first malignant cell, etc.
In the future, we intend to apply the model to additional second cancer data sets, where only a single estimate of radiation dose is available, but more information can be gained about age/time dependencies of radiation-induced risk. For example, some such studies with long follow-up times and large numbers of patients (e.g. Chaturvedi et al. 2007; Brown et al. 2007) suggest that these age/time dependencies may have different trends from the age/time dependencies suggested by Japanese atomic bomb survivors data, which is used here. Using data on older radiotherapy protocols optimally will be an important step in credibly projecting second cancer risks of current and future protocols decades into the future.
Acknowledgments
Research supported by National Cancer Institute grant 5T32-CA009529 (IS), Department Of Energy DE-FG02-03ER63668 and National Institutes of Health CA78496 (PH), National Aeronautics and Space Administration 03-OBPR-07-0059-0065(LH), National Aeronautics and Space Administration NSCOR NNJ04HJ12G/NNJ06HA28G (RKS), National Institutes of Health grants P41 EB002033-09 and P01 CA-49062 (DJB).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Footnotes
An erratum to this article is available at http://dx.doi.org/10.1007/s00411-011-0378-5.
Contributor Information
Igor Shuryak, Email: ishuryak@gmail.com.
David J. Brenner, Phone: +1-212-3059930, FAX: +1-212-3053229, Email: djb3@columbia.edu
References
- Armitage P. Multistage models of carcinogenesis. Environ Health Perspect. 1985;63:195–201. doi: 10.2307/3430046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armitage P, Doll R. The age distribution of cancer and a multi-stage theory of carcinogenesis. Br J Cancer. 1954;VIII:1–12. doi: 10.1038/bjc.1954.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett J, Little MP, Richardson S. Flexible dose–response models for Japanese atomic bomb survivor data: Bayesian estimation and prediction of cancer risk. Radiat Environ Biophys. 2004;43:233–245. doi: 10.1007/s00411-004-0258-3. [DOI] [PubMed] [Google Scholar]
- Bentzen SM, Ruifrok AC, Thames HD. Repair capacity and kinetics for human mucosa and epithelial tumors in the head and neck: clinical data on the effect of changing the time interval between multiple fractions per day in radiotherapy. Radiother Oncol. 1996;38:89–101. doi: 10.1016/0167-8140(95)01689-9. [DOI] [PubMed] [Google Scholar]
- Brenner DJ, Curtis RE, Hall EJ, Ron E. Second malignancies in prostate carcinoma patients after radiotherapy compared with surgery. Cancer. 2000;88:398–406. doi: 10.1002/(SICI)1097-0142(20000115)88:2<398::AID-CNCR22>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Brenner DJ, Shuryak I, Russo S, Sachs RK. Reducing second breast cancers: a potential role for prophylactic mammary irradiation. J Clin Oncol. 2007;25:4868–4872. doi: 10.1200/JCO.2007.11.0379. [DOI] [PubMed] [Google Scholar]
- Brown LM, Chen BE, Pfeiffer RM, Schairer C, Hall P, Storm H, Pukkala E, Langmark F, Kaijser M, Andersson M, Joensuu H, Fossa SD, Travis LB. Risk of second non-hematological malignancies among 376, 825 breast cancer survivors. Breast Cancer Res Treat. 2007;106:439–451. doi: 10.1007/s10549-007-9509-8. [DOI] [PubMed] [Google Scholar]
- Carr ZA, Kleinerman RA, Stovall M, Weinstock RM, Griem ML, Land CE. Malignant neoplasms after radiation therapy for peptic ulcer. Radiat Res. 2002;157:668–677. doi: 10.1667/0033-7587(2002)157[0668:MNARTF]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- Challeton C, Branea F, Schlumberger M, Gaillard N, de Vathaire F, Badie C, Antonini P, Parmentier C. Characterization and radiosensitivity at high or low dose rate of four cell lines derived from human thyroid tumors. Int J Radiat Oncol Biol Phys. 1997;37:163–169. doi: 10.1016/s0360-3016(96)00449-x. [DOI] [PubMed] [Google Scholar]
- Chaturvedi AK, Engels EA, Gilbert ES, Chen BE, Storm H, Lynch CF, Hall P, Langmark F, Pukkala E, Kaijser M, Andersson M, Fossa SD, Joensuu H, Boice JD, Kleinerman RA, Travis LB. Second cancers among 104, 760 survivors of cervical cancer: evaluation of long-term risk. J Natl Cancer Inst. 2007;99:1634–1643. doi: 10.1093/jnci/djm201. [DOI] [PubMed] [Google Scholar]
- Chen MF, Lin CT, Chen WC, Yang CT, Chen CC, Liao SK, Liu JM, Lu CH, Lee KD. The sensitivity of human mesenchymal stem cells to ionizing radiation. Int J Radiat Oncol Biol Phys. 2006;66:244–253. doi: 10.1016/j.ijrobp.2006.03.062. [DOI] [PubMed] [Google Scholar]
- Dasu A, Toma-Dasu I, Olofsson J, Karlsson M. The use of risk estimation models for the induction of secondary cancers following radiotherapy. Acta Oncol. 2005;44:339–347. doi: 10.1080/02841860510029833. [DOI] [PubMed] [Google Scholar]
- Denes J, Krewski D. An exact representation for the generating function for the Moolgavkar–Venzon–Knudson two-stage model of carcinogenesis with stochastic stem cell growth. Math Biosci. 1996;131:185–204. doi: 10.1016/0025-5564(95)00046-1. [DOI] [PubMed] [Google Scholar]
- Editorial (no authors listed). Cancer survivors: living longer, and now, better (2004) Lancet 364:2153–2154 [DOI] [PubMed]
- Fowler JF. Biological factors influencing optimum fractionation in radiation therapy. Acta Oncol. 2001;40:712–717. doi: 10.1080/02841860152619124. [DOI] [PubMed] [Google Scholar]
- Gilbert ES, Stovall M, Gospodarowicz M, Van Leeuwen FE, Andersson M, Glimelius B, Joensuu T, Lynch CF, Curtis RE, Holowaty E, Storm H, Pukkala E, van’t Veer MB, Fraumeni JF, Boice JD, Jr, Charke EA, Travis LB. Lung cancer after treatment for Hodgkin’s disease: focus on radiation effects. Radiat Res. 2003;159:161–173. doi: 10.1667/0033-7587(2003)159[0161:LCATFH]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- Heidenreich WF, Cullings HM, Funamoto S, Paretzke HG. Promoting action of radiation in the atomic bomb survivor carcinogenesis data? Radiat Res. 2007;168:750–756. doi: 10.1667/RR0919.1. [DOI] [PubMed] [Google Scholar]
- Hodgson DC, Koh ES, Tran TH, Heydarian M, Tsang R, Pintilie M, Xu T, Huang L, Sachs RK, Brenner DJ. Individualized estimates of second cancer risks after contemporary radiation therapy for Hodgkin lymphoma. Cancer. 2007;110:2576–2586. doi: 10.1002/cncr.23081. [DOI] [PubMed] [Google Scholar]
- Inskip PD, Monson RR, Wagoner JK, Stovall M, Davis FG, Kleinerman RA, Boice JD., Jr Cancer mortality following radium treatment for uterine bleeding. Radiat Res. 1990;123:331–344. doi: 10.2307/3577741. [DOI] [PubMed] [Google Scholar]
- Ivanov VK, Gorski AI, Tsyb AF, Ivanov SI, Naumenko RN, Ivanova LV. Solid cancer incidence among the Chernobyl emergency workers residing in Russia: estimation of radiation risks. Radiat Environ Biophys. 2004;43:35–42. doi: 10.1007/s00411-003-0223-6. [DOI] [PubMed] [Google Scholar]
- Ivanov VK, Gorsky AI, Kashcheev VV, Maksioutov MA, Tumanov KA (2009) Latent period in induction of radiogenic solid tumors in the cohort of emergency workers. Radiat Environ Biophys. doi:10.1007/s00411-009-0223-2 [DOI] [PubMed]
- Koh ES, Tran TH, Heydarian M, Sachs RK, Tsang RW, Brenner DJ, Pintilie M, Xu T, Chung J, Paul N, Hodgson DC. A comparison of mantle versus involved-field radiotherapy for Hodgkin’s lymphoma: reduction in normal tissue dose and second cancer risk. Radiat Oncol. 2007;2:13. doi: 10.1186/1748-717X-2-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp-Schneider A, Portier CJ. Distinguishing between models of carcinogenesis: the role of clonal expansion. Fundam Appl Toxicol. 1991;17:601–613. doi: 10.1016/0272-0590(91)90210-U. [DOI] [PubMed] [Google Scholar]
- Lindsay KA, Wheldon EG, Deehan C, Wheldon TE. Radiation carcinogenesis modelling for risk of treatment-related second tumours following radiotherapy. Br J Radiol. 2001;74:529–536. doi: 10.1259/bjr.74.882.740529. [DOI] [PubMed] [Google Scholar]
- Little MP. Comparison of the risks of cancer incidence and mortality following radiation therapy for benign and malignant disease with the cancer risks observed in the Japanese A-bomb survivors. Int J Radiat Biol. 2001;77:431–464. doi: 10.1080/09553000010022634. [DOI] [PubMed] [Google Scholar]
- Little MP (2009) Heterogeneity of variation of relative risk by age at exposure in the Japanese atomic bomb survivors. Radiat Environ Biophys [DOI] [PubMed]
- Little MP, Li G. Stochastic modelling of colon cancer: is there a role for genomic instability? Carcinogenesis. 2007;28:479–487 . doi: 10.1093/carcin/bgl173. [DOI] [PubMed] [Google Scholar]
- Little MP, Wright EG. A stochastic carcinogenesis model incorporating genomic instability fitted to colon cancer data. Math Biosci. 2003;183:111–134. doi: 10.1016/S0025-5564(03)00040-3. [DOI] [PubMed] [Google Scholar]
- Lundell M, Holm LE. Risk of solid tumors after irradiation in infancy. Acta Oncol. 1995;34:727–734. doi: 10.3109/02841869509127179. [DOI] [PubMed] [Google Scholar]
- Moolgavkar SH. The multistage theory of carcinogenesis and the age distribution of cancer in man. J Natl Cancer Inst. 1978;61:49–52. doi: 10.1093/jnci/61.1.49. [DOI] [PubMed] [Google Scholar]
- Moolgavkar S. Multistage models for carcinogenesis. J Natl Cancer Inst. 1980;65:215–216. [PubMed] [Google Scholar]
- Moolgavkar SH, Luebeck EG. Multistage carcinogenesis and the incidence of human cancer. Genes Chromosomes Cancer. 2003;38:302–306. doi: 10.1002/gcc.10264. [DOI] [PubMed] [Google Scholar]
- Neglia JP, Robison LL, Stovall M, Liu Y, Packer RJ, Hammond S, Yasui Y, Kasper CE, Mertens AC, Donaldson SS, Meadows AT, Inskip PD. New primary neoplasms of the central nervous system in survivors of childhood cancer: a report from the Childhood Cancer Survivor Study. J Natl Cancer Inst. 2006;98:1528–1537. doi: 10.1093/jnci/djj411. [DOI] [PubMed] [Google Scholar]
- Ogawa K, Utsunomiya T, Mimori K, Tanaka F, Haraguchi N, Inoue H, Murayama S, Mori M. Differential gene expression profiles of radioresistant pancreatic cancer cell lines established by fractionated irradiation. Int J Oncol. 2006;28:705–713. [PubMed] [Google Scholar]
- Phillips TM, McBride WH, Pajonk F. The response of CD24(−/low)/CD44+ breast cancer-initiating cells to radiation. J Natl Cancer Inst. 2006;98:1777–1785. doi: 10.1093/jnci/djj495. [DOI] [PubMed] [Google Scholar]
- Press WH. Numerical recipes: the art of scientific computing. Cambridge: Cambridge University Press; 1989. [Google Scholar]
- Preston DL, Ron E, Tokuoka S, Funamoto S, Nishi N, Soda M, Mabuchi K, Kodama K. Solid cancer incidence in atomic bomb survivors: 1958–1998. Radiat Res. 2007;168:1–64. doi: 10.1667/RR0763.1. [DOI] [PubMed] [Google Scholar]
- Rachidi W, Harfourche G, Lemaitre G, Amiot F, Vaigot P, Martin MT. Sensing radiosensitivity of human epidermal stem cells. Radiother Oncol. 2007;83:267–276. doi: 10.1016/j.radonc.2007.05.007. [DOI] [PubMed] [Google Scholar]
- Rew DA, Wilson GD. Cell production rates in human tissues and tumours and their significance Part II: clinical data. Eur J Surg Oncol. 2000;26:405–417. doi: 10.1053/ejso.1999.0907. [DOI] [PubMed] [Google Scholar]
- Ron E. Childhood cancer–treatment at a cost. J Natl Cancer Inst. 2006;98:1510–1511. doi: 10.1093/jnci/djj437. [DOI] [PubMed] [Google Scholar]
- Ronckers CM, Sigurdson AJ, Stovall M, Smith SA, Mertens AC, Liu Y, Hammond S, Land CE, Neglia JP, Donaldson SS, Meadows AT, Sklar CA, Robison LL, Inskip PD. Thyroid cancer in childhood cancer survivors: a detailed evaluation of radiation dose response and its modifiers. Radiat Res. 2006;166:618–628. doi: 10.1667/RR3605.1. [DOI] [PubMed] [Google Scholar]
- Sachs RK, Brenner DJ. Solid tumor risks after high doses of ionizing radiation. Proc Natl Acad Sci USA. 2005;102:13040–13045. doi: 10.1073/pnas.0506648102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachs RK, Shuryak I, Brenner D, Fakir H, Hlatky L, Hahnfeldt P (2007) Second cancers after fractionated radiotherapy: stochastic population dynamics effects. J Theor Biol [DOI] [PMC free article] [PubMed]
- Shimomatsuya T, Tanigawa N, Muraoka R. Proliferative activity of human tumors: assessment using bromodeoxyuridine and flow cytometry. Jpn J Cancer Res. 1991;82:357–362. doi: 10.1111/j.1349-7006.1991.tb01854.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuryak I, Sachs RK, Hlatky L, Little MP, Hahnfeldt P, Brenner DJ. Radiation-induced leukemia at doses relevant to radiation therapy: modeling mechanisms and estimating risks. J Natl Cancer Inst. 2006;98:1794–1806. doi: 10.1093/jnci/djj497. [DOI] [PubMed] [Google Scholar]
- Shuryak I, Hahnfeldt P, Hlatky L, Sachs RK, Brenner DJ (2009) A new view of radiation-induced cancer: integrating short- and long-term processes. Part I: approach. Radiat Environ Biophys. doi:10.1007/s00411-009-0230-3 [DOI] [PMC free article] [PubMed]
- Thames HD, Bentzen SM, Turesson I, Overgaard M, van den Bogaert W. Fractionation parameters for human tissues and tumors. Int J Radiat Biol. 1989;56:701–710. doi: 10.1080/09553008914551931. [DOI] [PubMed] [Google Scholar]
- Thames HD, Bentzen SM, Turesson I, Overgaard M, Van den Bogaert W. Time-dose factors in radiotherapy: a review of the human data. Radiother Oncol. 1990;19:219–235. doi: 10.1016/0167-8140(90)90149-Q. [DOI] [PubMed] [Google Scholar]
- Tokunaga M, Norman JE, Jr, Asano M, Tokuoka S, Ezaki H, Nishimori I, Tsuji Y. Malignant breast tumors among atomic bomb survivors, Hiroshima and Nagasaki, 1950–74. J Natl Cancer Inst. 1979;62:1347–1359. [PubMed] [Google Scholar]
- Travis LB, Curtis RE, Boice JD, Jr, Platz CE, Hankey BF, Fraumeni JF., Jr Second malignant neoplasms among long-term survivors of ovarian cancer. Cancer Res. 1996;56:1564–1570. [PubMed] [Google Scholar]
- Travis LB, Curtis RE, Storm H, Hall P, Holowaty E, Van Leeuwen FE, Kohler BA, Pukkala E, Lynch CF, Andersson M, Bergfeldt K, Clarke EA, Wiklund T, Stoter G, Gospodarowicz M, Sturgeon J, Fraumeni JF, Jr, Boice JD., Jr Risk of second malignant neoplasms among long-term survivors of testicular cancer. J Natl Cancer Inst. 1997;89:1429–1439. doi: 10.1093/jnci/89.19.1429. [DOI] [PubMed] [Google Scholar]
- Travis LB, Gospodarowicz M, Curtis RE, Clarke EA, Andersson M, Glimelius B, Joensuu T, Lynch CF, van Leeuwen FE, Holowaty E, Storm H, Glimelius I, Pukkala E, Stovall M, Fraumeni JF, Jr, Boice JD, Jr, Gilbert E. Lung cancer following chemotherapy and radiotherapy for Hodgkin’s disease. J Natl Cancer Inst. 2002;94:182–192. doi: 10.1093/jnci/94.3.182. [DOI] [PubMed] [Google Scholar]
- Travis LB, Hill DA, Dores GM, Gospodarowicz M, van Leeuwen FE, Holowaty E, Glimelius B, Andersson M, Wiklund T, Lynch CF, Van’t Veer MB, Glimelius I, Storm H, Pukkala E, Stovall M, Curtis R, Boice JD, Jr, Gilbert E. Breast cancer following radiotherapy and chemotherapy among young women with Hodgkin disease. JAMA. 2003;290:465–475. doi: 10.1001/jama.290.4.465. [DOI] [PubMed] [Google Scholar]
- Travis LB, Fossa SD, Schonfeld SJ, McMaster ML, Lynch CF, Storm H, Hall P, Holowaty E, Andersen A, Pukkala E, Andersson M, Kaijser M, Gospodarowicz M, Joensuu T, Cohen RJ, Boice JD, Jr, Dores GM, Gilbert ES. Second cancers among 40, 576 testicular cancer patients: focus on long-term survivors. J Natl Cancer Inst. 2005;97:1354–1365. doi: 10.1093/jnci/dji278. [DOI] [PubMed] [Google Scholar]
- Upton AC. The state of the art in the 1990’s: NCRP Report No. 136 on the scientific bases for linearity in the dose-response relationship for ionizing radiation. Health Phys. 2003;85:15–22. doi: 10.1097/00004032-200307000-00005. [DOI] [PubMed] [Google Scholar]
- van Leeuwen FE, Klokman WJ, Stovall M, Dahler EC, van’t Veer MB, Noordijk EM, Crommelin MA, Aleman BM, Broeks A, Gospodarowicz M, Travis LB, Russell NS. Roles of radiation dose, chemotherapy, and hormonal factors in breast cancer following Hodgkin’s disease. J Natl Cancer Inst. 2003;95:971–980. doi: 10.1093/jnci/95.13.971. [DOI] [PubMed] [Google Scholar]
- Walsh L (2009) Heterogeneity of variation of relative risk by age at exposure in the Japanese atomic bomb survivors. Radiat Environ Biophys [DOI] [PubMed]
- Williams JR, Zhang Y, Zhou H, Gridley DS, Koch CJ, Russell J, Slater JS, Little JB. A quantitative overview of radiosensitivity of human tumor cells across histological type and TP53 status. Int J Radiat Biol. 2008;84:253–264. doi: 10.1080/09553000801953342. [DOI] [PubMed] [Google Scholar]