

Abstract
TNF-related apoptosis-inducing ligand (TRAIL) is a potential anticancer agent due to its selectivity in killing transformed cells. However, TRAIL can also stimulate TRAIL-resistant cancer cells’ proliferation and metastasis. Thus, acquired TRAIL resistance during TRAIL therapy would shift the patient’s treatment from beneficial to detrimental. In this study we focused on the acquired TRAIL resistance mechanism and demonstrated that the elevated expression of the anti-apoptotic factor cellular FLICE-like inhibitory protein (c-FLIP) and the pro-survival Bcl-2 family member myeloid cell leukemia 1 (Mcl-1) underlie the main mechanism of this type of TRAIL resistance in lung cancer cells. Chronic exposure to TRAIL resulted in lung cancer cell resistance to TRAIL-induced cytotoxicity, and this resistance was associated with the increase in the cellular levels of c-FLIP L and Mcl-1L. Overexpresssion of c-FLIPL suppressed recruitment of caspase-8 to the death-inducing signaling complex (DISC) while increased Mcl-1L expression blunted the mitochondrial apoptosis pathway. The elevation of c-FLIP L and Mcl-1L expression was due to Akt-mediated stabilization of these proteins in TRAIL-resistant cells. Importantly, suppressing c-FLIPL and Mcl-1L expression by RNA interference collectively alleviated acquired TRAIL resistance. Taken together, these results identify c-FLIPL and Mcl-1L as the major determinants of acquired TRAIL resistance and could be molecular targets for improving TRAIL’s therapeutic value against lung cancer.
Keywords: TRAIL, c-FLIP, Mcl-1, Akt, apoptosis, lung cancer
Introduction
TNF-related apoptosis-inducing ligand (TRAIL) is regarded as the most promising anticancer agent in the TNF superfamily of cytokines because of its selective cytotoxicity in transformed cells (1, 2). However, numerous cancer cells are insensitive to TRAIL-induced cytotoxicity. The mechanism of this type of primary TRAIL resistance in cancer cells has been attributed to dysfunction of different steps in the TRAIL-induced apoptosis pathway and/or elevation of survival signals (3). In addition, cancer cells acquire TRAIL resistance during the course of TRAIL exposure; the mechanism of which is controversial (1, 4-7). Notably, TRAIL can promote proliferation and metastasis in TRAIL-resistant cancer cells (8). The acquired TRAIL resistance is particularly important for the clinical application of TRAIL because it may switch TRAIL’s effect from being beneficial to detrimental during the course of TRAIL treatment. Therefore, it is crucial to understand the mechanism of acquired TRAIL resistance in order to retain TRAIL’s cancer-killing activity while circumventing its cancer promoting potential.
TRAIL’s cellular effects are mediated by its functional receptors, death receptor (DR) 4/TRAIL-R1 and DR5/TRAIL-R2. Similar to TNF receptor 1 (TNF-R1), DR4 and DR5 are death receptors that contain a death domain in their cytoplasmic region to transduce cell death signals (1). They are also able to transmit signals leading to activation of the transcription factor nuclear factor-κB (NF-κB) and the c-Jun N-terminal kinase (JNK) (9, 10). Decoy receptors (DcR) 1/TRAIL-R3 and DcR2/TRAIL-R4, and osteoprotegerin (OPG) can also bind TRAIL. Lacking a cytoplasmic region, DcR1 does not transmit signals into the cell. DcR2 has a truncated death domain, making it incapable of transmitting death signals while retaining the ability to mediate NF-κB and JNK activation (1). Both DcR1 and DcR2 function as decoy receptors to block TRAIL-induced apoptosis (1). OPG is a secreted TRAIL receptor that can also inhibit the therapeutic activity of TRAIL (11).
TRAIL-induced pro-apoptotic signals are transduced by the death-inducing signal complex (DISC) that consists of the TRAIL receptors, TRADD, RIP, TRAF2, and FADD (1). RIP-dependent NF-κB activation induced by TRAIL is anti-apoptotic (9). Whether the role of TRAIL-induced JNK activation is apoptotic or anti-apoptotic is somewhat controversial and is believed to depend on the duration and extent of JNK activation (12, 13). TRAIL-induced apoptosis is executed through FADD-mediated recruitment of caspase-8 and subsequent activation of downstream effector caspase-3 and -7 (14). In addition, the apoptotic signal is further amplified through caspase-8-mediated cleavages of BID, a BH3-only member of the Bcl-2 family. The cleavage product, tBID, migrates to mitochondria and activates the mitochondrial apoptosis pathway. This process causes release of cytochrome C and Smac from mitochondria to the cytosol to induce caspase-9-mediated activation of effector caspases (14).
Several proteins act as negative regulators of TRAIL-induced apoptosis. The cellular FLICE-like inhibitory protein (c-FLIP), which is structurally similar to caspase-8, can be recruited to the DISC to inhibit the recruitment and activation of caspase-8 (3). In addition, the ratio of pro- (Bax, Bak, and Bik) and anti- (Bcl-2 and Bcl-xL) apoptotic Bcl-2 family members can determine the cell’s death or survival. Particularly, myeloid cell leukemia 1 (Mcl-1) recently has been attributed to TRAIL resistance in cancer cells (15, 16). Furthermore, the inhibitor of apoptosis proteins (IAP), including c-IAP-1, c-IAP-2, XIAP, and survivin, negatively regulate TRAIL-induced apoptosis through inhibiting caspases (14).
The purpose of this study was to elucidate the mechanism of acquired TRAIL resistance in lung cancer cells. We found that the expression of c-FLIP L and Mcl-1 L is increased in an Akt-dependent manner in cells with acquired TRAIL resistance. Knockdown of c-FLIP L and Mcl-1 L collectively alleviated TRAIL resistance. These results suggest that c-FLIP L and Mcl-1 L play a key role in acquired TRAIL resistance, making them promising molecular targets for sensitizing TRAIL-induced lung cancer cell death.
Materials and Methods
Reagents and Antibodies
Glutathione S-transferase (GST) TRAIL was prepared as described previously (9, 17). Human TNF was purchased from R&D Systems (Minneapolis, MN). MG-132 and LY294002 were from Calbiochem (La Jolla, CA). Adriamycin, etopside, and cycloheximide were from Sigma (St. Louis, MO). Antibodies against cIAP-1, cIAP-2, Bax, Bid, Akt, survivin, Bcl-2, caspase-3, and caspase-8 were from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-FADD, -DR4, -DR5, -DcR1, -DcR2, and - poly(ADP-ribose) polymerase (PARP) were from BD Biosciences (San Diego, CA). Anti-β-actin was purchased from Sigma. Antibodies for Bcl-xL, XIAP, BAD, cytochrome C, caspase-9, Akt, and phospho-Akt(Ser473) were from Cell Signaling (Beverly, MA). Antibodies against c-FLIP and Mcl-1 were from Alexis (San Diego, CA) and Biovision (Mountain View, CA), respectively.
Cell Culture and Induction of TRAIL-Resistant Cells
H460, H1568, and A549 cells were obtained from American Type Culture Collection (Manassas, VA) and grown in RPMI 1640 supplemented with 10% fetal bovine serum, 1 mmol/L glutamate, 100 units/mL penicillin, and 100 μg/mL streptomycin. To exclude the possibility that a population of TRAIL-resistant cells were selected during the course of establishment of TRAIL resistance, we cloned the cells and obtained five TRAIL-sensitive clones (which could be completely killed by treatment with 80 ng/ml TRAIL for 3 days) and pooled them for induction of TRAIL resistance. To induce TRAIL resistance, the TRAIL-sensitive H460 cells (designated as H460) were treated initially with a nonapoptotic dose (0.125 ng/ml) of TRAIL for 3 days and then split with fresh medium, with the TRAIL dose doubling every 3 days to a final dose of 128 ng/mL. The resulting cells were designated as H460 TRAIL-resistant (H460-TR) cells and maintained in medium containing 50 ng/mL of TRAIL. For A549 and H1568 cells, the starting and final concentrations of TRAIL were 1 ng/mL and 512 ng/mL, respectively. These TRAIL-resistant cells were designated as A549-TR and H1568-TR.
Cytotoxicity Assay
A cytotoxicity assay based on the release of lactate dehydrogenase (LDH) was conducted using a cytotoxicity detection kit (Roche, Penzberg, Germany). Cells were seeded in 24-well plates at 70% to 80% confluence. After overnight culture, cells were treated as indicated in each figure legend. LDH release was determined as described previously (18, 19).
Western Blotting
Cell lysates were prepared by suspending cells in M2 buffer (20 mmol/L Tris-HCl [pH 7.6], 0.5% NP40, 250 mmol/L NaCl, 3 mmol/L EDTA, 3 mmol/L EGTA, 2 mmol/L DTT, 0.5 mmol/L phenylmethylsulfonyl fluoride, 20 mmol/L β-glycerophosphate, 1 mmol/L sodium vanadate, and 1 μg/mL leupeptin). Equal amounts of protein from each cell lysate were resolved by 10%, 12%, or 15% SDS-PAGE and analyzed by Western blot. Cytosol was prepared as described (20). Cell fractions were prepared as described previously to detect cytochrome C release from mitochondria (20). The proteins were visualized by enhanced chemiluminescence following the manufacturer’s instruction (Amersham, Piscataway, NJ). Each experiment was repeated at least thrice and representative results are shown. NIH image 1.62 was employed to quantify protein expression levels.
GST-Pull-Down and DISC Analysis
Cells (1 × 108) were treated with 1 μg/mL of GST-TRAIL for 20 min or left untreated. Cells were then washed twice with ice-cold PBS and lysed with 1 mL M2 buffer. The soluble fraction was incubated with 30 μl of (50%) glutathione-sepharose beads (Pharmacia,) for 3 h on a rotator at 4°C. GST-TRAIL (1 μg/mL) was added to the lysates prepared from nonstimulated cells to precipitate the nonstimulated receptors. After five washes with M2 buffer, the bound proteins were eluted by boiling for 3 min in SDS-PAGE loading buffer and resolved in 10% SDS-PAGE. The presence of DR4, DR5, FADD, caspase-8, and c-FLIP was then determined by Western blot.
Knockdown of c-FLIP, Mcl-1, and Akt by RNAi
Short-interfering RNAs for cFLIP and Mcl-1 and the nontargeting siRNA (Silencer® negative control #1 siRNA) were obtained from Ambion (Austin, TX). The two siRNAs had the following sense strand sequences: c-FLIP, GGGACCUUCUGGAUAUUUUtt; Mcl-1, GGACUUUUAUACCUGUUAUtt. siRNAs against Akt1, Akt2, and Akt3 (siGENOME SMARTpool) were purchased from Dharmacon (Lafayette, CO). H460-TR cells were seeded in a 12-well plate the day before transfection at ∼ 50% confluency. SiRNA was transfected with siRNA INTERFERin™ siRNA transfection reagent (Polyplus-transfection, New York, NJ). Thirty hours after transfection the Mcl-1 and c-FLIP protein levels were measured by Western blot. TRAIL (80 ng/ml) was added to the culture 30 h (for Mcl-1 and c-FLIP siRNA) or 48 h (for Akt siRNA) post-transfection and incubated for 24 h to examine TRAIL-induced cytotoxicity.
Results
Establishment of Acquired TRAIL Resistance in H460 Cells
The human lung cancer-derived cell line H460 was used as a model to investigate the mechanism of acquired TRAIL resistance because these cells are relatively susceptible to TRAIL-induced cytotoxicity (Fig. 1A). H460 cells were initially treated with a nontoxic low dose (0.125 ng/ml) of TRAIL followed by exposure to two-fold increased TRAIL concentrations every 3 days to a final dose of 128 ng/ml. The experiment was conducted with TRAIL-sensitive cells (see Materials and Methods) and there was little cell death during the course of treatment; therefore it is likely that TRAIL resistance is acquired rather than due to selection of a sub-population of TRAIL-resistant cells. The H460-TR cells were remarkably insensitive to TRAIL-induced death (Fig. 1A), suggesting that acquired TRAIL resistance in H460 cells has been established by chronic exposure to nontoxic doses of TRAIL.
Figure 1. Chronic TRAIL exposure induces acquired resistance to TRAIL-induced cytotoxicity in H460 lung cancer cells.
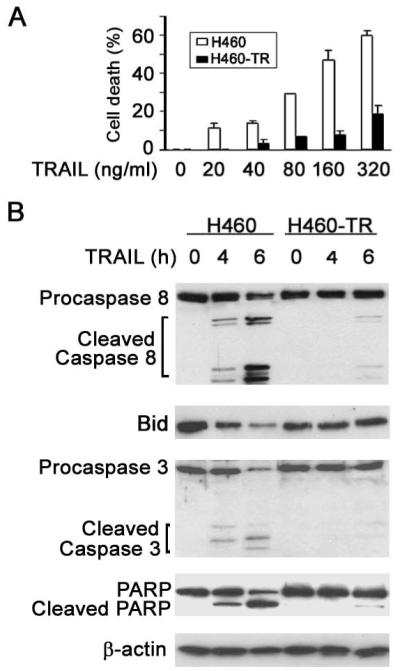
A, H460 and H460-TR cells were treated with indicated concentrations of TRAIL for 24 h. Cell death was measured by an LDH release assay. Columns, mean of three experiments; bars, SD. B, H460 and H460-TR cells were treated with 80 ng/mL TRAIL for the indicated times. Caspase-8, Bid, caspase-3, and PARP were detected by Western blot with specific antibodies against each of these proteins. β-actin was detected as an input control.
TRAIL-Induced Caspase-8 Activation Is Impaired in H460-TR Cells
To address the mechanism of acquired TRAIL resistance, we first examined caspase activation in H460-TR cells. Because caspase-8 is the initiator caspase that is essential for TRAIL-induced apoptosis, we compared activation of this key caspase between H460-TR and H460 cells. TRAIL treatment caused marked activation of caspase-8 activation in H460 cells as detected by cleavage of this caspase starting at 2 h post-treatment (Fig. 1B, data not shown). In contrast, activation of caspase-8 was significantly impaired in H460-TR cells (Fig. 1B). The activation of caspase-3, as well as cleavage of Bid and PARP by caspases-8 and -3, respectively, was also suppressed in H460-TR cells (Fig. 1B). These results suggest that acquired TRAIL resistance in H460 cells may be, at least in part, due to suppression of caspase-8 activation, which occurs at an early stage of TRAIL receptor-mediated apoptosis signaling.
Increased c-FLIP Expression and Decreased Recruitment of Caspase-8 to DISC in H460-TR Cells
Because caspase-8 is recruited to and activated at the TRAIL-DISC during initiation of apoptosis, we next examined whether there is a defect in expression of the TRAIL-DISC components (1). There were no detectable changes in expression levels of TRAIL receptors (DR4, DR5, DcR1, and DcR2), TRADD, FADD, caspase-8, RIP, or TRAF2 in H460-TR cells. However, the expression of c-FLIPL and c-FLIPS was significantly increased in H460-TR cells (Fig. 2A). Because c-FLIP suppresses death receptor-mediated apoptosis by inhibiting the recruitment to and activation of caspase-8 at the DISC (3, 21), we examined DISC formation in H460-TR cells by a GST pull-down assay (see Materials and Methods). Pull-down of both DR4 and DR5 with GST-TRAIL was comparable between H460 and H460-TR cells (Fig. 2B, data not shown), suggesting that these receptors’ exposure on the cytoplasm membrane and binding to TRAIL were normally retained in H460-TR cells. Similarly, FADD’s recruitment to the DISC was comparable in H460 and H460-TR cells (Fig. 2B). In contrast, caspase-8 recruitment to the DISC was dramatically reduced in H460-TR cells. This reduction was accompanied by a dramatic increase in the recruitment of the modified form of c-FLIPL (p43) to the DISC (Fig. 2B). It should be noted that after TRAIL treatment the c-FLIPL protein was modified and became a 43kd band (p43) as detected by Western blot, which is consistent with previous reports (22, 23). The recruitment of c-FLIPS was unchanged in H460-TR cells (Fig. 2B). These results suggest that increased expression and recruitment of c-FLIPL, but not c-FLIPS, to the DISC contributes to blocking caspase-8 activation in H460-TR cells. Therefore, we focused our studies on c-FLIPL.
Figure 2. Increased c-FLIPL expression and suppressed recruitment of caspase-8 to DISC in H460-TR cells.
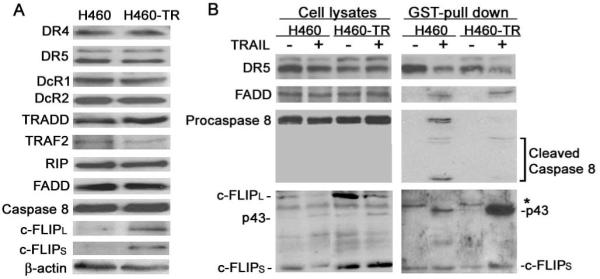
A, The expression of DISC components in H460 and H460-TR cells was detected by Western blot with specific antibodies against each protein. β-actin was detected as an input control. B, The DISC formation detected by GSTpull-down assay. H460 and H460-TR cells were treated with 1 μg/mL GST-TRAIL for 20 min or left untreated. The cells were lyzed in M2 buffer. GST-TRAIL (1 μg/mL) was added to lysates from unstimulated cells to preciptate unstimulated TRAIL receptors. GST-TRAIL-bound protein complexes were analyzed for the presence of DR5, FADD, caspase-8, and c-FLIP by Western blot. *, non-specific band.
c-FLIP expression is regulated at the transcriptional and post-translational levels (24). The increase of c-FLIPL protein in H460-TR cells is unlikely through higher transcription rates because no significant difference in mRNA expression of c-FLIPL was observed between H460 and H460-TR cells (data not shown). However, when protein synthesis was blocked by cycloheximide, c-FLIPL protein rapidly turned over in H460-TR cells; this turnover could be reversed by the proteasome inhibitor MG132, suggesting that proteasomal degradation is pivotal in regulating c-FLIPL expression in these cells (Fig. 3A). Therefore, we compared the stability of c-FLIPL in H460 and H460-TR cells by shutting off protein synthesis with cycloheximide and chasing c-FLIPL expression for up to 4 h. The half-life of c-FLIPL in H460-TR cells was ∼ 180 min, much longer than that of the H460 cells (∼ 75 min) (Fig. 3B, 3C). These results imply that increased protein stability in H460-TR cells contributes to increases in c-FLIPL protein levels, which leads to the suppression of caspase-8 recruitment and activation during TRAIL-induced apoptotic signaling in H460-TR cells.
Figure 3. Increased protein stability of c-FLIPL in H460-TR cells.
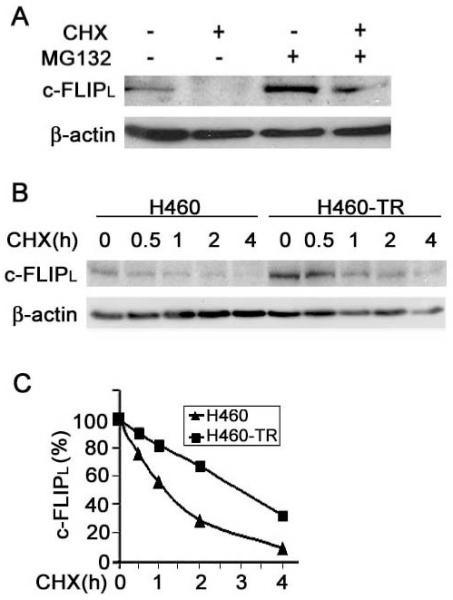
A, H460-TR cells were treated with either 10 μg/mL cycloheximide (CHX) or 10 μM MG132 or both for 4 h. c-FLIPL was detected by Western blot. β-actin was detected as an input control. B, H460 and H460-TR cells were treated with 10 μg/mL CHX for indicated times. c-FLIPL protein was detected by Western blot. β-actin was detected as an input control. C, The results of c-FLIP expression,shown in panel B, were quantified and normalized to β-actin.
Elevated Expression of Mcl-1L and Impaired Mitochondrial Apoptosis Pathway in H460-TR cells
Several proteins such as members of the IAP and Bcl-2 families also affect TRAIL-induced cell death (3). Therefore, we examined the expression levels of these proteins by Western blot analysis. As shown in Fig. 4A, no significant changes in expression levels of IAP family members, including survivin, XIAP, cIAP1, and cIAP2, were observed in H460-TR cells. Among the Bcl-2 family proteins examined, only the expression level of Mcl-1L was dramatically increased. Interestingly, the expression of Mcl-1S remained unchanged (Fig. 4A). Although minor changes of Bcl-XL and BAD were seen occasionally, this observation was inconstant; we thus focused on Mcl-1L. Because Mcl-1L negatively regulates the mitochondria-mediated apoptosis pathway, we examined whether this pathway is blocked in H460-TR cells. Indeed, TRAIL-induced release of cytochrome C from mitochondria and activation of caspase-9 were greatly reduced in H460-TR cells (Fig. 4B, 4C). These results suggest that blocking the intrinsic apoptosis pathway by increased Mcl-1L expression may underlie another mechanism of acquired TRAIL resistance in H460-TR cells.
Figure 4. Increased Mcl-1L expression and suppressed mitochondrial apoptosis pathway in H460-TR cells.

A, Expression of Bcl-2 family and IAP family proteins in H460 and H460-TR cells was detected by Western blot analysis. β-actin was detected as an input control. B, H460 and H460-TR cells were treated with 80 ng/mL TRAIL for the indicated times. The cytosolic fraction of cell extracts was probed for cytochrome C (upper panel). Total cell extracts from TRAIL-treated (80 ng/ml) H460 and H460-TR cells were detected for caspase-9 by Western blot. C, H460-TR cells were treated with either 10 μg/mL cycloheximide (CHX) or 10 μM MG132 or both for 4 h. Mcl-1L protein was detected by Western blot. β-actin was detected as an input control. D, H460 and H460-TR cells were treated with 10 μg/mL CHX for indicated times. Mcl-1L protein was analyzed by Western blot. β-actin was detected as an input control (upper panel). The results of Mcl-1L expression were quantified and normalized to β-actin (lower panel).
We further investigated the mechanism of the Mcl-1L increase in H460-TR cells and detected no alteration in expression of Mcl-1L mRNA (data not shown). Proteasomal degradation appeared to be important in controlling the expression level of Mcl-1L in H460-TR cells because MG132 effectively blocked the decrease of Mcl-1L levels when protein synthesis was blocked (Fig. 4D). The Mcl-1L protein has a prolonged half-life (80 min) in H460-TR cells compared with that of the H460 cells (30 min) (Fig. 4D). These results indicate that increased protein stability led to the upregulation of Mcl-1L, contributing to the suppression of mitochondria-mediated apoptosis in H460-TR cells.
Suppression of c-FLIPL and Mcl-1L Expression Concurrently Alleviates Acquired TRAIL Resistance
We further confirmed the role of c-FLIPL and Mcl-1L in acquired TRAIL resistance by reducing the expression of these proteins. Because both c-FLIPL and Mcl-1L are short-lived proteins that can be effectively suppressed by protein synthesis inhibitors (Figs. 3B, 4D), we assessed the effect of cycloheximide on TRAIL-induced cytotoxicity in H460-TR cells (Fig. 5A). Cycloheximide restored TRAIL-induced cytotoxicity, concomitant to a dramatic reduction of c-FLIPL or Mcl-1L expression in H460-TR cells. Additionally, knockdown of c-FLIPL and Mcl-1L expression was further used to verify the role of these proteins in acquired TRAIL resistance (Fig. 5B). Transfection of control siRNA had no detectable effect; however, suppressing expression of either c-FLIPL or Mcl-1L partially alleviated acquired TRAIL resistance (Fig. 5C). Importantly, concurrent knockdown of c-FLIPL and Mcl-1L cooperatively sensitized H460-TR cells to TRAIL-induced cytotoxicity (Fig. 5C), indicating that both c-FLIPL and Mcl-1 are important in acquired TRAIL resistance.
Figure 5. Suppressing c-FLIPL and Mcl-1L expression in H460-TR cells alleviates acquired TRAIL resistance.
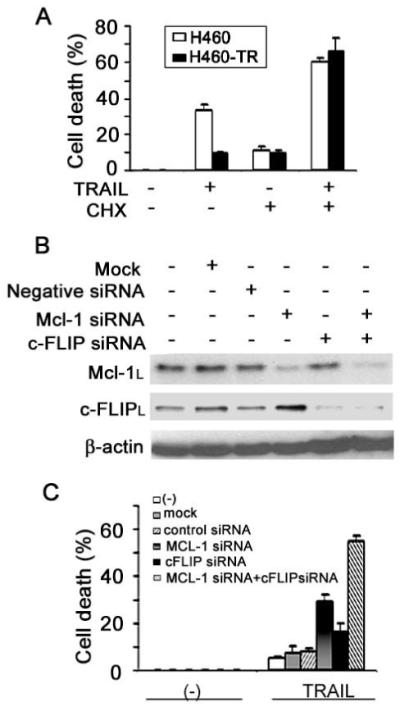
A, H460-TR cells were left untreated or treated with 10 μg/mL cycloheximide (CHX) for 30 min followed by treatment with 80 ng/mL TRAIL for 24 h. Cell death was measured by LDH assay. Columns, mean of three experiments; bars, SD. B, H460-TR cells were transfected with either Mcl-1 siRNA, c-FLIP siRNA, or both. Mcl-1L and c-FLIPL were assessed by Western blot 30 h after transfection. Nontransfected, mock, or negative siRNA-transfected cells also are shown as controls. C, H460-TR cells were transfected with indicated siRNA. Thirty hours after transfection, cells were treated with 80 ng/mL TRAIL for another 24 h and cell death was detected as described in A.
Elevated Akt Activity Contributes to Stabilization of Mcl-1L and c-FLIPL
Akt is a master kinase involved in cell survival (25). Recently it was reported that Akt positively regulates Mcl-1 and c-FLIP expression (26-28). Interestingly, Akt activity was markedly increased in H460-TR cells (Fig. 6A). To investigate whether the elevated Akt activity contributes to the increased protein stability of Mcl-1L and c-FLIPL, H460-TR cells were treated with the specific PI3-kinase inhibitor LY294002 to suppress Akt activity. Blocking Akt by LY294002 reduced Mcl-1L and c-FLIPL expression and alleviated TRAIL resistance of H460-TR cells (Fig. 6B, data not shown). To further confirm the role of Akt in regulating Mcl-1L and c-FLIPL protein levels, RNAi was used to knock down Akt expression by concurrently targeting all of its three isoforms, Akt1, Akt2, and Akt3, in H460-TR cells. Marked downregulation of Mcl-1L and c-FLIPL was achieved by suppressing Akt expression and activity (Fig. 6C). Moreover, knockdown of Akt significantly restored sensitivity to TRAIL-induced cytotoxicity in H460-TR cells (Fig. 6D), indicating that Akt contributes substantially to acquired TRAIL resistance in lung cancer cells through regulating Mcl-1L and c-FLIPL expression.
Figure 6. Elevated Akt activity contributes to stabilization of Mcl-1L and c-FLIPL.
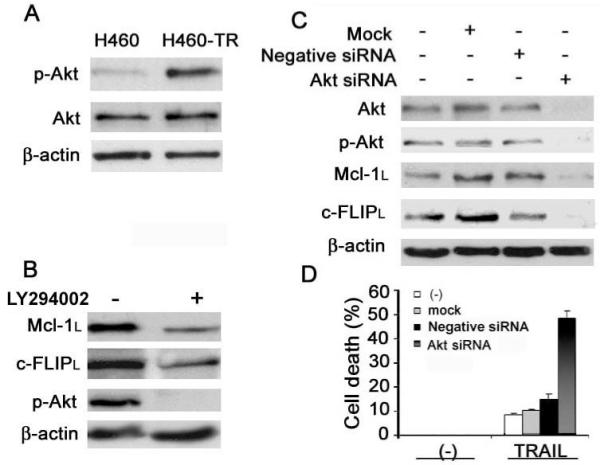
A, Phosphorylated-Akt (p-Akt) and total Akt in H460 and H460-TR cells were measured by Western blot. β-actin was detected as an input control. B, H460-TR cells were treated with 10 μM LY294002 overnight. Mcl-1L, c-FLIPL, and p-Akt were detected by Western blot. β-actin was detected as an input control. C, Pooled siRNA against Akt1, Akt2, and Akt3 was used to knock down Akt in H460-TR cells. p-Akt, total Akt, Mcl-1L, and c-FLIPL were measured by Western blot. D, Nontransfected, mock, and negative siRNA-transfected cells are shown as in H460-TR cells. Control cells were treated with 80 ng/mL TRAIL for 24 h and cell death was detected by LDH assay. Columns, mean of three experiments; bars, SD.
Discussion
In this study we demonstrated that the acquired TRAIL resistance is associated with enhanced expression of c-FLIPL and Mcl-1L in H460 lung cancer cells. Modified c-FLIPL was preferentially recruited to the TRAIL DISC and suppressed recruitment and activation of caspase-8, thereby inhibiting the extrinsic apoptosis pathway. The increased Mcl-1L level contributes to suppressing the mitochondrial apoptosis pathway. Inhibition of FLIPL and Mcl-1L expression by RNA interference cooperatively alleviated the acquired TRAIL resistance. Thus, acquired TRAIL resistance to TRAIL-induced cytotoxicity was due to suppressing both the extrinsic and intrinsic apoptosis pathways. We further demonstrated that elevated Akt activity is crucial for overexpression of c-FLIPL and Mcl-1L. Similar observations were also made in H1568 and A549 cells (data not shown). Collectively, data from this study demonstrate that the Akt-mediated increase of c-FLIPL and Mcl-1L levels underlies the main mechanism of acquired TRAIL resistance in lung cancer. Therefore, Akt, c-FLIPL, and Mcl-1L could be useful targets for circumventing acquired TRAIL resistance in lung cancer cells.
As a structural homologue of caspase-8, c-FLIP binds FADD, competing with caspase-8 for recruitment to DISC. c-FLIP suppression is assumed to be involved in mediating the sensitization of cancer cells to TNF- or TRAIL-induced apoptosis by cycloheximide or the transcription inhibitor actinomycin D (29). In several types of cancers, increased c-FLIP expression is correlated with resistance to TRAIL-, Fas- or TNF-induced apoptosis (21, 23, 30-32). However, the mechanisms underlying the c-FLIP expression increase have not been well elucidated. Post-translational modification and degradation of c-FLIP as well as increased mRNA levels have been suggested to be involved in increasing c-FLIP expression (24). In this study chronic TRAIL exposure induced c-FLIPL expression in lung cancer cells, likely through post-transcriptional regulation. Although both the long- and short-forms of c-FLIP were increased, only c-FLIPL was preferentially recruited to the TRAIL DISC. Because the recruited c-FLIPL was the proteolyzed form (p43) and the extent of c-FLIPL recruitment to the DISC was enhanced in relation to the protein expression levels (Fig. 3), it is plausible that an unidentified mechanism that modifies c-FLIPL may have been activated during chronic TRAIL exposure to facilitate recruitment of c-FLIPL to the DISC. Furthermore, it is noteworthy that although both c-FLIPL and c-FLIPs are apoptosis inhibitors, their contributions to apoptosis regulation, specifically to acquired TRAIL resistance, could be distinct. c-FLIPL was reported to be more significant in cancer cells’ resistance to therapy (21, 30). Interestingly, increased c-FLIPs did not block apoptosis induced by TRAIL plus the proteasome inhibitor bortezomib (33).
Increased expression of anti-apoptotic Bcl-2 family members such as Bcl-xL and Mcl-1 is associated with cancer cells’ resistance to therapy, suggesting that the intrinsic pathway also plays a role in resistance to therapeutics (34). Mcl-1 is involved in lung cancer cells’ resistance to apoptosis induced by chemotherapeutics, ionizing radiation, and TRAIL (35, 36). Mcl-1L can block the tBid-mediated activation of the mitochondrial apoptosis pathway induced by TRAIL (15). Of the two Mcl-1 isoforms, Mcl-lL is widely regarded as an anti-apoptosis factor while Mcl-1S is found to be proapoptotic (37). Thus, an increase in the ratio of Mcl-1L to Mcl-1S would elevate the apoptosis threshold in cancer cells. Interestingly, we show that the expression of Mcl-1L but not Mcl-1S was dramatically increased after chronic exposure to TRAIL. The increased Mcl-1L expression was correlated with inhibiting TRAIL-induced cytochrome C release from mitochondria and activation of caspase-9. Knockdown of Mcl-1L partially alleviated TRAIL resistance, indicating that suppressing the mitochondrial apoptosis pathway by Mcl-1L at least is partly involved in the mechanism of acquired TRAIL resistance.
We further found that increased Akt activity is accompanied by acquired TRAIL resistance in lung cancer cells. Suppressing Akt activity with the PI3K inhibitor LY294002 or Akt siRNA suppressed expression of both c-FLIPL and Mcl-1L. This finding is consistent with other reports showing that Akt regulates these two survival factors in cancer cells (26, 38) and with our recent findings that inhibited Akt activity dramatically sensitizes lung cancer cells to TNF- or TRAIL-induced cytotoxicity (17, 39). While our data suggest that Akt may enhance the expression of c-FLIPL and Mcl-1L by affecting the protein degradation pathway, the exact mechanisms underlying such enhancement is not clear currently. As a glycogen-synthase kinase 3 (GSK3) inhibitor, Akt may increase the stability of Mcl-1 by suppressing GSK3β. Phosphorylation of Mcl-1 by GSK3 recently has been shown to enhance Mcl-1 protein turnover (40). Akt activation also was associated with suppressing GSK3 and increasing the c-FLIP expression level (41). Whether the regulation of Mcl-1 and c-FLIP is activated through the same mechanism and whether it involves GSK3 remains to be elucidated.
Direct evidence showing that increased expression of c-FLIPL and Mcl-1L is the main determinant of acquired TRAIL resistance in lung cancer cells came from the siRNA knockdown experiment in which concurrent knockdown of these two proteins additively sensitized H460-TR cells to TRAIL-induced cytotoxicity. Therefore, c-FLIPL and Mcl-1L could be ideal molecular targets for attenuating acquired TRAIL resistance to lung cancer. It is remarkable that TRAIL can stimulate TRAIL-resistant cancer cells’ proliferation and metastasis (8). If it arises during therapy, such acquired TRAIL resistance would shift the TRAIL treatment from beneficial to detrimental. Thus, preventing or attenuating acquired TRAIL resistance by targeting c-FLIPL and Mcl-1L could be crucial in retaining TRAIL’s cancer-killing activity and circumventing its cancer promoting potential. In vivo experiments with animal models are needed to verify the effect of this approach in sensitizing TRAIL for lung cancer therapy.
Acknowledgements
We thank Vicki Fisher for help in preparing this manuscript. This study is partly supported by a grant (R03CA125796) from the National Cancer Institute.
Financial support:
R03CA125796, National Cancer Institute/NIH
Abbreviations
- c-FLIP
cellular FLICE-like inhibitory protein
- DISC
death-inducing signal complex
- DR
death receptor
- H460-TR
TRAIL-resistant H460 cells
- IAP
inhibitor of apoptosis proteins
- JNK
c-Jun N-terminal kinase
- LDH
lactate dehydrogenase
- Mcl-1
myeloid cell leukemia 1
- NF-κB
nuclear factor-κB
- OPG
osteoprotegerin
- GSK3
glycogen-synthase kinase 3
- GST
glutathione S-transferase
- PARP
poly(ADP-ribose) polymerase
- PI3-K
PI3-kinase
- TNF-R1
TNF receptor 1
- DcR
decoy receptor
- TRAIL
TNF-related apoptosis-inducing ligand
References
- 1.Wajant H, Gerspach J, Pfizenmaier K. Tumor therapeutics by design: targeting and activation of death receptors. Cytokine Growth Factor Rev. 2005;16:55–76. doi: 10.1016/j.cytogfr.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 2.Bouralexis S, Findlay DM, Evdokiou A. Death to the bad guys: targeting cancer via Apo2L/TRAIL. Apoptosis. 2005;10:35–51. doi: 10.1007/s10495-005-6060-0. [DOI] [PubMed] [Google Scholar]
- 3.Zhang L, Fang B. Mechanisms of resistance to TRAIL-induced apoptosis in cancer. Cancer Gene Ther. 2005;12:228–37. doi: 10.1038/sj.cgt.7700792. [DOI] [PubMed] [Google Scholar]
- 4.Lane D, Cote M, Grondin R, Couture MC, Piche A. Acquired resistance to TRAIL-induced apoptosis in human ovarian cancer cells is conferred by increased turnover of mature caspase-3. Mol Cancer Ther. 2006;5:509–21. doi: 10.1158/1535-7163.MCT-05-0362. [DOI] [PubMed] [Google Scholar]
- 5.Lee TJ, Lee JT, Park JW, Kwon TK. Acquired TRAIL resistance in human breast cancer cells are caused by the sustained cFLIP(L) and XIAP protein levels and ERK activation. Biochem Biophys Res Commun. 2006;351:1024–30. doi: 10.1016/j.bbrc.2006.10.163. [DOI] [PubMed] [Google Scholar]
- 6.Song JJ, An JY, Kwon YT, Lee YJ. Evidence for two modes of development of acquired tumor necrosis factor-related apoptosis-inducing ligand resistance. Involvement of Bcl-xL. J Biol Chem. 2007;282:319–28. doi: 10.1074/jbc.M608065200. [DOI] [PubMed] [Google Scholar]
- 7.Wenger T, Mattern J, Penzel R, et al. Specific resistance upon lentiviral TRAIL transfer by intracellular retention of TRAIL receptors. Cell Death Differ. 2006;13:1740–51. doi: 10.1038/sj.cdd.4401867. [DOI] [PubMed] [Google Scholar]
- 8.Malhi H, Gores GJ. TRAIL resistance results in cancer progression: a TRAIL to perdition? Oncogene. 2006;25:7333–5. doi: 10.1038/sj.onc.1209765. [DOI] [PubMed] [Google Scholar]
- 9.Lin Y, Devin A, Cook A, et al. The death domain kinase RIP is essential for TRAIL (Apo2L)-induced activation of IkappaB kinase and c-Jun N-terminal kinase. Mol Cell Biol. 2000;20:6638–45. doi: 10.1128/mcb.20.18.6638-6645.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wajant H. TRAIL and NFkappaB signaling--a complex relationship. Vitam Horm. 2004;67:101–32. doi: 10.1016/S0083-6729(04)67007-5. [DOI] [PubMed] [Google Scholar]
- 11.Holen I, Shipman CM. Role of osteoprotegerin (OPG) in cancer. Clin Sci (Lond) 2006;110:279–91. doi: 10.1042/CS20050175. [DOI] [PubMed] [Google Scholar]
- 12.Lin A, Dibling B. The true face of JNK activation in apoptosis. Aging Cell. 2002;1:112–6. doi: 10.1046/j.1474-9728.2002.00014.x. [DOI] [PubMed] [Google Scholar]
- 13.Ventura JJ, Cogswell P, Flavell RA, Baldwin AS, Jr., Davis RJ. JNK potentiates TNF-stimulated necrosis by increasing the production of cytotoxic reactive oxygen species. Genes Dev. 2004;18:2905–15. doi: 10.1101/gad.1223004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang X. The expanding role of mitochondria in apoptosis. Genes Dev. 2001;15:2922–33. [PubMed] [Google Scholar]
- 15.Clohessy JG, Zhuang J, de Boer J, Gil-Gomez G, Brady HJ. Mcl-1 interacts with truncated Bid and inhibits its induction of cytochrome c release and its role in receptor-mediated apoptosis. J Biol Chem. 2006;281:5750–9. doi: 10.1074/jbc.M505688200. [DOI] [PubMed] [Google Scholar]
- 16.Weng C, Li Y, Xu D, Shi Y, Tang H. Specific cleavage of Mcl-1 by caspase-3 in tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis in Jurkat leukemia T cells. J Biol Chem. 2005;280:10491–500. doi: 10.1074/jbc.M412819200. [DOI] [PubMed] [Google Scholar]
- 17.Chen W, Wang X, Zhuang J, Zhang L, Lin Y. Induction of Death Receptor 5 and Suppression of Survivin Contribute to Sensitization of TRAIL-induced cytotoxicity by Quercetin in Non-small Cell Lung Cancer Cells. Carcinogenesis. 2007;28:2114–21. doi: 10.1093/carcin/bgm133. [DOI] [PubMed] [Google Scholar]
- 18.Wang X, Ju W, Renouard J, Aden J, Belinsky SA, Lin Y. 17-allylamino-17-demethoxygeldanamycin synergistically potentiates tumor necrosis factor-induced lung cancer cell death by blocking the nuclear factor-kappaB pathway. Cancer Res. 2006;66:1089–95. doi: 10.1158/0008-5472.CAN-05-2698. [DOI] [PubMed] [Google Scholar]
- 19.Ju W, Wang X, Shi H, Chen W, Belinsky SA, Lin Y. A critical role of luteolin- induced reactive oxygen species in blockage of tumor necrosis factor-activated nuclear factor-kappaB pathway and sensitization of apoptosis in lung cancer cells. Mol Pharmacol. 2007;71:1381–8. doi: 10.1124/mol.106.032185. [DOI] [PubMed] [Google Scholar]
- 20.Sitailo LA, Tibudan SS, Denning MF. The protein kinase C delta catalytic fragment targets Mcl-1 for degradation to trigger apoptosis. J Biol Chem. 2006;281:29703–10. doi: 10.1074/jbc.M607351200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wilson TR, McLaughlin KM, McEwan M, et al. c-FLIP: A key regulator of colorectal cancer cell death. Cancer Res. 2007;67:5754–62. doi: 10.1158/0008-5472.CAN-06-3585. [DOI] [PubMed] [Google Scholar]
- 22.Hietakangas V, Poukkula M, Heiskanen KM, Karvinen JT, Sistonen L, Eriksson JE. Erythroid differentiation sensitizes K562 leukemia cells to TRAIL-induced apoptosis by downregulation of c-FLIP. Mol Cell Biol. 2003;23:1278–91. doi: 10.1128/MCB.23.4.1278-1291.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ricci MS, Kim SH, Ogi K, et al. Reduction of TRAIL-induced Mcl-1 and cIAP2 by c-Myc or sorafenib sensitizes resistant human cancer cells to TRAIL-induced death. Cancer Cell. 2007;12:66–80. doi: 10.1016/j.ccr.2007.05.006. [DOI] [PubMed] [Google Scholar]
- 24.Dutton A, Young LS, Murray PG. The role of cellular FLICE inhibitory protein (c- FLIP) in the pathogenesis and treatment of cancer. Expert Opin Ther Targets. 2006;10:27–35. doi: 10.1517/14728222.10.1.27. [DOI] [PubMed] [Google Scholar]
- 25.Chen YL, Law PY, Loh HH. Inhibition of PI3K/Akt signaling: an emerging paradigm for targeted cancer therapy. Curr Med Chem Anticancer Agents. 2005;5:575–89. doi: 10.2174/156801105774574649. [DOI] [PubMed] [Google Scholar]
- 26.Kobayashi S, Werneburg NW, Bronk SF, Kaufmann SH, Gores GJ. Interleukin-6 contributes to Mcl-1 up-regulation and TRAIL resistance via an Akt-signaling pathway in cholangiocarcinoma cells. Gastroenterology. 2005;128:2054–65. doi: 10.1053/j.gastro.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 27.Kang YC, Kim KM, Lee KS, et al. Serum bioactive lysophospholipids prevent TRAIL-induced apoptosis via PI3K/Akt-dependent cFLIP expression and Bad phosphorylation. Cell Death Differ. 2004;11:1287–98. doi: 10.1038/sj.cdd.4401489. [DOI] [PubMed] [Google Scholar]
- 28.Maurer U, Charvet C, Wagman AS, Dejardin E, Green DR. Glycogen synthase kinase-3 regulates mitochondrial outer membrane permeabilization and apoptosis by destabilization of MCL-1. Mol Cell. 2006;21:749–60. doi: 10.1016/j.molcel.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 29.Kreuz S, Siegmund D, Scheurich P, Wajant H. NF-kappaB inducers upregulate cFLIP, a cycloheximide-sensitive inhibitor of death receptor signaling. Mol Cell Biol. 2001;21:3964–73. doi: 10.1128/MCB.21.12.3964-3973.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sharp DA, Lawrence DA, Ashkenazi A. Selective knockdown of the long variant of cellular FLICE inhibitory protein augments death receptor-mediated caspase-8 activation and apoptosis. J Biol Chem. 2005;280:19401–9. doi: 10.1074/jbc.M413962200. [DOI] [PubMed] [Google Scholar]
- 31.Oyarzo MP, Medeiros LJ, Atwell C, et al. c-FLIP confers resistance to FAS- mediated apoptosis in anaplastic large-cell lymphoma. Blood. 2006;107:2544–7. doi: 10.1182/blood-2005-06-2601. [DOI] [PubMed] [Google Scholar]
- 32.Liu X, Yue P, Schonthal AH, Khuri FR, Sun SY. Cellular FLICE-inhibitory protein down-regulation contributes to celecoxib-induced apoptosis in human lung cancer cells. Cancer Res. 2006;66:11115–9. doi: 10.1158/0008-5472.CAN-06-2471. [DOI] [PubMed] [Google Scholar]
- 33.Liu X, Yue P, Chen S, et al. The proteasome inhibitor PS-341 (bortezomib) up-regulates DR5 expression leading to induction of apoptosis and enhancement of TRAIL-induced apoptosis despite up-regulation of c-FLIP and survivin expression in human NSCLC cells. Cancer Res. 2007;67:4981–8. doi: 10.1158/0008-5472.CAN-06-4274. [DOI] [PubMed] [Google Scholar]
- 34.Thomadaki H, Scorilas A. BCL2 family of apoptosis-related genes: functions and clinical implications in cancer. Crit Rev Clin Lab Sci. 2006;43:1–67. doi: 10.1080/10408360500295626. [DOI] [PubMed] [Google Scholar]
- 35.Song L, Coppola D, Livingston S, Cress D, Haura EB. Mcl-1 regulates survival and sensitivity to diverse apoptotic stimuli in human non-small cell lung cancer cells. Cancer Biol Ther. 2005;4:267–76. doi: 10.4161/cbt.4.3.1496. [DOI] [PubMed] [Google Scholar]
- 36.Taniai M, Grambihler A, Higuchi H, et al. Mcl-1 mediates tumor necrosis factor-related apoptosis-inducing ligand resistance in human cholangiocarcinoma cells. Cancer Res. 2004;64:3517–24. doi: 10.1158/0008-5472.CAN-03-2770. [DOI] [PubMed] [Google Scholar]
- 37.Le Gouill S, Podar K, Harousseau JL, Anderson KC. Mcl-1 regulation and its role in multiple myeloma. Cell Cycle. 2004;3:1259–62. doi: 10.4161/cc.3.10.1196. [DOI] [PubMed] [Google Scholar]
- 38.Bortul R, Tazzari PL, Cappellini A, et al. Constitutively active Akt1 protects HL60 leukemia cells from TRAIL-induced apoptosis through a mechanism involving NF- kappaB activation and cFLIP(L) up-regulation. Leukemia. 2003;17:379–89. doi: 10.1038/sj.leu.2402793. [DOI] [PubMed] [Google Scholar]
- 39.Wang X, Chen W, Lin Y. Sensitization of TNF-induced cytotoxicity in lung cancer cells by concurrent suppression of the NF-kappaB and Akt pathways. Biochem Biophys Res Commun. 2007;355:807–12. doi: 10.1016/j.bbrc.2007.02.030. [DOI] [PubMed] [Google Scholar]
- 40.Zhao Y, Altman BJ, Coloff JL, et al. Glycogen synthase kinase 3alpha and 3beta mediate a glucose-sensitive antiapoptotic signaling pathway to stabilize Mcl-1. Mol Cell Biol. 2007;27:4328–39. doi: 10.1128/MCB.00153-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Panka DJ, Mano T, Suhara T, Walsh K, Mier JW. Phosphatidylinositol 3-kinase/Akt activity regulates c-FLIP expression in tumor cells. J Biol Chem. 2001;276:6893–6. doi: 10.1074/jbc.C000569200. [DOI] [PubMed] [Google Scholar]