

Abstract
Purpose
The immunophilin protein FKBP8 interacts with Bcl2/Bcl-XL and is essential for mouse eye development. The purpose of this study was to define the expression of the FKBP8 gene in cultured human RPE cells and explore its involvement in the control of apoptosis.
Methods
Rapid amplification of cDNA ends (RACE) was performed on RNA isolated from human RPE cells. The existence of FKBP8 and a splice variant was confirmed by RT-PCR. The interaction between Bcl-XL and FKBP8 was measured by coimmunoprecipitation. ARPE-19 cells stably overexpressing FKBP8 and its splice variant were established. Their responses to thapsigargin and t-butyl hydroperoxide–induced cell death were measured by flow cytometry. Apoptosis was determined by terminal deoxyribonucleotidyl transferase-mediated fluorescein-dUTP nick-end labeling (TUNEL) assay. The activities of the nuclear factor of activated T cells (NFAT) were measured by reporter assay after transient transfection.
Results
RACE and RT-PCR identified a splice variant of FKBP8 lacking exons 3, 4, and 5 in human RPE cells. Both the full-length and the short form of FKBP8 proteins showed mitochondrial distribution and directly interacted with Bcl-XL. Overexpression of FKBP8 caused increased sensitivity to apoptosis induced by thapsigargin. The transcriptional activity of NFAT was not affected by FKBP8.
Conclusions
FKBP8 and its novel splice variant are Bcl-XL-interacting proteins and regulate the apoptotic signaling pathways in the RPE.
The human retinal pigment epithelial (RPE) cells have low turnover rate and often survive for an individual’s lifespan.1 However, when there is loss of RPE cells via apoptosis, which may occur in chronic eye diseases such as age-related macular degeneration (AMD), retinal function can be compromised. Patchy loss of RPE is one of the classic features of AMD2 and it has been shown that this process involves apoptosis.3 In eyes with wet AMD, apoptotic cells are present in choroidal neovascular membranes.4 The molecular mechanisms controlling mitochondria- and death receptor-mediated apoptosis have been studied extensively5-7 and it has been clearly demonstrated that the Bcl2 family of proteins are central regulators of apoptosis.8-10 Results from recent studies indicate that an immunophilin protein, FKBP8, interacts with Bcl2/ Bcl-XL and regulates the downstream signaling events.11-14
FK506-binding proteins (FKBPs) belong to a major family of immunophilins that bind to immunosuppressive drugs, FK506 and rapamycin, and convey their effects on immune responses and apoptosis.15 At least 15 FKBPs have been identified in humans.16,17 Despite the distinct subcellular localization, all FKBP proteins have a conserved motif, the FK506-binding domain, that mediates peptidyl-prolyl cis-trans isomerase (PPIase) activity and FK506 binding.16,17 FKBPs often function as chaperones or co-chaperones by directly interacting with other proteins. For instance, the prototypical FKBP family member FKBP12 binds to ryanodine receptor (RyR) and inositol 1,4,5-trisphosphate (IP3) receptor, both of which are major intracellular calcium channels.18,19 Accordingly, it plays a key regulatory role in intracellular Ca2+ signaling, and most FKBP12-knockout mice die during embryonic development because of severe cardiomyopathy.20
FKBP8 (also known as FKBP38) was originally isolated from a T-cell cDNA library by a degenerative PCR approach.21 It is expressed in a variety of tissues with the highest abundance in the brain.21 The gene is located on chromosome 19, area p12, and consists of 9 exons.22 Compared with other FKBP family proteins, FKBP8 displays unique features in FK506 binding ability and PPIase activity. Instead of being constitutively active or inactive, its PPIase activity requires prior binding of calcium and calmodulin (CaM) via a CaM binding domain.13 FKBP8 binds to FK506 only in the presence of Ca/CaM.13,23 FKBP8-knockout mice died at embryonic day (E)13.5 and showed defects in eye development.24 Another unique function of FKBP8 is that it interacts with Bcl2 and Bcl-XL.11-13 Although controversial, such interaction has been demonstrated in different types of cells and is believed to be important for the localization and protein stability of Bcl2/Bcl-XL.11-14
The high abundance of FKBP8 in the retina and the requirement of FKBP8 for normal eye development24 suggest that the protein performs important functions in this structure. In the present study, we showed that the FKBP8 gene is expressed in cultured human RPE cells, along with a splice variant lacking the FK506-binding domain. Overexpression of both the full-length and the short form of FKBP8 sensitized RPE cells to thapsigargin-induced apoptosis, but did not affect the transcriptional function of the nuclear factor of activated T cells (NFAT). These data indicate that, by interacting with Bcl2 family proteins, FKBP8 may be a functional component of the apoptotic signaling network in the RPE cells and that such an effect could be independent of FK506.
Materials and Methods
Cell Culture
Cultures of human retinal pigment epithelial (hRPE) cells were established as described previously.25 ARPE-19 cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA). The cells were cultured in Dulbecco’s modified Eagle’s medium with 10% fetal bovine serum in a humidified CO2 incubator at 37°C.
RNA Isolation, cDNA Synthesis, and Rapid Amplification of cDNA Ends
Total RNA was prepared from human RPE cells (RNeasy Mini Kit; Qiagen, Valencia, CA) and was reverse transcribed into cDNA by using random hexamer primers (Applied Biosystems, Inc. [ABI], Foster City, CA). To perform 5′ rapid amplification of cDNA ends (RACE), we used a primer (5′-GCA GCA AGG AAC TCT CGG GCC AGG G-3′) corresponding to the region of 301-325 (GenBank accession number NM_012181; http://www.ncbi.nlm.nih.gov/Genbank; provided in the public domain by the National Center for Biotechnology Information, Bethesda, MD) to amplify the 5′ end of FKBP8 cDNA (Smart RACE cDNA amplification kit; BD-Clontech, Mountain View, CA). PCR products were ligated into the TOPO TA cloning vector (Invitrogen, Carlsbad, CA), and multiple clones were chosen for sequencing.
Construction and Overexpression of FLAG-Tagged FKBP8 in ARPE-19 Cells
The open reading frames of FKBP8 and its splice variant were amplified with the primer pairs: forward, 5′-AGC AGC ATG GAC TAC AAA GAT GAT GAC GAC AAG GCA TCG TGT GCT GAA CCC TC-3′; and reverse, 5′-GTG GTC AGT TCC TGG CAG CG-3′. Both the full-length and the splice variant were cloned into the pQCXIP vector (BD-Clontech), and the inserts were verified by DNA sequencing. The plasmids containing FKBP8 or the splice variant (FKBP8s) were transfected into 293T viral cells, along with the viral packaging plasmids pCGP and pVSV (Takara, Madison, WI; FuGENE 6 transfection reagent; Roche Applied Science, Indianapolis, IN).26 Two days after transfection, the culture medium containing the replication-deficient virus was collected, filtered through a 0.45-μm filter, and used to infect ARPE-19 cells. Cells were then selected with 1 μg/mL puromycin for 2 days, and the surviving cells were pooled for further experiments.
Measurement of FKBP8 Localization by Confocal Microscopy
ARPE-19 cells stably expressing FLAG-FKBP8 and FLAG-FKBP8s were seeded onto four-well glass culture chamber slides (Nunc Inc., Rochester, NY). The mitochondria were stained with 100 nM red fluorescent tracer (MitoTracker Red; Invitrogen-Molecular Probes, Eugene, OR).27 The cells were then fixed with 2% formaldehyde followed by permeabilization with 100% methanol. The recombinant FLAG-FKBP8 and FLAG-FKBP8s were stained with anti-FLAG antibody (Sigma-Aldrich, St. Louis, MO). Images were acquired with a confocal microscope (LSM 510; Carl Zeiss Meditec, Inc., Dublin, CA)28 provided by the Vanderbilt Cell Imaging Shared Resource.
Coimmunoprecipitation
The cells were grown on a 100-mm dish for 48 hours before they were harvested. FLAG-tagged FKBP8 and FKBP8s were immunoprecipitated (FLAG Tagged protein immunoprecipitation kit; Sigma-Aldrich), according to the manufacturer’s protocol. Briefly, the cells were lysed with 1 mL of lysis buffer provided with the kit and incubated with the anti-FLAG M2 affinity gel overnight. After stringent washing, immunoprecipitates were eluted by 3× FLAG peptide. To immunoprecipitate Bcl-XL, the cell lysates were incubated with antibodies to Bcl-XL (Santa Cruz Biotechnology, Santa Cruz, CA) and protein G-sepharose beads. The immunoprecipitates were boiled in 2× SDS loading buffer and subjected to Western blot analysis.11
Cell Viability and TUNEL Assay
After treatment with thapsigargin and t-butyl hydroperoxide (tBH), the attached cells were released by tryptic digestion and pooled with floating cells. After being washed with phosphate-buffered saline (PBS), the cells were stained with a cell viability–toxicity kit (LIVE/DEAD; Invitrogen-Molecular Probes) for 30 minutes at room temperature and analyzed using flow cytometry (BD Immunocytometry Systems, San Jose, CA).28 For TUNEL staining, harvested cells were fixed in 4% formaldehyde and permeabilized with 0.1% Triton X-100. A cell-viability assay (In Situ Cell Death Detection Kit; Roche Applied Sciences) was used for terminal deoxyribonucleotidyl transferase (TdT)-mediated DNA nick-end labeling, followed by flow cytometry.25
Expression and Transcriptional Activity Measurement of NFAT
The mRNA of different NFAT genes was amplified by RT-PCR with gene-specific primers (Table 1) and RNA isolated from ARPE-19 cells. The PCR products were resolved by 1% agarose gel electrophoresis. To measure the transactivation of NFAT in the RPE, the cells were transiently transfected with an NFAT-luciferase reporter plasmid (Stratagene, La Jolla, CA) together either FKBP8 or FKBP8s subcloned in the pcDNA3.1 expression vector. A pRL-CMV Vector (Promega, Madison, WI) was cotransfected as an internal control. After 24 hours, the cells were stimulated with 1 μg/mL ionomycin and 60 μg/mL phorbol 12-myristate 13-acetate (PMA) for 6 hours. To inhibit NFAT activation, we pretreated the cells with 30 μM FK506. The luciferase activity was measured by using a dual-luciferase reporter assay system (Promega).29
Table 1.
Primers Used for PCR Amplifications
| Gene | Primer Sequence | Product Size (bp) |
|---|---|---|
| NFAT-c1 | F: 5′-AGC CGA ATT CTC TGG TGG TTG AGA-3′ | 247 |
| R: 5′-AGC TGC TGG CTG TAG TAT GGT CTT-3′ | ||
| NFAT-c2 | F: 5′-AAC TGT CAC CAC CAC CAG CTA TGA-3′ | 321 |
| R: 5′-AGG CAG CTG TCT GTG TCT TGT CTT-3′ | ||
| NFAT-c3 | F: 5′-TCA AGT CAC ACC AAC ACC TCC TGT-3′ | 264 |
| R: 5′-TGA GGC AGG GTA TGC ACA GAA TGA-3′ | ||
| NFAT-c4 | F: 5′-AGA CCC TGC TTG CGA AAC TCC TTA-3′ | 316 |
| R: 5′-ATG CAC ATC ACT CTG GGA AGG GAA-3′ | ||
| NFAT5 | F: 5′-TGC TGT GTT ACA GGG CTC TTC AGT-3′ | 239 |
| R: 5′-GTT GTG CCT GTT CTT GGG AAA GCA-3′ | ||
| FKBP8 | F: 5′-ATG GCA TCG TGT GCT GAA CCC TC-3′ | Full length: 1239; Variant: 759 |
| Primer pair 1 | R: 5′-GTT CCT GGC AGC GAT GAC CAC ACA-3′ | |
| FKBP8 | F: 5′-CAT GGG ACA ACC CCC GGC GGA G-3′ | 133 |
| Primer pair 2 | R: 5′-CGT CAT GTC CAC CAG AAT GTC C-3′ | |
| FKBP8 | F: 5′-GGA CAT TCT GGT GGA CAT GAC G-3′ | 478 |
| Primer pair 3 | R: 5′-GTT CCT GGC AGC GAT GAC CAC ACA-3′ |
Results
Human RPE Expression of FKBP8 and a Novel Splice Variant
Results from several previous studies have been inconsistent in defining the open reading frame and the mRNA sequence of the FKBP8 gene.22,30 To determine the RPE-specific expression and to obtain more information on the 5′ end of FKBP8 mRNA, we performed 5′ RACE with total RNA isolated from cultured human RPE cells. The reaction used an FKBP8-specific primer and an adaptor primer that was provided with the RACE kit. The longest sequence obtained from the amplification reaction encodes 412 amino acids and is a good match with one of the sequences deposited in GenBank (NM_012181).
To confirm the existence of FKBP8 transcripts in the RPE, we designed primers to perform RT-PCR amplification of the entire open reading frame of FKBP8 (Table 1). As shown in Figure 1, a strong band with size greater than 1000 bp was amplified from human RPE cells. An additional product of approximately 750 bp was also detected. Both bands were gel-purified and sequenced. The larger product corresponded to the full-length FKBP8 transcript. Sequencing of the smaller band revealed a new splice variant lacking exons 3, 4, and 5 (Fig. 2). The presence of the splice variant was further verified by RT-PCR by using primers specific to the unique junction area of the isoform (Fig. 1, lanes 2, 3; Table 1). The same mRNA splice variant was also detected in cultured human neuroblastoma cells (data not shown). The absence of exons 3, 4, and 5 in the splice variant did not generate frame shift mutation. Although the FK506-binding domain is missing in the predicted protein sequence, the variant still has the CaM-binding domain, transmembrane (TM) domain, and most of the protein–protein interaction domain containing the tetratricopeptide repeat (TPR; Fig. 2).
Figure 1.
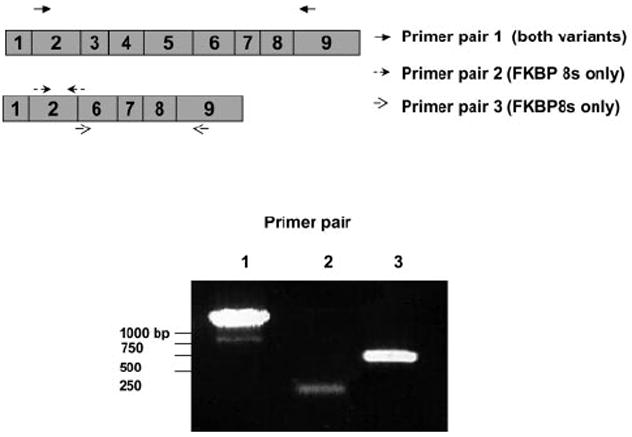
Measurement of FKBP8 expression in human RPE cells. The transcripts of the FKBP8 gene were amplified by RT-PCR with specific primers (Table 1). PCR products were run on 1% agarose gels, stained with ethidium bromide, and visualized under UV light. The data presented are representative of results of two separate experiments.
Figure 2.
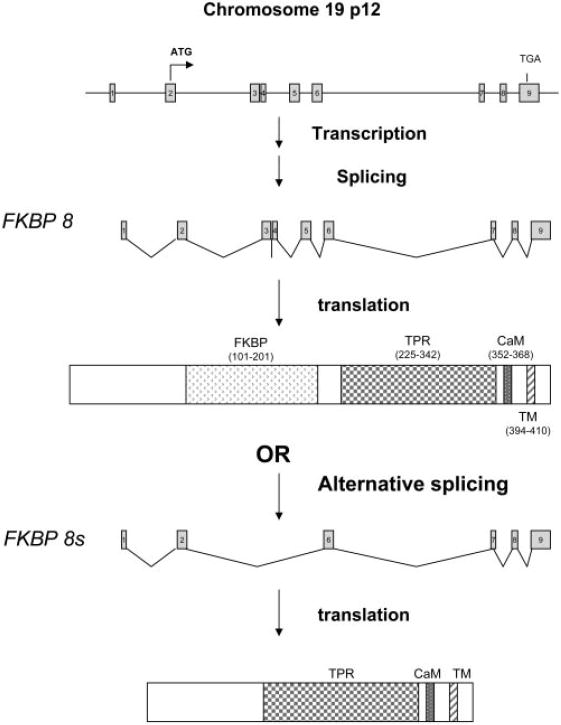
Schematic presentation of human FKBP8 gene and the splice variant. The alternative mRNA splicing resulted in a variant (FKBP8s) lacking the exons 3, 4, and 5, which encode for the FK506-binding domain.
FKBP8 contains a TM domain at its C terminus that is essential for its distribution in the mitochondrial outer membrane.11 To determine the subcellular distribution of FKBP8 proteins in the RPE, ARPE-19 cells were stably transfected with FLAG-tagged FKBP8 or FKBP8s and stained with anti-FLAG antibody followed by confocal microscopy (Fig. 3B). The expressions of the tagged-proteins were verified by Western blot analysis (Fig. 3A). Consistent with the previous study,11 part of the FLAG-tagged FKBP8 showed colocalization with red fluorescence (MitoTracker Red; Invitrogen-Molecular Probes; Fig. 3B), indicating mitochondrial distribution. Most of the splice variant, FKBP8s, was also localized to the mitochondria.
Figure 3.
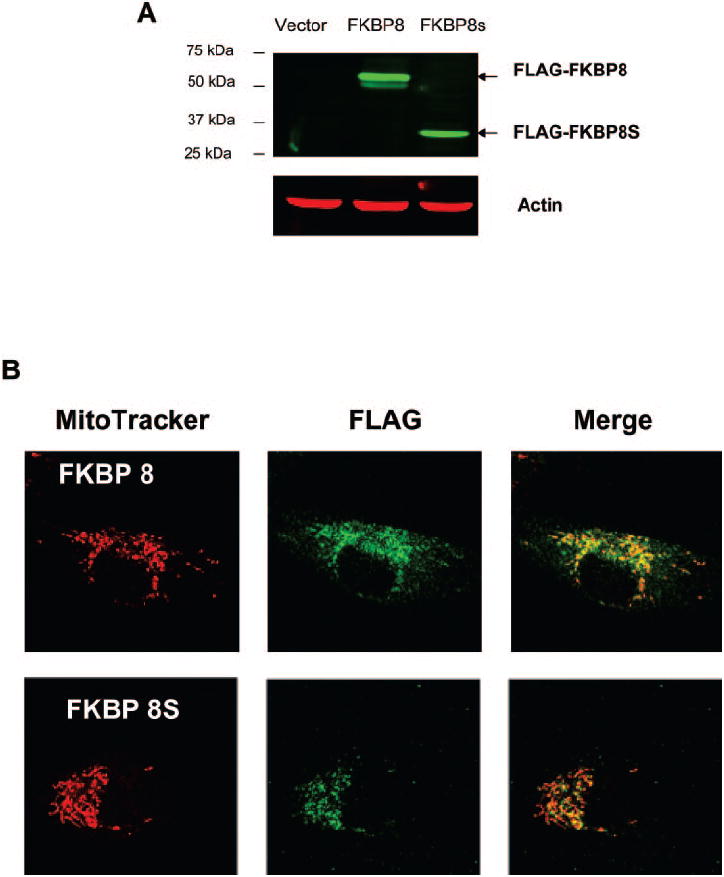
Subcellular localization of FKBP8 and FKBP8s. ARPE-19 cells were transduced with FLAG-tagged FKBP8, FKBP8s, or empty vector, and the expression was verified by Western blot analysis (A). The distribution of epitope-tagged proteins was indicated by immunostaining with anti-FLAG antibody and mitochondria were stained with a fluorescent tracker (MitoTracker Red; Invitrogen-Molecular Probes, Eugene, OR) (B). Top: cells transduced with FKBP8; bottom: represents cells transduced with FKBP8s.
FKBP8 Interaction with Bcl-XL and Increased Sensitivity to Thapsigargin in the RPE
To determine the interaction between FKBP8 and BCL-XL in cultured RPE cells, we performed a coimmunoprecipitation (Co-IP) assay using ARPE-19 cells stably transduced with empty vector, FLAG-FKBP8, or FLAG-FKBP8s. When FLAG-tagged FKBPs were immunoprecipitated using the anti-FLAG antibody, BCL-XL was found to be associated with either FLAG-FKBP8 or FLAG-FKBP8s, but not in vector control cells (Fig. 4A). A reciprocal Co-IP assay using anti-BCL-XL showed that both FLAG-FKBP8 and FLAG-FKBP8s bound to Bcl-XL, but not with the control rabbit IgG (Fig. 4B). These results suggest that the splice variant of FKBP8 is still capable of binding to BCL-XL despite the absence of the FK506-binding domain.
Figure 4.
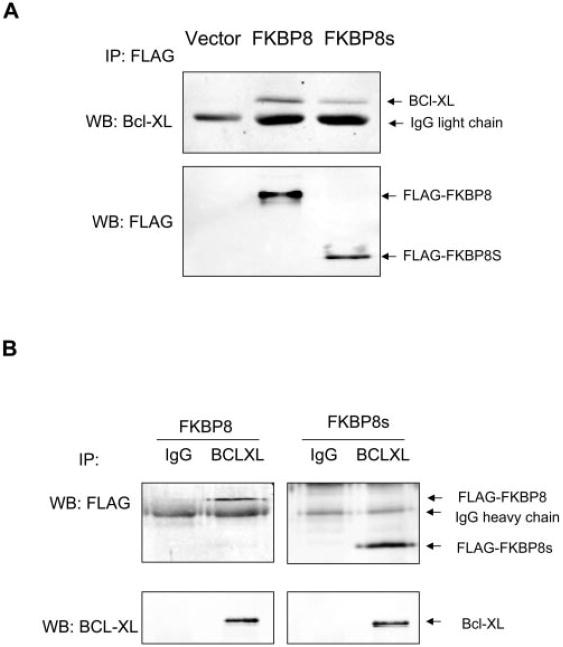
FKBP8 and the splice variant FKBP8s interacted with Bcl-XL in RPE cells. ARPE-19 cells stably overexpressing FKBP8 or FKBP8s were subjected to co-IP with either anti-FLAG (A) or anti-Bcl-XL (B) antibody. The precipitated immune complexes were subjected to Western blot analyses (WB) with anti-Bcl-XL and anti-FLAG.
To determine functional consequences of the interaction between Bcl-XL and FKBP8 in the RPE, thapsigargin and tBH were used to induce apoptosis in ARPE-19 cells stably overexpressing either FKBP8 or FKBP8s. Increased sensitivity to thapsigargin was observed in cells overexpressing FKBP8s (Fig. 5A). After a 72-hour treatment, no significant change in cell viability was observed in vector-transduced ARPE cells. However, a nearly 40% loss of viability was detected in cells overexpressing FKBP8s exposed to 3 μM thapsigargin. At the same concentration, full length FKBP8 also potentiated the toxicity of thapsigargin. All three cell lines showed comparable responses to tBH-induced cell death (Fig. 5B). Thapsigargin-induced apoptosis and DNA strand breaks were also detected by TUNEL assay (Fig. 6A). The quantification results from the TUNEL assay were comparable to the viability data (Fig. 6B).
Figure 5.
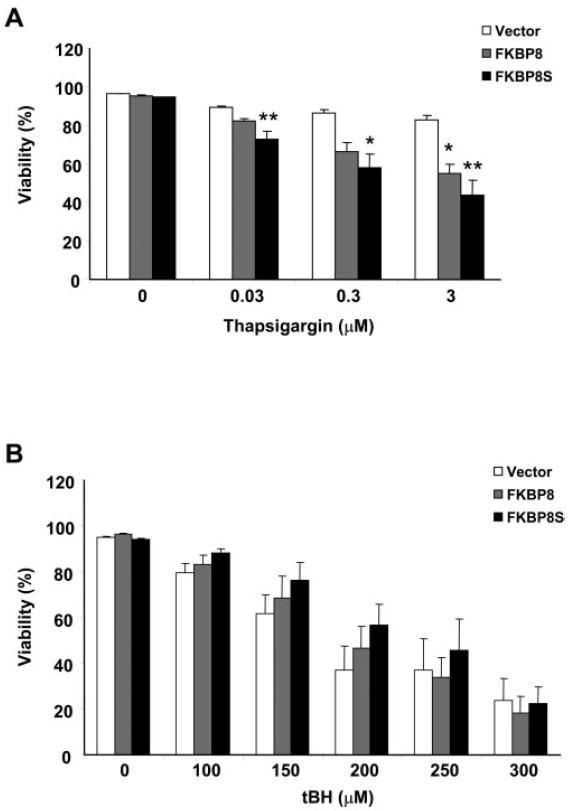
FKBP8 and FKBP8s had similar regulatory roles in RPE apoptosis. ARPE-19 cells transduced with vector, FKBP8, and FKBP8s were treated with different concentrations of thapsigargin for 72 hours (A) or tBH for 16 hours (B). The viability was analyzed with a cytotoxicity kit. Data are the averages of results in five separate experiments (mean ± SE). *P < 0.05 and **P < 0.01, significant differences from vector, determined by one-way ANOVA and the Dunnett multiple comparison test.
Figure 6.
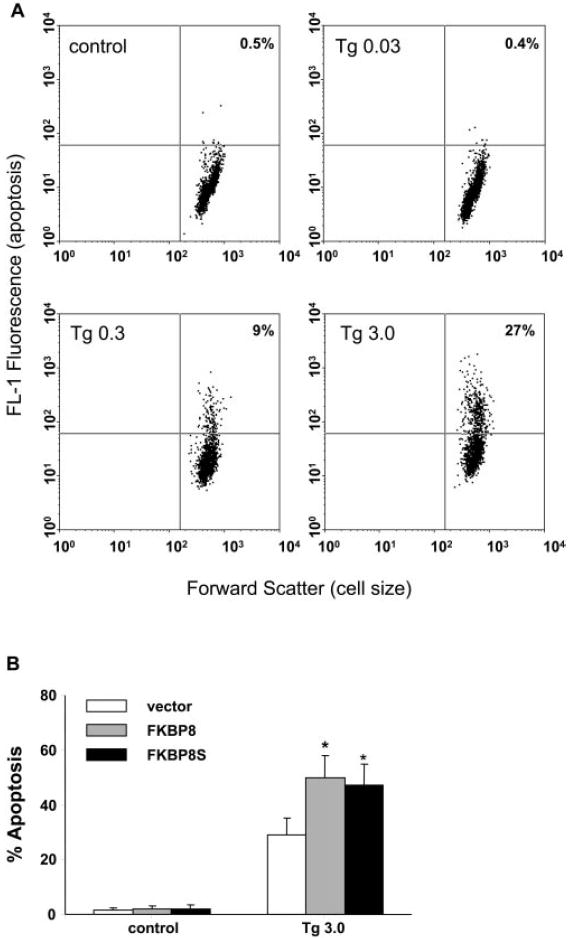
Thapsigargin-induced apoptosis in cultured ARPE-19 cells. (A) ARPE cells stably overexpressing FKBP8 were treated with 0.03 (Tg0.03), 0.3 (Tg 0.3), and 3 (Tg 3.0) μM thapsigargin for 72 hours. Apoptotic cells were stained by TUNEL and analyzed by flow cytometry. The populations of cells in the upper right quadrants were apoptotic. (B) Apoptosis in ARPE cells transduced with vector, FKBP8 and FKBP8s after exposure to thapsigargin for 72 hours. Data represented are the averages of three separate experiments (mean ± SE). *P < 0.05, significant differences from vector, determined by one-way ANOVA and Dunnett multiple comparison test.
FK506 exerts its immunosuppressive activities by binding to FKBPs and calcineurin (CaN) and consequently modulates the CaN/NFAT pathway.16,17 To determine whether FKBP8 affects NFAT function, we used a reporter assay to measure the transcriptional activity of NFAT after exposure to phorbol ester and ionomycin. Cultured RPE cells expressed the calcium-dependent NFAT-c1, -c3, and c4 and calcium-independent NFAT5 (Fig. 7A). When stimulated with PMA/ionomycin, the NFAT activity was increased by nearly threefold in vector control cells and FK506 completely inhibited NFAT activation (Fig. 7B). Cotransfection of either the full-length or splice variant of FKBP8 did not significantly alter the function of NFAT (Fig. 7B).
Figure 7.
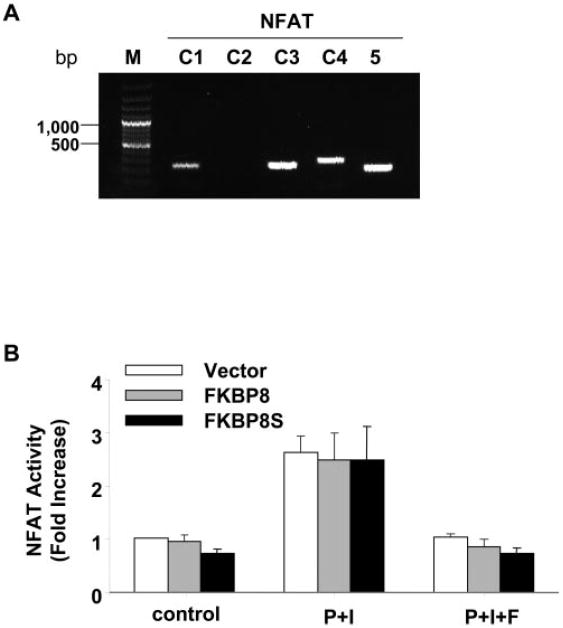
Effects of FKBP8 on NFAT activation in the ARPE cells. (A) The mRNA expression of different NFAT genes was analyzed by RT-PCR. (B) The transcriptional activity of NFAT was measured by a reporter assay after transient cotransfection with FKBP8 or FKBP8s. NFAT was activated by ionomycin and PMA treatment (P+I) and was inhibited by FK506 (P+I+F). Data presented are the average of results in three separate experiments. No significant differences were observed in the three cell lines.
Discussion
The development and progression of AMD involves multiple factors, including genetic variations,31,32 inflammatory and immune response,33-35 and environmental oxidative stress.36-39 All these risk factors can affect the apoptotic signaling pathways and result in the loss of RPE cells by programmed cell death. The Bcl2/Bcl-XL pathway of apoptosis is largely conserved from nematode to human.40 However, in higher organisms, various tissue-specific regulatory mechanisms have evolved to control the functions of the Bcl2 family proteins. Thus, characterization of the Bcl-XL-associated signaling network will facilitate the understanding of the molecular mechanisms implicated in the degeneration process of the retina and the RPE.
In the present study, we performed initial characterization of a Bcl-XL-interacting protein, FKBP8, in cultured human RPE cells. The human FKBP8 gene contains 9 exons, and two potential ATG start codons exist in the second exon. Using the 5′-RACE approach, we have provided experimental evidence defining the mRNA transcripts that use the first ATG as the translational initiation codon. Furthermore, we identified a novel splice variant that is expressed in human RPE cells (Figs. 1, 2). It encodes a short form of FKBP8 protein, which lacks the FK506-binding domain but retains protein–protein interaction TPR, CaM, and TM domains (Fig. 2).
As anticipated from the function of the TM domain, FKBP8s had mitochondrial localization (Fig. 3). The interaction between FKBP8 and BCL-XL appears to require multiple domains of FKBP8.11 In the present study, we showed that FKBP8s lacking the FK506-binding domain was still able to bind BCL-XL (Fig. 4) and had functions as similar to those of full-length FKBP8 in RPE apoptosis (Figs. 5, 6). Results from the previous studies also showed that the binding with FK506, although essential in regulating immune responses, was dispensable for the interaction between FKBP8 and BCL-XL.11 In fact, a more potent FKBP inhibitor, GPI1046, was able to release Bcl2 from FKBP8.13 It appears that the FK506-binding domain is involved in the dissociation of FKBP8 from Bcl2/Bcl-XL, which may partly explain why overexpression of FKBP8s had more pronounced effects than FKBP8 in response to thapsigargin treatment (Fig. 5A).
FKBP8 exerts anti- or proapoptotic effects in a cell-type–dependent manner.11,13,14 In HeLa cells, manipulation of FKBP8 expression by overexpression or RNAi indicates an antiapoptotic function of FKBP8.11,14 However, knockdown of FKBP8 confers protection against apoptosis in neuroblastoma cells.13 Our study showed that in RPE cells, FKBP8 has a proapoptotic function similar to that in neuroblastoma cells. The binding between FKBP8 and Bcl2 can be antiapoptotic by inhibiting phosphorylation or increasing the stability of Bcl2.14,41 Conversely, the binding of FKBP8 to Bcl2 can prevent Bcl2 from interacting with BAD and CaN and therefore can prevent the antiapoptotic functions of Bcl2.13,23 How FKBP8 regulates the cell death program in RPE cells in response to pathologically relevant stimuli remains to be characterized in the future.
The Bcl2 family of proteins have subcellular locations on the mitochondrial outer membrane, nuclear envelope, and endoplasmic reticulum.42,43 Bcl2 and Bcl-XL interact with the IP3 receptors and modulate the release of calcium from the endoplasmic reticulum.44-46 They also interact with calmodulin-dependent protein kinase and control the formation of autophagy.47 Thus, by interacting with Bcl-XL, FKBP8 may have various cellular effects that are not associated with apoptosis and remain to be defined further. Results from the NFAT activity measurements (Fig. 7) indicated that FKBP8 may not be a major determinant of NFAT-mediated transcriptional regulation of genes involved in cellular adaptive responses or the immune response. It will be interesting to determine how the ratio between the mRNA and protein levels of the full-length and the splice variant of FKBP8 can be changed in response to conditions of chronic and acute oxidative stress. Such changes may contribute to the sensitivity of the RPE and other types of neuronal or epithelial cells to various stimuli of apoptosis.
As a unique member of the FKBP family of immunophilins, FKBP8 appears to have important but not yet well-defined functions in age-related degenerative diseases. For instance, Presenilins 1 and 2, suggested as causative molecules for familial Alzheimer’s disease, promote apoptosis at least partly through altering the subcellular distribution of FKBP8 and Bcl2.12 The neuroprotective effects of FK506 and its derivatives have been shown in models of Parkinson’s disease and other conditions of neurotoxicity.48 Thus, FKBP8 and other FKBPs are potential targets of intervention for diseases like AMD, which involves dysfunction in the control of apoptosis as well as the immune response.
Acknowledgments
Supported by the American Health Association Foundation, the Alston Callahan Postdoctoral Scholar Award (YC) from International Retinal Research Foundation, National Eye Institute Grants EY07892 and EY08126, National Institute of Environmental Health Sciences Grant ES014668, and Research to Prevent Blindness, Inc.
Footnotes
Disclosure: Y. Chen, None; P. Sternberg, None; J. Cai, None
References
- 1.Machemer R, Laqua H. Pigment epithelium proliferation in retinal detachment (massive periretinal proliferation) Am J Ophthalmol. 1975;80:1–23. doi: 10.1016/0002-9394(75)90862-4. [DOI] [PubMed] [Google Scholar]
- 2.Sarks JP, Sarks SH, Killingsworth MC. Evolution of geographic atrophy of the retinal pigment epithelium. Eye. 1988;2:552–577. doi: 10.1038/eye.1988.106. [DOI] [PubMed] [Google Scholar]
- 3.Dunaief JL, Dentchev T, Ying GS, Milam AH. The role of apoptosis in age-related macular degeneration. Arch Ophthalmol. 2002;120:1435–1442. doi: 10.1001/archopht.120.11.1435. [DOI] [PubMed] [Google Scholar]
- 4.Hinton DR, He S, Lopez PF. Apoptosis in surgically excised choroidal neovascular membranes in age-related macular degeneration. Arch Ophthalmol. 1998;116:203–209. doi: 10.1001/archopht.116.2.203. [DOI] [PubMed] [Google Scholar]
- 5.Saikumar P, Dong Z, Mikhailov V, Denton M, Weinberg JM, Venkatachalam MA. Apoptosis: definition, mechanisms, and relevance to disease. Am J Med. 1999;107:489–506. doi: 10.1016/s0002-9343(99)00259-4. [DOI] [PubMed] [Google Scholar]
- 6.Gupta S. Molecular signaling in death receptor and mitochondrial pathways of apoptosis (review) Int J Oncol. 2003;22:15–20. [PubMed] [Google Scholar]
- 7.Orrenius S, Gogvadze V, Zhivotovsky B. Mitochondrial oxidative stress: implications for cell death. Annu Rev Pharmacol Toxicol. 2007;47:143–183. doi: 10.1146/annurev.pharmtox.47.120505.105122. [DOI] [PubMed] [Google Scholar]
- 8.Chan SL, Yu VC. Proteins of the bcl-2 family in apoptosis signalling: from mechanistic insights to therapeutic opportunities. Clin Exp Pharmacol Physiol. 2004;31:119–128. doi: 10.1111/j.1440-1681.2004.03975.x. [DOI] [PubMed] [Google Scholar]
- 9.Gustafsson AB, Gottlieb RA. Bcl-2 family members and apoptosis, taken to heart. Am J Physiol. 2007;292:C45–C51. doi: 10.1152/ajpcell.00229.2006. [DOI] [PubMed] [Google Scholar]
- 10.Zhang N, Peairs JJ, Yang P, et al. The importance of Bcl-xL in the survival of human RPE cells. Invest Ophthalmol Vis Sci. 2007;48:3846–3853. doi: 10.1167/iovs.06-1145. [DOI] [PubMed] [Google Scholar]
- 11.Shirane M, Nakayama KI. Inherent calcineurin inhibitor FKBP38 targets Bcl-2 to mitochondria and inhibits apoptosis. Nat Cell Biol. 2003;5:28–37. doi: 10.1038/ncb894. [DOI] [PubMed] [Google Scholar]
- 12.Wang HQ, Nakaya Y, Du Z, et al. Interaction of presenilins with FKBP38 promotes apoptosis by reducing mitochondrial Bcl-2. Hum Mol Genet. 2005;14:1889–1902. doi: 10.1093/hmg/ddi195. [DOI] [PubMed] [Google Scholar]
- 13.Edlich F, Weiwad M, Erdmann F, et al. Bcl-2 regulator FKBP38 is activated by Ca2+/calmodulin. EMBO J. 2005;24:2688–2699. doi: 10.1038/sj.emboj.7600739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kang CB, Feng L, Chia J, Yoon HS. Molecular characterization of FK-506 binding protein 38 and its potential regulatory role on the anti-apoptotic protein Bcl-2. Biochem Biophys Res Commun. 2005;337:30–38. doi: 10.1016/j.bbrc.2005.09.023. [DOI] [PubMed] [Google Scholar]
- 15.Harrar Y, Bellini C, Faure JD. FKBPs: at the crossroads of folding and transduction. Trends Plant Sci. 2001;6:426–431. doi: 10.1016/s1360-1385(01)02044-1. [DOI] [PubMed] [Google Scholar]
- 16.Dumont FJ. FK506, an immunosuppressant targeting calcineurin function. Curr Med Chem. 2000;7:731–748. doi: 10.2174/0929867003374723. [DOI] [PubMed] [Google Scholar]
- 17.Barik S. Immunophilins: for the love of proteins. Cell Mol Life Sci. 2006;63:2889–2900. doi: 10.1007/s00018-006-6215-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Timerman AP, Ogunbumni E, Freund E, Wiederrecht G, Marks AR, Fleischer S. The calcium release channel of sarcoplasmic reticulum is modulated by FK-506-binding protein: dissociation and reconstitution of FKBP-12 to the calcium release channel of skeletal muscle sarcoplasmic reticulum. J Biol Chem. 1993;268:22992–22999. [PubMed] [Google Scholar]
- 19.Cameron AM, Steiner JP, Sabatini DM, Kaplin AI, Walensky LD, Snyder SH. Immunophilin FK506 binding protein associated with inositol 1,4,5-trisphosphate receptor modulates calcium flux. Proc Natl Acad Sci USA. 1995;92:1784–1788. doi: 10.1073/pnas.92.5.1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shou W, Aghdasi B, Armstrong DL, et al. Cardiac defects and altered ryanodine receptor function in mice lacking FKBP12. Nature. 1998;391:489–492. doi: 10.1038/35146. [DOI] [PubMed] [Google Scholar]
- 21.Lam E, Martin M, Wiederrecht G. Isolation of a cDNA encoding a novel human FK506-binding protein homolog containing leucine zipper and tetratricopeptide repeat motifs. Gene. 1995;160:297–302. doi: 10.1016/0378-1119(95)00216-s. [DOI] [PubMed] [Google Scholar]
- 22.Nielsen JV, Mitchelmore C, Pedersen KM, Kjaerulff KM, Finsen B, Jensen NA. Fkbp8: novel isoforms, genomic organization, and characterization of a forebrain promoter in transgenic mice. Genomics. 2004;83:181–192. doi: 10.1016/j.ygeno.2003.07.001. [DOI] [PubMed] [Google Scholar]
- 23.Weiwad M, Edlich F, Erdmann F, et al. A reassessment of the inhibitory capacity of human FKBP38 on calcineurin. FEBS Lett. 2005;579:1591–1596. doi: 10.1016/j.febslet.2004.12.098. [DOI] [PubMed] [Google Scholar]
- 24.Bulgakov OV, Eggenschwiler JT, Hong DH, Anderson KV, Li T. FKBP8 is a negative regulator of mouse sonic hedgehog signaling in neural tissues. Development. 2004;131:2149–2159. doi: 10.1242/dev.01122. [DOI] [PubMed] [Google Scholar]
- 25.Jiang S, Wu MW, Sternberg P, Jones DP. Fas mediates apoptosis and oxidant-induced cell death in cultured hRPE cells. Invest Ophthalmol Vis Sci. 2000;41:645–655. [PubMed] [Google Scholar]
- 26.Cai J, Kirlin WG, Chen Y, Yan X, Jones DP, Sartorelli AC. Overexpression of heat shock factor 1 inhibits butyrate-induced differentiation in colon cancer cells. Cell Stress Chaperones. 2006;11:199–207. doi: 10.1379/CSC-180R.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jiang S, Cai J, Wallace DC, Jones DP. Cytochrome c-mediated apoptosis in cells lacking mitochondrial DNA: signaling pathway involving release and caspase 3 activation is conserved. J Biol Chem. 1999;274:29905–29911. doi: 10.1074/jbc.274.42.29905. [DOI] [PubMed] [Google Scholar]
- 28.Chen Y, Cai J, Murphy TJ, Jones DP. Overexpressed human mitochondrial thioredoxin confers resistance to oxidant-induced apoptosis in human osteosarcoma cells. J Biol Chem. 2002;277:33242–33248. doi: 10.1074/jbc.M202026200. [DOI] [PubMed] [Google Scholar]
- 29.Ha KN, Chen Y, Cai J, Sternberg P., Jr Increased glutathione synthesis through an ARE-Nrf2-dependent pathway by zinc in the RPE: implication for protection against oxidative stress. Invest Ophthalmol Vis Sci. 2006;47:2709–2715. doi: 10.1167/iovs.05-1322. [DOI] [PubMed] [Google Scholar]
- 30.Okamoto T, Nishimura Y, Ichimura T, et al. Hepatitis C virus RNA replication is regulated by FKBP8 and Hsp90. EMBO J. 2006;25:5015–5025. doi: 10.1038/sj.emboj.7601367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moshfeghi DM, Blumenkranz MS. Role of genetic factors and inflammation in age-related macular degeneration. Retina. 2007;27:269–275. doi: 10.1097/IAE.0b013e31802e3e9b. [DOI] [PubMed] [Google Scholar]
- 32.DeWan A, Bracken MB, Hoh J. Two genetic pathways for age-related macular degeneration. Curr Opin Genet Dev. 2007;17:228–233. doi: 10.1016/j.gde.2007.04.004. [DOI] [PubMed] [Google Scholar]
- 33.Donoso LA, Kim D, Frost A, Callahan A, Hageman G. The role of inflammation in the pathogenesis of age-related macular degeneration. Surv Ophthalmol. 2006;51:137–152. doi: 10.1016/j.survophthal.2005.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Anderson DH, Mullins RF, Hageman GS, Johnson LV. A role for local inflammation in the formation of drusen in the aging eye. Am J Ophthalmol. 2002;134:411–431. doi: 10.1016/s0002-9394(02)01624-0. [DOI] [PubMed] [Google Scholar]
- 35.Hageman GS, Luthert PJ, Victor Chong NH, Johnson LV, Anderson DH, Mullins RF. An integrated hypothesis that considers drusen as biomarkers of immune-mediated processes at the RPE-Bruch’s membrane interface in aging and age-related macular degeneration. Prog Retin Eye Res. 2001;20:705–732. doi: 10.1016/s1350-9462(01)00010-6. [DOI] [PubMed] [Google Scholar]
- 36.Yildirim O, Ates NA, Tamer L, et al. Changes in antioxidant enzyme activity and malondialdehyde level in patients with age-related macular degeneration. Ophthalmologica. 2004;218:202–206. doi: 10.1159/000076845. [DOI] [PubMed] [Google Scholar]
- 37.Cai J, Nelson KC, Wu M, Sternberg P, Jr, Jones DP. Oxidative damage and protection of the RPE. Prog Retin Eye Res. 2000;19:205–221. doi: 10.1016/s1350-9462(99)00009-9. [DOI] [PubMed] [Google Scholar]
- 38.Beatty S, Koh H, Phil M, Henson D, Boulton M. The role of oxidative stress in the pathogenesis of age-related macular degeneration. Surv Ophthalmol. 2000;45:115–134. doi: 10.1016/s0039-6257(00)00140-5. [DOI] [PubMed] [Google Scholar]
- 39.AREDS study group. A randomized, placebo-controlled, clinical trial of high-dose supplementation with vitamins C and E and beta carotene for age-related cataract and vision loss: AREDS report no. 9. Arch Ophthalmol. 2001;119:1439–1452. doi: 10.1001/archopht.119.10.1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Reed JC. Bcl-2 family proteins. Oncogene. 1998;17:3225–3236. doi: 10.1038/sj.onc.1202591. [DOI] [PubMed] [Google Scholar]
- 41.Kang CB, Tai J, Chia J, Yoon HS. The flexible loop of Bcl-2 is required for molecular interaction with immunosuppressant FK-506 binding protein 38 (FKBP38) FEBS Lett. 2005;579:1469–1476. doi: 10.1016/j.febslet.2005.01.053. [DOI] [PubMed] [Google Scholar]
- 42.Krajewski S, Tanaka S, Takayama S, Schibler MJ, Fenton W, Reed JC. Investigation of the subcellular distribution of the bcl-2 oncoprotein: residence in the nuclear envelope, endoplasmic reticulum, and outer mitochondrial membranes. Cancer Res. 1993;53:4701–4714. [PubMed] [Google Scholar]
- 43.Akao Y, Otsuki Y, Kataoka S, Ito Y, Tsujimoto Y. Multiple subcellular localization of bcl-2: detection in nuclear outer membrane, endoplasmic reticulum membrane, and mitochondrial membranes. Cancer Res. 1994;54:2468–2471. [PubMed] [Google Scholar]
- 44.Li C, Wang X, Vais H, Thompson CB, Foskett JK, White C. Apoptosis regulation by Bcl-x(L) modulation of mammalian inositol 1,4,5-trisphosphate receptor channel isoform gating. Proc Natl Acad Sci USA. 2007;104:12565–12570. doi: 10.1073/pnas.0702489104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xu L, Kong D, Zhu L, Zhu W, Andrews DW, Kuo TH. Suppression of IP3-mediated calcium release and apoptosis by Bcl-2 involves the participation of protein phosphatase 1. Mol Cell Biochem. 2007;295:153–165. doi: 10.1007/s11010-006-9285-5. [DOI] [PubMed] [Google Scholar]
- 46.Pinton P, Rizzuto R. Bcl-2 and Ca2+ homeostasis in the endoplasmic reticulum. Cell Death Differ. 2006;13:1409–1418. doi: 10.1038/sj.cdd.4401960. [DOI] [PubMed] [Google Scholar]
- 47.Hoyer-Hansen M, Bastholm L, Szyniarowski P, et al. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol Cell. 2007;25:193–205. doi: 10.1016/j.molcel.2006.12.009. [DOI] [PubMed] [Google Scholar]
- 48.Gold BG, Nutt JG. Neuroimmunophilin ligands in the treatment of Parkinson’s disease. Curr Opin Pharmacol. 2002;2:82–86. doi: 10.1016/s1471-4892(01)00125-4. [DOI] [PubMed] [Google Scholar]