

Abstract
Objective:
Mutations in both alleles of parkin have been shown to result in Parkinson disease (PD). However, it is unclear whether haploinsufficiency (presence of a mutation in only 1 of the 2 parkin alleles) increases the risk for PD.
Methods:
We performed comprehensive dosage and sequence analysis of all 12 exons of parkin in a sample of 520 independent patients with familial PD and 263 controls. We evaluated whether presence of a single parkin mutation, either a sequence (point mutation or small insertion/deletion) or dosage (whole exon deletion or duplication) mutation, was found at increased frequency in cases as compared with controls. We then compared the clinical characteristics of cases with 0, 1, or 2 parkin mutations.
Results:
We identified 55 independent patients with PD with at least 1 parkin mutation and 9 controls with a single sequence mutation. Cases and controls had a similar frequency of single sequence mutations (3.1% vs 3.4%, p = 0.83); however, the cases had a significantly higher rate of dosage mutations (2.6% vs 0%, p = 0.009). Cases with a single dosage mutation were more likely to have an earlier age at onset (50% with onset at ≤45 years) compared with those with no parkin mutations (10%, p = 0.00002); this was not true for cases with only a single sequence mutation (25% with onset at ≤45 years, p = 0.06).
Conclusions:
Parkin haploinsufficiency, specifically for a dosage mutation rather than a point mutation or small insertion/deletion, is a risk factor for familial PD and may be associated with earlier age at onset.
GLOSSARY
- ADL
= Activities of Daily Living;
- GDS
= Geriatric Depression Scale;
- MLPA
= multiplex ligation-dependent probe amplification;
- MMSE
= Mini-Mental State Examination;
- PD
= Parkinson disease;
- UPDRS
= Unified Parkinson’s Disease Rating Scale.
Parkinson disease (PD; MIM 168600) is the second most common neurodegenerative disease. Mutations in 5 genes have been identified to segregate with PD in a mendelian fashion. Alterations in 3, PRKN (parkin), DJ-1, and PINK1, are typically transmitted with autosomal recessive inheritance, whereas alterations in the other 2, SNCA and LRRK2, are inherited in an autosomal dominant fashion. Mutations in these 5 genes seem to be causative in less than 5% of patients.1
More than 100 different parkin mutations have been reported and are found in all 12 exons of the gene.2 Point mutations as well as copy number variation (i.e., deletions and duplications of entire exons, hereafter referred to as “dosage” changes) have been identified as pathogenic mutations.3–11 Harboring 2 parkin mutations typically leads to juvenile-onset (ages 15–25 years) or early-onset (<45 years) forms of PD.5,6 Several previous studies have hypothesized that haploinsufficiency, or the presence of a mutation in only 1 of 2 parkin alleles, may lead to an increased risk of PD. If true, this would suggest that parkin may be both a causative and a susceptibility gene.11–14
We have performed both sequencing and dosage analysis of all parkin exons in a sample of controls and sequentially recruited familial PD cases, allowing us to obtain unbiased estimates of the frequency of parkin mutations in controls as well as cases with a family history of PD.
METHODS
Subjects screened (n = 520).
PD families were ascertained as part of an ongoing genetic study through a pair of siblings, both of whom were reported to have PD. The study was approved at each site’s institutional review board. The first 520 families out of a total of 735 recruited were included in the present study, and a single affected individual from each family, typically the proband, was selected to be comprehensively screened for parkin mutations. Results of parkin screening for 127 of these individuals have been previously reported.9,11 All participants were seen by a movement disorder specialist at 1 of 59 Parkinson Study Group sites located throughout North America. Each participant completed a uniform clinical assessment that included the Unified Parkinson’s Disease Rating Scale (UPDRS) Parts II (Activities of Daily Living) and III (Motor Exam),15 Schwab and England score,16 Hoehn and Yahr stage,17 the Mini-Mental State Examination (MMSE),18 the Geriatric Depression Scale (GDS),19 and the Blessed Functional Activity Scale (Blessed).20 In addition, a diagnostic checklist with inclusion criteria consisting of clinical features highly associated with autopsy-confirmed PD and exclusion criteria consisting of clinical features highly associated with non-PD pathologic diagnoses was used to classify individuals as having either verified PD (90%) or nonverified PD (10%).21 DNA was obtained from peripheral blood.
Cases used in association analyses (n = 420).
To avoid bias from population stratification, association analyses were limited to self-reported white, non-Hispanic individuals who met criteria for verified PD and did not harbor a known LRRK2 mutation.22–24 Of the 520 individuals screened, 29 were not of white, non-Hispanic descent, 50 did not meet criteria for verified PD, and 33 had previously been identified to harbor an LRRK2 mutation.22–24 Some PD cases fulfilled more than 1 of these exclusion criteria. The final analyzed data set included 420 independent cases.
Controls screened (n = 263).
The control sample consisted of 270 neurologically normal individuals obtained from the National Institute of Neurological Disorders and Stroke Human Genetics DNA and Cell Line Repository (Camden, NJ; DNA panels NDPT002, NDPT006, NDPT009). Data from a prior genome-wide association study verified that these individuals were unrelated.25 Individuals not of white, non-Hispanic descent (n = 2) and controls reporting a family history of PD in a first or second degree relative (n = 5) were excluded. All controls provided appropriate written informed consent for storage and future use of their samples. The final analyzed data set included 263 independent controls.
Parkin sequencing.
PCR and sequencing primers were designed using the chromosome 6 genomic contig sequence NT_007422.13 enabling PCR/sequencing of all 12 coding exons and corresponding intron/exon boundaries of parkin. Typically, 80 ng of each genomic DNA was PCR amplified in a 40-μL reaction using conditions empirically determined for each primer pair as previously described.26 The resulting PCR products were purified using the QIAquick 96 PCR purification kit (QIAGEN, Santa Clara, CA) and sequenced on an ABI 3730 DNA analyzer using the Applied Biosystems (Foster City, CA) BigDye Terminator Kit, version 1.1. DNA sequences were aligned and analyzed using Sequencher 4.5 (Gene Codes Corporation, Ann Arbor, MI). All missense mutations were analyzed using SIFT (http://blocks.fhcrc.org/sift/SIFT.html) to predict in silico whether the amino acid change would be deleterious or benign.
Parkin dosage analysis using MLPA.
Multiplex ligation-dependent probe amplification (MLPA) was performed with 100 ng of genomic DNA according to manufacturer’s instructions using the P051 and P052 Salsa MLPA Parkinson probe sets (MRC-Holland, Amsterdam, The Netherlands). This probe set includes probes for all parkin exons, and 10 of the 12 exons have a duplicate probe. Probe amplification products were run on an ABI 3730xl DNA Analyzer using GS500 size standard (Applied Biosystems). MLPA peak plots were visualized using Genemapper Software version 3.7 (Applied Biosystems), and nonnormalized values for peak height and peak area were then exported to an Excel template. Normalization of data and calculation of dosage ratios was performed as described at www.mrc-holland.com/MLPA%20analysis.htm. A dosage ratio value of ≤0.7 was used as the boundary for deletions, and ≥1.35 was used as the boundary for duplications.
Parkin dosage analysis using real-time PCR.
All deletions and duplications identified using MLPA were verified using real-time PCR. Applied Biosystems’ Assay by Design service was used to design fam-labeled TaqMan gene expression assays for each exon of parkin. Genomic DNA samples were quantitated by Pico Green fluorescence in triplicate with the Quant-iT PicoGreen dsDNA Kit (Molecular Probes, Eugene, OR). After quantitation, 50 ng of genomic DNA was used in a real-time absolute quantitation assay for the parkin exon in question, performed on the 7300 Real Time PCR System (Applied Biosystems). Assays were performed as 25-μL reactions in triplicate, with each genomic DNA sample being done in duplicate for the parkin exon in question. After real-time PCR, data were analyzed with the ABI Sequence Detection Software, RQ Study upgrade, version 1.2.3 (Applied Biosystems). Quantitation of the target amount in DNA samples was accomplished by measuring the Ct value and comparing it with a standard curve. Quantitation for study samples was then compared with those of control samples known not to have exonic deletions/duplications.
Statistical analysis.
The common variants S167N, V380L, and D394N, previously shown to be benign polymorphisms,5,27,28 were not coded as mutations in these analyses.
The Fisher exact test was used to evaluate the first hypothesis: the presence of a single parkin mutation increased the risk of PD. Analyses were also performed classifying the single parkin mutation as either a dosage or sequence mutation and tested whether the presence of each of these different types of mutations individually increased the risk of PD.
The second hypothesis tested was that individuals with parkin mutations (either 1 or 2) had different clinical characteristics (i.e., age at onset, UPDRS subscores, MMSE, GDS, Hoehn and Yahr stage) compared with those cases without a parkin mutation. The groups with 2 parkin mutations and 1 parkin mutation were compared independently with the control group. Linear and logistic regression models were used, where relevant.
RESULTS
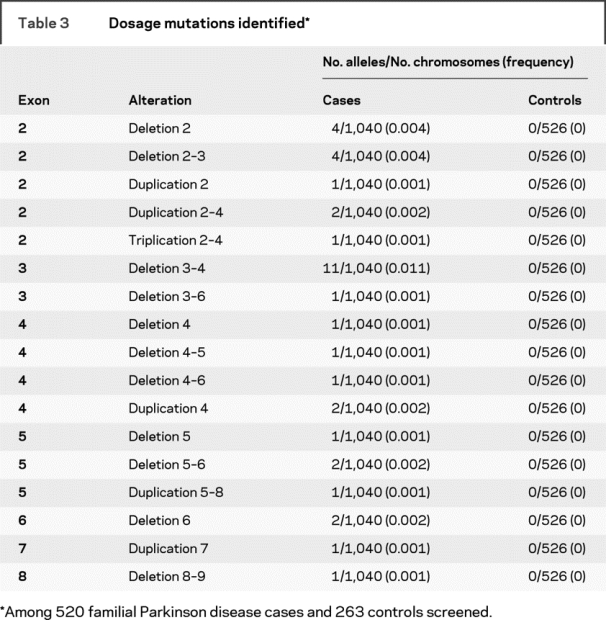
A total of 520 consecutive, independent, familial PD cases were screened for parkin mutations using both direct sequencing and MLPA analysis (tables 1 and 2). Mutations, defined as any variant found in <1% of all control chromosomes and that change the amino acid sequence, were identified in 55 patients (10.6% of familial PD cases) (table 3). The percentage of individuals with parkin mutations was higher when symptoms began in earlier decades: 33% for those with onset between ages 11 and 20 years, 47% for onset between 21 and 30 years, 52% for onset between 31 and 40 years, 14% for onset between 41 and 50 years, 2% for onset between 51 and 60 years, 7% for onset between 61 and 70 years, and 5% for onset at age ≥71 years (tables e-1 and e-2 on the Neurology® Web site at www.neurology.org). Of those harboring at least 1 parkin mutation, 27 had a mutation in both alleles (11 homozygous, 16 compound heterozygous), and 28 had a mutation detected in only 1 of the 2 alleles (12 dosage mutations, 16 sequence mutations). The most common mutation detected in our PD cohort was the cis deletion of exons 3–4 (13% of observed mutations), followed by P437L (11%) and R275W (10%). All other mutations each represented less than 10% of the total number of mutations observed (tables e-1, e-2, 2, and 3).
Table 1 Sample demographics
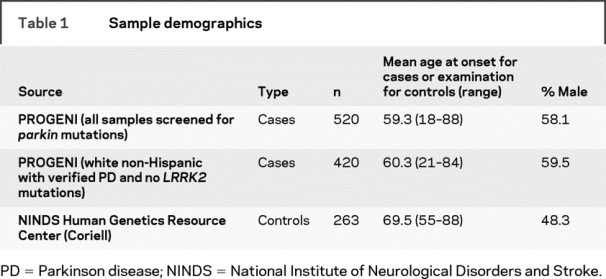
Table 2 Sequence mutations identified
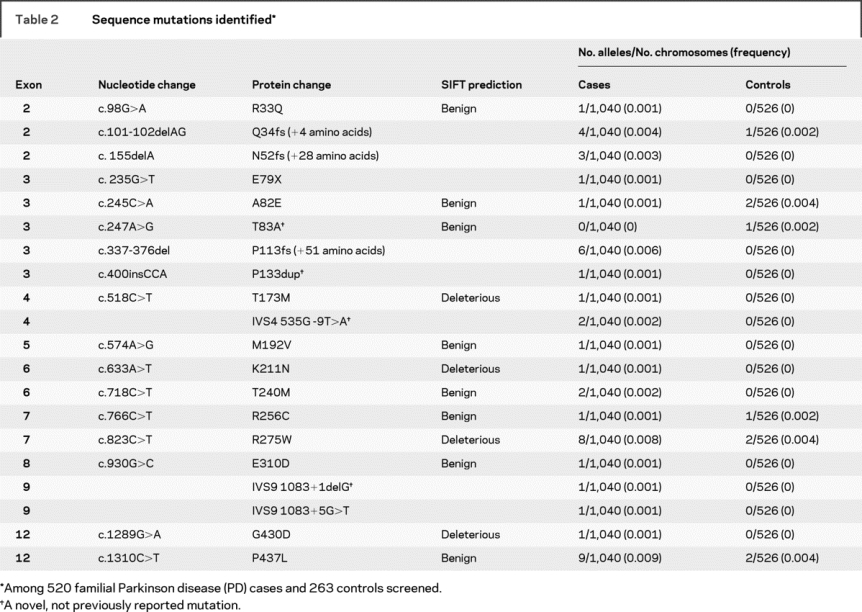
Table 3 Dosage mutations identified
Nine of the 263 controls were heterozygous carriers of a single parkin sequence mutation (table 2). The most common mutations detected in the control sample were A82E, R275W, and P437L, with 2 instances of each. MLPA analysis yielded no parkin dosage changes in the controls (tables e-1 and e-2). Controls carrying a parkin variant did not have a significantly earlier age at examination (69.4 years vs 69.5 years for those not harboring a parkin mutation); however, those carrying a variant predicted to be deleterious by the program SIFT (table 2) tended to be younger at examination (57, 60, 73 years) (table e-1).
Within the 420 unrelated white, non-Hispanic, familial PD cases, the frequency of mutations in only 1 of the 2 parkin alleles was 5.7% (24 PD cases), with 3.1% having a single change detected by sequence analysis and 2.6% having a single dosage change identified by MLPA (table 4). In comparison, only 9 controls carried a parkin mutation (3.4% of controls), with none being a dosage mutation. Overall, cases were not more likely than controls to carry a mutation in only 1 of 2 parkin alleles (5.7% vs 3.4%, p = 0.20). However, when the type of parkin mutation was compared, there was a higher frequency of a single parkin dosage mutation in the cases than in the controls (2.6% vs 0%, p = 0.009). The frequency of each individual sequence and dosage mutation was too low to provide sufficient power to test for association with each mutation separately.
Table 4 Association between parkin carrier status and Parkinson disease
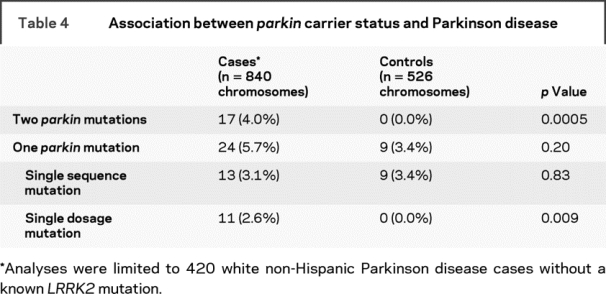
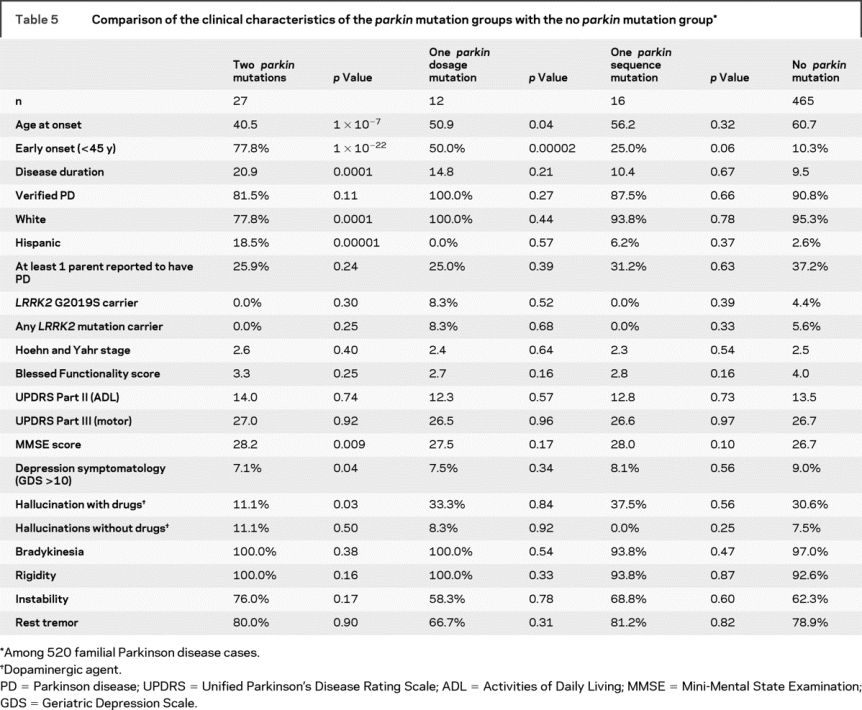
The clinical features of the parkin mutation-positive and mutation-negative PD individuals were remarkably similar (table 5). The core symptoms of PD (bradykinesia, rest tremor, muscular rigidity, and postural instability) were all found at comparable rates. There was no difference in the proportion of individuals classified as verified PD (87.3% of mutation carriers vs 90.8% of noncarriers, p = 0.46). PD cases with a single dosage mutation were more likely to have an earlier age at onset (<45 years) compared with those with no parkin mutations (p = 0.00002), but the same could not be said for those with only a single sequence mutation (p = 0.06). Individuals with 2 mutations had a significantly earlier onset than those with only 1 mutation (p = 0.0008), though this too was different for sequence (p = 0.001) and dosage mutations (p = 0.02). No other clinical feature (other than ethnicity) differed between those that carried 2 mutations vs 1 mutation. As shown previously,29 there was no association between harboring 1 or 2 parkin mutations and depressive symptomatology (GDS score ≥ 10) when individuals showing signs of cognitive impairment (MMSE < education-specific threshold) were excluded from analysis (p = 0.19).
Table 5 Comparison of the clinical characteristics of the parkin mutation groups with the no parkin mutation group
DISCUSSION
Interpreting the clinical significance of parkin mutations has been problematic because of the large number of different mutations that have been identified in the gene.1 Truncating or frame-shift mutations likely lead to a loss of enzyme function, whereas the effect of missense, synonymous and variants near splice sites may not have as obvious an effect on enzyme function. It has not been conclusively shown whether a mutation on only 1 of the 2 alleles affects the risk for PD.11–14,27 Here we provide evidence that certain types of mutations in the heterozygous state are more likely to increase risk than others. Specifically, a single copy of a deletion or duplication was found in 2.9% of cases and none of the controls (p = 0.009). PD cases carrying a single dosage change were more likely to have onset before age 45 years (50% compared with 10% of those without parkin mutations, p = 0.00002); however, cases carrying a single sequence mutation were no more likely to have onset before age 45 years than those without a mutation (25% vs 10%, p = 0.06). These data suggest that dosage changes in parkin play a larger role in disease susceptibility than do sequence mutations.
There is also heterogeneity of effect with regard to missense mutations. At one end of the spectrum are several common variants previously shown to be benign polymorphisms (S167N, V380L, D394N)5,27,28 that were therefore excluded from analysis in the current study. Other observed variants included in our analyses may also be benign, but they are found at such a low frequency that it is difficult to draw firm conclusions about their pathogenicity. One such variant, P437L, is predicted by the program SIFT to be benign. This variant is found in 6 independent cases within this familial cohort, and the 3 PD cases that were homozygous for the variant tended to have a later age at onset (ages 45, 70, 81 years; table 2). If the allele is pathogenic, it would seem to result in later onset PD as compared with other missense mutations. When P437L and all other variants categorized as “benign” by the SIFT algorithm were excluded from analyses, the same conclusions are reached—namely, single sequence mutations are equally common in cases and controls (p = 0.75), single sequence mutations are not associated with early age at onset (22% with onset at ≤45 years compared with 10% among those without a parkin mutation; p = 0.26), and single dosage changes are more common in cases than controls (p = 0.009). However, with benign mutations excluded and the 2 types of mutations (sequence and dosage) combined, cases had a higher frequency of a single parkin mutation than controls (p = 0.02). These results suggest that the variants predicted by SIFT as being benign are likely to be correct; however, additional studies are required to confirm the in silico predictions.
Individuals with 2 parkin mutations tended to have earlier onset and consequently tended to have a longer duration of disease. Significant differences in other clinical characteristics were not seen in this study. Hispanics made up a disproportionately higher percentage of the individuals with 2 parkin mutations as compared with their representation in the rest of the sample (12.1% of cases with 2 parkin mutations vs 2.4% of cases with no parkin mutations, p = 0.00001). All 6 Hispanic cases harboring parkin mutations were from Puerto Rico. Particular alleles were more common in the Puerto Rican cases; of the 12 parkin alleles observed, 4 were cis deletions of exons 3–4, 3 were N52fs (+28 amino acids) frameshift mutations, 2 were R275W, 2 were cis deletions of exons 2–3, and 1 was the wild-type allele. If parkin mutations are in fact more common in Puerto Rico or in the greater Hispanic population at large, it is especially important that any genetic association analyses be performed within a single population to avoid bias due to population stratification. Overrepresentation of parkin mutations in Hispanic populations has been noted previously,30 and the cis deletion of exons 3 and 4 was found to be common among Puerto Rican cases.2
One subject with PD harbored both a heterozygous deletion of exon 2 and an LRRK2 G2019S allele (table 5). This individual met criteria for verified PD and had onset at age 41 years. One sibling of this individual had onset at 68 years and had the same 2 mutant alleles at LRRK2 and parkin, whereas the other sibling had onset at 83 years and inherited only the G2019S allele, suggesting that the mutant parkin allele contributed to earlier disease onset.
In this study, we have used MLPA to screen for whole exon deletions and duplications and confirmed every dosage change using real-time PCR. Previously, we used semiquantitative fluorescent multiplex PCR to identify parkin dosage mutations.9,11 Several dosage changes reported in our previous studies9,11 did not replicate using the newer more robust methods, including what seemed to be a common deletion in exon 8. We have now determined that one of the primers used to amplify this region annealed to a region containing a previously unknown SNP, which resulted in the erroneously observed exon deletion. In contrast, only 2 mutations flagged by MLPA (deletion of exon 4, duplication of exon 9) did not validate using real-time PCR.
Phase could be determined conclusively for 89% of individuals (including all heterozygous rearrangements involving consecutive exons), and identity by descent estimates from prior linkage data provided supportive evidence for an additional 8% of individuals (tables e-1 and e-2). Should any of the inconclusive compound heterozygotes with dosage mutations prove to have both variants on a single allele, this would only strengthen the evidence that heterozygous dosage mutations are associated with disease.
In the past, most studies of parkin have focused on either sporadic or early-onset cases. Our study is relatively unique in its ascertainment of familial PD, and it is therefore likely to be enriched for a genetic etiology. In addition, we have not limited our screening to cases below an age at onset threshold. For this reason, we were able to show that parkin mutations are present in individuals with an older age at onset (tables e-1 and e-2). Therefore, a mutation in the parkin gene should not be ruled out when evaluating patients with a later age at onset, especially if they have a family history of PD.
We have completed the largest study to perform both sequencing and dosage analysis in cases and controls. Given the high frequency of exon rearrangements in our sample, it is apparent that thorough screening of the parkin gene must include dosage studies. In this study, we were able to show that while the frequency of single sequence mutations is similar between cases and controls (even when variants predicted to be benign were excluded), cases are much more likely to harbor dosage changes, indicating that this type of mutation may be more pathogenic.
AUTHOR CONTRIBUTIONS
All statistical analyses were performed by N. Pankratz.
ACKNOWLEDGMENT
Control samples and clinical data were provided from the National Institute of Neurological Disorders and Stroke Human Genetics DNA and Cell Line Repository (http://ccr.coriell.org/ninds). The authors thank Dr. Lorraine Clark for her helpful suggestions in drafting the manuscript. The authors thank all subjects for their participation in this research study.
DISCLOSURE
Dr. Marder serves as an editorial board member of Neurology®. Dr. Pfeiffer serves on the Center Review Board of the National Parkinson Foundation; serves as Co-editor-in-Chief of Parkinsonism and Related Disorders; receives royalties from publishing Neurogastroenterology [Butterworth Heinemann(Elsevier) 2008], Parkinson’s Disease [CRC Press (Taylor & Francis) 2008], and Parkinson’s Disease and Nonmotor Dysfunction (Humana Press, 2008); has served on an external advisory committee for the Udall Center; has received speaker honoraria from the Parkinson Association of Southwest Florida, the Black Hills Neurology Clinic, Mayo Clinic Jacksonville, Jackson-Madison County Memorial Hospital, Indian Physicians Medical Group (Memphis, TN), and Yale University; serves on scientific advisory boards for Kyowa, Solvay, and Ipsen; serves on speakers’ bureaus of GlaxoSmithKline, Boehringer-Ingelheim, Novartis, Teva, and UCB/Schwarz; receives research support from Boehringer-Ingelheim, Kyowa, Novartis, Eisai, UCB/ Schwarz, and Santhera; and has served as an expert consultant to law firms Spriggs & Hollingsworth and Davis Graham & Stubbs.
Supplementary Material
APPENDIX
Parkinson Study Group Investigators:
PROGENI Steering Committee: University of Tennessee Health Science Center: R.F. Pfeiffer (Chair); University of Rochester: F. Marshall, D. Oakes, A. Rudolph, A. Shinaman; Columbia University Medical Center: K. Marder; Indiana University School of Medicine: P.M. Conneally, T. Foroud, C. Halter; University of Kansas Medical Center: K. Lyons; Eli Lilly and Company: E. Siemers; Medical College of Ohio: L. Elmers; University of California, Irvine: N. Hermanowicz.
PSG-PROGENI Investigators and Coordinators: Albany Medical College: S. Factor, D. Higgins, S. Evans; Barrow Neurological Institute: H. Shill, M. Stacy, J. Danielson, L. Marlor, K. Williamson; Baylor College of Medicine: J. Jankovic, C. Hunter; Beth Israel Deaconess Medical Center: D. Simon, P. Ryan, L. Scollins; Beth Israel Medical Center: R. Saunders-Pullman, K. Boyar, C. Costan-Toth, E. Ohmann; Brigham and Women’s Hospital: L. Sudarsky, C. Joubert; Brown University (Memorial Hospital of Rhode Island): J. Friedman, K. Chou, H. Fernandez, M. Lannon; Cleveland Clinic Florida-Weston: N. Galvez-Jimenez, A. Podichetty, K. Thompson; Clinical Neuroscience Center: P. Lewitt, M. DeAngelis; Colorado Neurological Institute: C. O’Brien, L. Seeberger, C. Dingmann, D. Judd; Columbia University Medical Center: K. Marder, J. Fraser, J. Harris; Creighton University: J. Bertoni, C. Peterson; Evanston Northwestern Healthcare: M. Rezak, G. Medalle; Hotel-Dieu Hospital-Chum: S. Chouinard, M. Panisset, J. Hall, H. Poiffaut; Hunter Homes McGuire Veterans Medical Center: V. Calabrese, P. Roberge; Indiana University School of Medicine: J. Wojcieszek, J. Belden; Institute for Neurodegenerative Disorders: D. Jennings, K. Marek, S. Mendick; Johns Hopkins University: S. Reich, B. Dunlop; London Health Sciences Centre: M. Jog, C. Horn; Mayo Clinic Jacksonville: R. Uitti, M. Turk; McFarland Neurosciences: T. Ajax, J. Mannetter; Medical College of Georgia: K. Sethi, J. Carpenter, B. Dill, L. Hatch, K. Ligon, S. Narayan; Medical College of Wisconsin: K. Blindauer, K. Abou-Samra, J. Petit; Medical University of Ohio: L. Elmer, E. Aiken, K. Davis, C. Schell, S. Wilson; Mount Sinai School of Medicine: M. Velickovic, W. Koller (deceased), S. Phipps; North Shore-LIJ Health System: A. Feigin, M. Gordon, J. Hamann, E. Licari, M. Marotta-Kollarus, B. Shannon, R. Winnick; Northwestern University: T. Simuni, A. Videnovic, A. Kaczmarek, K. Williams, M. Wolff; Ochsner Clinic Foundation: J. Rao, M. Cook; Ohio State University: M. Fernandez, S. Kostyk, J. Hubble, A. Campbell, C. Reider, A. Seward; Oregon Health and Science University: R. Camicioli, J. Carter, J. Nutt, P. Andrews, S. Morehouse, C. Stone; Ottawa Hospital Civic Site: T. Mendis, D. Grimes, C. Alcorn-Costa, P. Gray, K. Haas, J. Vendette; Pacific Neuroscience Medical Group: J. Sutton, B. Hutchinson, J. Young; Saskatoon District Health Board Royal University Hospital: A. Rajput, A. Rajput, L. Klassen, T. Shirley; Scott and White Hospital/Texas A&M University: B. Manyam, P. Simpson, J. Whetteckey, B. Wulbrecht; The Parkinson’s and Movement Disorder Institute: D. Truong, M. Pathak, K. Frei, N. Luong, T. Tra, A. Tran, J. Vo; Toronto Western Hospital, University Health: A. Lang, G. Kleiner-Fisman, A. Nieves, L. Johnston, J. So; UMDNJ-School of Osteopathic Medicine: G. Podskalny, L. Giffin; University of Alabama at Birmingham: P. Atchison, C. Allen; University of Alberta: W. Martin, M. Wieler; University of Calgary: O. Suchowersky, M. Klimek; University of California Irvine: N. Hermanowicz, S. Niswonger; University of California San Diego: C. Shults (deceased), D. Fontaine; University of California San Francisco: M. Aminoff, C. Christine, M. Diminno, J. Hevezi; University of Chicago: A. Dalvi, U. Kang, J. Richman, S. Uy, J. Young; University of Cincinnati: A. Dalvi, A. Sahay, M. Gartner, D. Schwieterman; University of Colorado Health Sciences Center: D. Hall, M. Leehey, S. Culver, T. Derian; University of Connecticut: T. Demarcaida, S. Thurlow; University of Iowa: R. Rodnitzky, J. Dobson; University of Kansas Medical Center: K. Lyons, R. Pahwa, T. Gales, S. Thomas; University of Maryland School of Medicine: L. Shulman, S. Reich, W. Weiner, K. Dustin; University of Miami: K. Lyons, C. Singer, W. Koller (deceased), W. Weiner, L. Zelaya; University of Minnesota: P. Tuite, V. Hagen, S. Rolandelli, R. Schacherer, J. Kosowicz; University of New Mexico: P. Gordon, J. Werner; University of Puerto Rico School of Medicine: C. Serrano, S. Roque; University of Rochester: R. Kurlan, D. Berry, I. Gardiner; University of South Florida: R. Hauser, J. Sanchez-Ramos, T. Zesiewicz, H. Delgado, K. Price, P. Rodriguez, S. Wolfrath; University of Tennessee Health Science Center: R. Pfeiffer, L. Davis, B. Pfeiffer; University of Texas Southwestern Medical Center: R. Dewey, B. Hayward, A. Johnson, M. Meacham, B. Estes; Wake Forest University School of Medicine: F. Walker, V. Hunt, C. O’Neill; Washington University: B. Racette, L. Good, M. Rundle.
Biostatistics and Clinical Trials Coordination Centers Staff: A. Watts, A. Wang, T. Ross, S. Bennett, D. Kamp, E. Julian-Baros, S. Daigneault, R. Doolan.
Address correspondence and reprint requests to Dr. Tatiana Foroud, Medical and Molecular Genetics, Indiana University, School of Medicine, Hereditary Genomics Division, 410 W. 10th St., MI-4000, Indianapolis, IN 46202 tforoud@iupui.edu
Supplemental data at www.neurology.org
*Parkinson Study Group–PROGENI Investigators are listed in the appendix.
Supported by NIH R01 NS37167 (N.P., D.K.K., M.W.P., C.A.H., A.R., R.F.P., K.S.M., T.F., W.C.N.) and M01 RR-00750.
Disclosure: Author disclosures are provided at the end of the article.
Received January 14, 2009. Accepted in final form April 17, 2009.
REFERENCES
- 1.Pankratz N, Foroud T. Genetics of Parkinson disease. Genet Med 2007;9:801–811. [DOI] [PubMed] [Google Scholar]
- 2.Hedrich K, Eskelson C, Wilmot B, et al. Distribution, type, and origin of Parkin mutations: review and case studies. Mov Disord 2004;19:1146–1157. [DOI] [PubMed] [Google Scholar]
- 3.Kitada T, Asakawa S, Hattori N, et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998;392:605–608. [DOI] [PubMed] [Google Scholar]
- 4.Lucking CB, Abbas N, Durr A, et al. Homozygous deletions in parkin gene in European and North African families with autosomal recessive juvenile parkinsonism. The European Consortium on Genetic Susceptibility in Parkinson’s Disease and the French Parkinson’s Disease Genetics Study Group. Lancet 1998;352:1355–1356. [DOI] [PubMed] [Google Scholar]
- 5.Abbas N, Lucking CB, Ricard S, et al. A wide variety of mutations in the parkin gene are responsible for autosomal recessive parkinsonism in Europe. French Parkinson’s Disease Genetics Study Group and the European Consortium on Genetic Susceptibility in Parkinson’s Disease. Hum Mol Genet 1999;8:567–574. [DOI] [PubMed] [Google Scholar]
- 6.Lucking CB, Durr A, Bonifati V, et al. Association between early-onset Parkinson’s disease and mutations in the parkin gene. N Engl J Med 2000;342:1560–1567. [DOI] [PubMed] [Google Scholar]
- 7.Hedrich K, Marder K, Harris J, et al. Evaluation of 50 probands with early-onset Parkinson’s disease for Parkin mutations. Neurology 2002;58:1239–1246. [DOI] [PubMed] [Google Scholar]
- 8.Kann M, Jacobs H, Mohrmann K, et al. Role of parkin mutations in 111 community-based patients with early-onset parkinsonism. Ann Neurol 2002;51:621–625. [DOI] [PubMed] [Google Scholar]
- 9.Nichols WC, Pankratz N, Uniacke SK, et al. Linkage stratification and mutation analysis at the Parkin locus identifies mutation positive Parkinson’s disease families. J Med Genet 2002;39:489–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.West A, Periquet M, Lincoln S, et al. Complex relationship between Parkin mutations and Parkinson disease. Am J Med Genet 2002;114:584–591. [DOI] [PubMed] [Google Scholar]
- 11.Foroud T, Uniacke SK, Liu L, et al. Heterozygosity for a mutation in the parkin gene leads to later onset Parkinson disease. Neurology 2003;60:796–801. [DOI] [PubMed] [Google Scholar]
- 12.Farrer M, Chan P, Chen R, et al. Lewy bodies and parkinsonism in families with parkin mutations. Ann Neurol 2001;50:293–300. [DOI] [PubMed] [Google Scholar]
- 13.Klein C, Pramstaller PP, Kis B, et al. Parkin deletions in a family with adult-onset, tremor-dominant parkinsonism: expanding the phenotype. Ann Neurol 2000;48:65–71. [PubMed] [Google Scholar]
- 14.Sun M, Latourelle JC, Wooten GF, et al. Influence of heterozygosity for parkin mutation on onset age in familial Parkinson disease: the GenePD study. Arch Neurol 2006;63:826–832. [DOI] [PubMed] [Google Scholar]
- 15.Fahn S, Elton RL; UPDRS Development Committee. Unified Parkinson’s disease rating scale. In: Fahn S, Marsden CD, Goldstein M, eds. Recent Developments in Parkinson’s Disease. New York: Macmillan Healthcare Information; 1987:153–163. [Google Scholar]
- 16.Schwab R, England A. Projection technique for evaluating surgery in Parkinson’s disease. In: Gillingham F, Donaldson I, eds. Third Symposium on Parkinson’s Disease. Edinburgh: E&S Livingstone; 1969:152–157. [Google Scholar]
- 17.Hoehn MM, Yahr MD. Parkinsonism: onset, progression and mortality. Neurology 1967;17:427–442. [DOI] [PubMed] [Google Scholar]
- 18.Folstein MF, Folstein SE, McHugh PR. “Mini-Mental State”: a practical method for grading the cognitive state of patients for the clinician J Psychiatr Res 1975;12:189–198. [DOI] [PubMed] [Google Scholar]
- 19.Yesavage JA, Brink TL, Rose TL, et al. Development and validation of a geriatric depression screening scale: a preliminary report. J Psychiatr Res 1982;17:37–49. [DOI] [PubMed] [Google Scholar]
- 20.Blessed G, Tomlinson BE, Roth M. The association between quantitative measures of dementia and of senile change in the cerebral grey matter of elderly subjects. Br J Psychiatry 1968;114:797–811. [DOI] [PubMed] [Google Scholar]
- 21.Pankratz N, Nichols WC, Uniacke SK, et al. Genome screen to identify susceptibility genes for Parkinson disease in a sample without parkin mutations. Am J Hum Genet 2002;71:124–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nichols WC, Pankratz N, Hernandez D, et al. Genetic screening for a single common LRRK2 mutation in familial Parkinson’s disease. Lancet 2005;365:410–412. [DOI] [PubMed] [Google Scholar]
- 23.Pankratz N, Pauciulo MW, Elsaesser VE, et al. Mutations in LRRK2 other than G2019S are rare in a north American-based sample of familial Parkinson’s disease. Mov Disord 2006;21:2257–2260. [DOI] [PubMed] [Google Scholar]
- 24.Nichols WC, Elsaesser VE, Pankratz N, et al. LRRK2 mutation analysis in Parkinson disease families with evidence of linkage to PARK8. Neurology 2007;69:1737–1744. [DOI] [PubMed] [Google Scholar]
- 25.Pankratz N, Wilk JB, Latourelle JC, et al. Genomewide association study for susceptibility genes contributing to familial Parkinson disease. Hum Genet 2009;124:593–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pankratz N, Pauciulo MW, Elsaesser VE, et al. Mutations in DJ-1 are rare in familial Parkinson disease. Neurosci Lett 2006;408:209–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kay DM, Moran D, Moses L, et al. Heterozygous parkin point mutations are as common in control subjects as in Parkinson’s patients. Ann Neurol 2007;61:47–54. [DOI] [PubMed] [Google Scholar]
- 28.Wang M, Hattori N, Matsumine H, et al. Polymorphism in the parkin gene in sporadic Parkinson’s disease. Ann Neurol 1999;45:655–658. [DOI] [PubMed] [Google Scholar]
- 29.Pankratz N, Marder KS, Halter CA, et al. Clinical correlates of depressive symptoms in familial Parkinson’s disease. Mov Disord 2008;23:2216–2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marder KS, Clark L, Mejia-Santana H, et al. Features associated with parkin mutation status in probands in the Core-PD Study. Ann Neurol 2007;62(suppl):S38. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.