

Abstract
Objective:
To investigate the relationship between baseline MRI and CSF biomarkers and subsequent change in continuous measures of cognitive and functional abilities in cognitively normal (CN) subjects and patients with amnestic mild cognitive impairment (aMCI) and Alzheimer disease (AD) and to examine the ability of these biomarkers to predict time to conversion from aMCI to AD.
Methods:
Data from the Alzheimer's Disease Neuroimaging Initiative, which consists of CN, aMCI, and AD cohorts with both CSF and MRI, were used. Baseline CSF (t-tau, Aβ1–42, and p-tau181P) and MRI scans were obtained in 399 subjects (109 CN, 192 aMCI, 98 AD). Structural Abnormality Index (STAND) scores, which reflect the degree of AD-like features in MRI, were computed for each subject.
Results:
Change on continuous measures of cognitive and functional performance was modeled as average Clinical Dementia Rating–sum of boxes and Mini-Mental State Examination scores over a 2-year period. STAND was a better predictor of subsequent cognitive/functional change than CSF biomarkers. Single-predictor Cox proportional hazard models for time to conversion from aMCI to AD showed that STAND and log (t-tau/Aβ1–42) were both predictive of future conversion. The age-adjusted hazard ratio for an interquartile change (95% confidence interval) of STAND was 2.6 (1.7, 4.2) and log (t-tau/Aβ1–42) was 2.0 (1.1, 3.4). Both MRI and CSF provided information about future cognitive change even after adjusting for baseline cognitive performance.
Conclusions:
MRI and CSF provide complimentary predictive information about time to conversion from amnestic mild cognitive impairment to Alzheimer disease and combination of the 2 provides better prediction than either source alone. However, we found that MRI was a slightly better predictor of future clinical/functional decline than the CSF biomarkers tested.
GLOSSARY
- AD
= Alzheimer disease;
- ADNI
= Alzheimer's Disease Neuroimaging Initiative;
- aMCI
= amnestic mild cognitive impairment;
- CDR-SB
= Clinical Dementia Rating–sum of boxes score;
- CI
= confidence interval;
- CN
= cognitively normal;
- HR
= hazard ratio;
- MMSE
= Mini-Mental State Examination;
- NFT
= neurofibrillary tangle;
- STAND
= Structural Abnormality Index.
Given that degenerative pathologic changes in Alzheimer disease (AD) occur before clinical symptoms,1 it would be useful to have diagnostic indicators of the underlying biology to complement clinical assessment in evaluating the risk of future clinical decline. Two core biochemical and imaging biomarkers, CSF and structural MRI, reflect different aspects of the pathology of AD. Two promising CSF biomarkers for AD are total tau (t-tau) and Aβ1–42.2–6 We have also included analyses of phospho-tau181P (p-tau181P) since it reflects phosphorylated tau and has been postulated to closely mirror neurofibrillary tangle (NFT) formation.7
Structural MRI captures disease-related structural changes in the brain by measuring loss of brain volume, the direct result of loss of neurons, synapses, and supporting cellular structures.8,9 There is ample evidence in the literature supporting that MRI is a good surrogate of neurodegeneration, NFT stage, and neuron and synapse loss. A technique developed in our laboratory condenses the degree and location of AD-related atrophy on the 3-dimensional MRI scan into a single number which is called Structural Abnormality Index (STAND) score10 and STAND scores based on antemortem MRI scans correlate well with Braak stage at autopsy.11 To date, only a handful of studies have compared MRI and CSF biomarkers for assessing future cognitive decline along the cognitively normal (CN)–amnestic mild cognitive impairment (aMCI)–AD spectrum.12–14
In this article, we evaluate the following 2 questions using Alzheimer's Disease Neuroimaging Initiative (ADNI) data: 1) What is the association between baseline biomarkers (MRI and CSF) and subsequent change on continuous cognitive and functional measures (Clinical Dementia Rating–sum of boxes [CDR-SB] and Mini-Mental State Examination [MMSE]) by clinical group? 2) Do baseline biomarkers predict time to conversion from aMCI to AD?
METHODS
ADNI is a longitudinal multisite observational study of elderly individuals who are CN or have aMCI or AD collected from 56 participating institutes15 (http://www.ADNI-info.org). Baseline MRI was obtained on all subjects and baseline CSF was obtained in approximately 55% of the cohort. In this study, all subjects with both CSF biomarker data at baseline and usable MRI scans were considered. Our cohort consists of 399 subjects (109 CN, 192 aMCI, 98 AD). We used cognitive data from screening and all follow-up visits available and biomarkers collected at baseline.
Standard protocol approvals, registrations, and patient consents.
Written informed consent was obtained for participation in these studies, as approved by the Institutional Review Board at each of the participating centers.
Clinical and cognitive assessment.
We used MMSE16 and CDR-SB17 as overall indices of general cognitive performance and global functional status. Two sets of clinical and cognitive assessments were used: longitudinal CDR-SB and MMSE change in all 3 clinical groups and time of conversion of MCI to AD from baseline. In addition to being collected at screening, CDR-SB and MMSE were collected at 6-, 12-, 18- (only for subjects classified as aMCI at screening), and 24-month visits. Time-to-event analysis was conducted on 186 subjects with aMCI with at least one follow-up. Sixty subjects with aMCI converted to AD over a median time of 1.5 years (taking into account the entire follow-up in each subject). Six of the 186 with a baseline diagnosis of MCI had a subsequent diagnosis of CN at some point in their follow-up (i.e., “backwash” phenomenon) who were treated as nonconverters in the time-to-event analysis. There was one subject with aMCI who progressed to semantic dementia, whom we considered to have aMCI since the subject did not convert to AD. Only 4 CN subjects converted to aMCI in the follow-up period available; therefore, prediction of conversion in CN subjects was not considered here.
The CSF and MRI methods and estimation of STAND scores are explained in detail in appendix e-1 on the Neurology® Web site at www.neurology.org.
Statistical analysis.
Biomarker prediction of future clinical change.
We used linear mixed-effects models to estimate the longitudinal change in mean CDR-SB and MMSE.18 All mixed models were fit using restricted maximum likelihood estimation. The models specified a random subject-specific intercept and a random subject-specific slope. This allows for heterogeneity among subjects in baseline values and rates of change and accounts for correlation among repeated measurements on the same subject. We included the following fixed effects of primary interest: years since baseline examination, diagnosis group, and a single biomarker along with all 2-way and 3-way interactions. Additionally, we adjusted for age by including baseline age and its interaction with diagnosis as fixed effects. These models allow for average baseline value to depend on baseline age, clinical group, and biomarker. The models allow average slope of cognitive measures to depend on baseline clinical group and to be modified by biomarker.
We also looked at the information each biomarker provides in addition to baseline MMSE and CDR-SB. The longitudinal CDR-SB models were adjusted for baseline MMSE by including the interaction between baseline MMSE, diagnosis, and time. Similarly, MMSE models were adjusted for baseline CDR-SB by including interaction between baseline CDR-SB, diagnosis, and time. These adjustments allow for the linear trajectory of the response to depend on baseline functional or cognitive performance that differs by group. Note that longitudinal CDR-SB models were adjusted for baseline MMSE and vice versa to somewhat avoid the issue of circularity.
In all models, biomarker variables were modeled as continuous measures with t-tau, p-tau181P, and t-tau/Aβ1–42 transformed using the natural logarithm to reduce skewness. We used model estimates to graphically represent change in mean CDR-SB and MMSE over time for 25th, 50th, and 75th percentile of biomarker measurements within group. Slopes are shown at the median age within group. The mixed effects models were found to fit the data well with good agreement between observed and predicted value along with approximately normally distributed residuals.
Baseline prediction of time to conversion from aMCI to AD.
We assessed the effect of each biomarker variable on time to a diagnosis of AD among subjects diagnosed with aMCI at baseline using Cox proportional hazards models that included age as an adjustment covariate. Baseline visit was considered time zero. Subjects who remained free of AD throughout all follow-up visits were censored at their last visit date. For subjects who converted to AD, the date of event was calculated as the midpoint between their last visit without AD and their first visit with AD. The biomarker variables were modeled using a restricted cubic spline with 3 knots in order to account for potential nonlinear effects. Results are summarized using hazard ratios (HRs) comparing 75th to 25th percentiles and presented graphically as estimated survivorship functions for 25th, 50th, and 75th percentiles of the biomarker. These survivorship functions are related to the Kaplan-Meier method but obtained from the Cox models and an estimated baseline hazard.19 Also note that we do not divide the sample into quartiles and then report HRs based on comparison of upper vs lower quartiles. The HRs and survivorship functions assume a subject 75 years old at baseline.
All data manipulation and analysis was performed using SAS version 9.1.3 and R version 2.7.1. Mixed effects models were fit using the NLME package in R.
RESULTS
Patient characteristics.
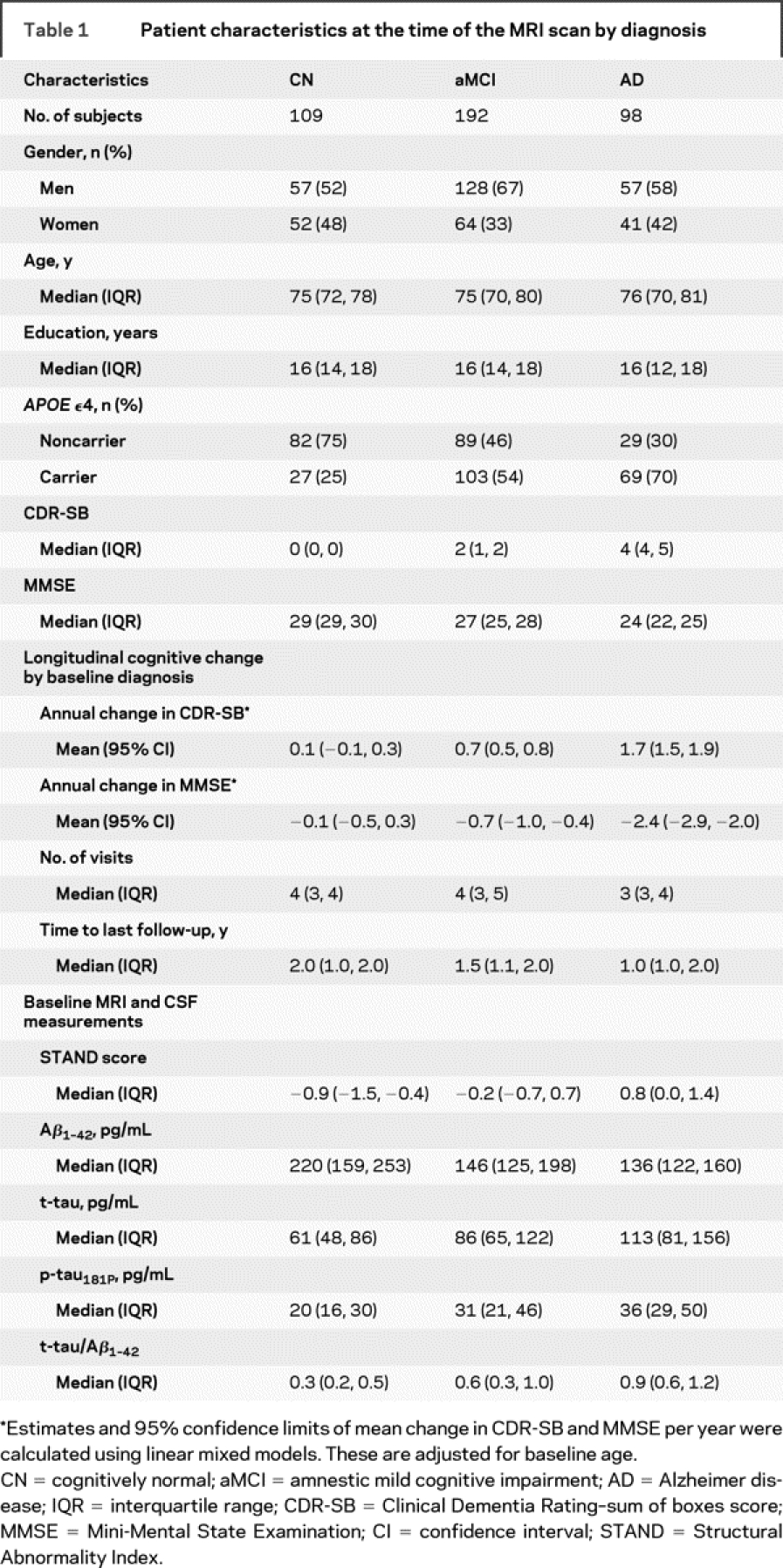
Demographics, clinical summary, longitudinal clinical data, and MRI and CSF biomarker summary are presented in table 1. Since the sampling interval for cognitive assessment is different among clinical groups, the estimates and 95% confidence limits of mean change in CDR-SB and MMSE per year were calculated using linear mixed models. These estimates are shown along with median follow-up time and number of visits by baseline diagnosis in table 1. All pairwise differences in rates of change in CDR-SB and MMSE are significant at 0.001 level except for CN vs aMCI on MMSE, which was significant at 0.01 level. Differences between CN and AD, CN and aMCI, and aMCI and AD were significant for all biomarkers.
Table 1 Patient characteristics at the time of the MRI scan by diagnosis
Biomarker prediction of future clinical change.
Average CDR-SB over time by diagnosis for 25th, 50th, and 75th percentiles of baseline Aβ1–42, t-tau, STAND score, ratio, and CDR-SB at baseline is plotted in figure 1. Among CN subjects, there was no appreciable relationship between baseline MRI or CSF biomarkers and subsequent change in CDR-SB. In contrast, CDR-SB worsened over time in aMCI and AD. Slopes of change in CDR-SB with time are progressively greater for each STAND score quartile from low to high, indicating appropriately ordered sensitivity of this measure to subsequent cognitive change; i.e., greater baseline atrophy is associated with greater subsequent cognitive decline. In contrast, although there is evidence that biologically worse t-tau and Aβ1–42 are associated with greater subsequent change on CDR-SB, scaling of change in CDR-SB by quartiles of baseline CSF biomarkers is not as clearcut.

Figure 1 Estimated average value of Clinical Dementia Rating–sum of boxes score (CDR-SB) over time by diagnosis group for the 25th, 50th, and 75th percentiles of each biomarker or baseline CDR-SB after accounting for baseline age
The percentiles for each biomarker are calculated within group. Curves assume a baseline age of 75 years for cognitively normal (CN), 75 years for amnestic mild cognitive impairment (aMCI), and 76.5 years for Alzheimer disease (AD), the median values observed in our sample. STAND = Structural Abnormality Index
We evaluated additive effects of these predictors in terms of explaining variability in CDR-SB over time. We found that when STAND score is in the longitudinal prediction model, Aβ1–42 did not contribute significantly (p = 0.19), nor did log(t-tau) (p = 0.25). We used natural logarithm transform to correct for skewness. On the other hand, a model with Aβ1–42 did improve when STAND was added (p < 0.001). Similarly, a model with log(t-tau) improved when STAND was added (p < 0.001). We found no evidence that log(t-tau) added to Aβ1–42 (p = 0.38) but some evidence that Aβ1–42 adds to log(t-tau) (p = 0.12). STAND adds to a model with t-tau/Aβ1–42 (p < 0.001) and there is some evidence that the ratio adds to a model with STAND (p = 0.12). A model with baseline CDR-SB improved when STAND was added (p < 0.001) and Aβ1–42 was added (p = 0.04); there was evidence that t-tau (p = 0.08) and ratio (p = 0.06) add to CDR-SB. However, baseline CDR-SB added to each of the biomarkers individually (p < 0.001). Average MMSE over time by diagnosis for 25th, 50th, and 75th percentiles of Aβ1–42, t-tau, and STAND score is shown in appendix e-2A, where we found similar relationships.
Estimated average value of CDR-SB over time by diagnosis group for the 25th, 50th, and 75th percentiles of each biomarker after accounting for baseline MMSE and age is shown in appendix e-2B. This figure illustrates the fact that STAND score provides additional information regarding future clinical change even after adjusting for baseline cognitive performance. A similar figure for the estimated average value of MMSE over time with respect to biomarkers after accounting for baseline CDR-SB and age is shown in appendix e-2C.
Biomarker prediction of time to conversion from aMCI to AD.
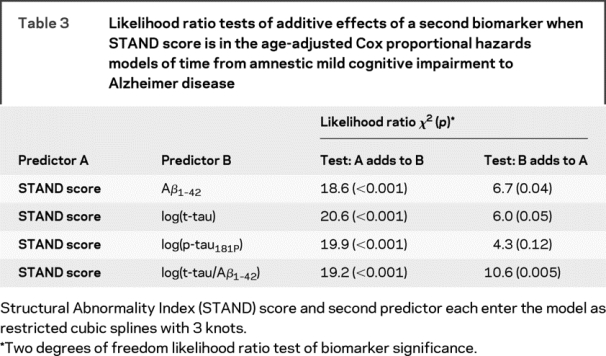
Estimated survivorship functions by 25th, 50th, and 75th percentile of baseline STAND and CSF biomarker values based on single-predictor Cox proportional hazard models modeling time to conversion from aMCI to AD are shown in figure 2 and model summaries in table 2. The relationship between time to conversion from aMCI to AD scaled appropriately by quartiles of STAND, i.e., worse baseline STAND was associated with shorter time to conversion. Aβ1–42 did not have an ordered relationship of time to conversion by quartiles that were biologically sensible, which might be due to the significant nonlinear relationship between Aβ1–42 and time to conversion in this sample. Aβ1–42, t-tau, and p-tau181 were not strongly associated with time to conversion from aMCI to AD. STAND and log(t-tau/Aβ1–42) were both found to be predictive of future conversion from aMCI to AD. The age-adjusted HR for an interquartile change (95% confidence interval [CI]) for STAND was 2.6 (1.7, 4.2) and log(t-tau/Aβ1–42) was 2.0 (1.1, 3.4). The difference in magnitudes of the HRs and greater χ2 statistic (table 2) provides evidence that STAND score is a better predictor than CSF of time to AD. We also estimated the concordance index (C-index), which in a survival model is analogous to AUROC in a logistic model and represents the estimated probability the model will correctly predict which of 2 patients has longer time to conversion. STAND score has a C-index of 0.69, log(t-tau) has a C-index of 0.60, ratio and Aβ1–42 have a C-index of 0.62. Based on 95% bootstrap CIs, the estimated differences in the C-index were STAND vs Aβ1–42 (−0.01 to +0.17); STAND vs log(t-tau) (+0.01 to +0.20); log(p-tau181P) (−0.01 to +0.17); and ratio (−0.02 to +0.16). While STAND appears to better discriminate than log(t-tau), the other CIs include 0, implying that at p = 0.05 level we cannot conclude that STAND provides better discrimination than Aβ1–42, log(p-tau181P), and the ratio. However, the trends point in that direction. The likelihood ratio tests of additive effects of time to conversion predictors when STAND is in the model are presented in table 3. STAND score added information to each of the CSF biomarkers including t-tau/Aβ1–42 (p < 0.001). t-tau, p-tau181P, and Aβ1–42 individually did not add information at the p = 0.01 level to STAND score; however, log(t-tau/Aβ1–42) added information (p < 0.01) to STAND score. When the models were adjusted for baseline CDR-SB, the effect of CDR-SB adjustment was minimal even though CDR-SB was itself significant. This indicates that MRI and CSF biomarkers provide predictive information complementary to clinical assessments.
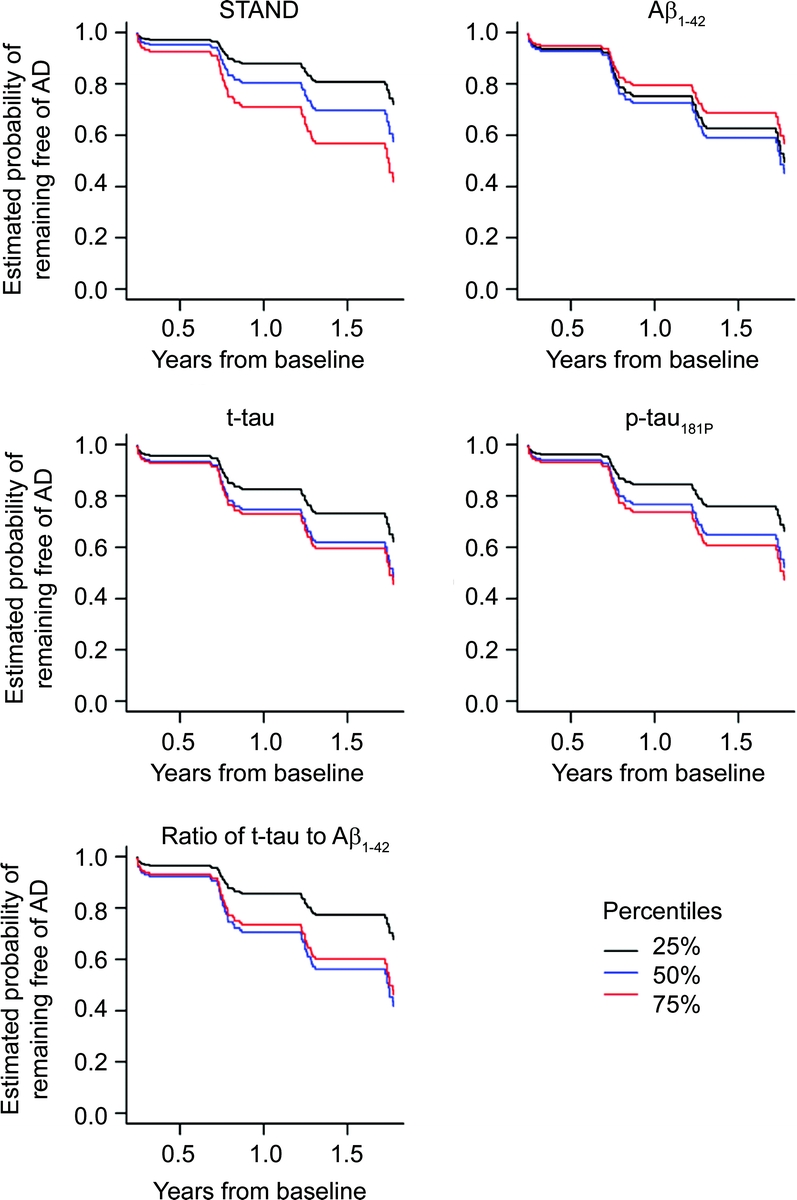
Figure 2 Estimated survivorship functions by quartiles based on age-adjusted Cox proportional hazard models modeling the imaging or CSF predictor with a restricted cubic spline with 3 knots
Estimates assume a subject age of 75 years at baseline. AD = Alzheimer disease; STAND = Structural Abnormality Index
Table 2 Model summaries from age-adjusted Cox proportional hazards models of time from amnestic mild cognitive impairment to Alzheimer disease
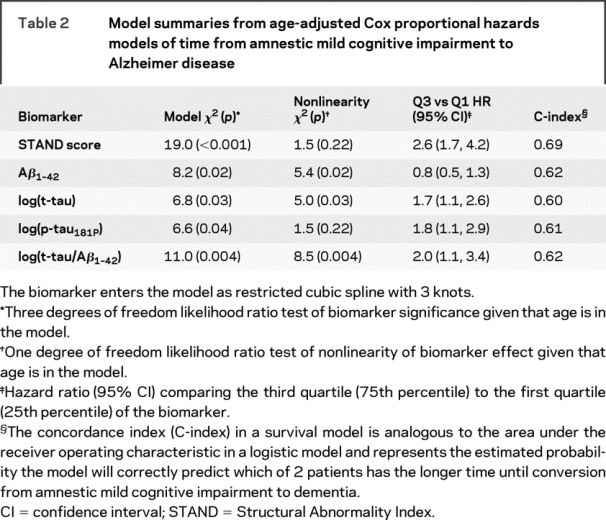
Table 3 Likelihood ratio tests of additive effects of a second biomarker when STAND score is in the age-adjusted Cox proportional hazards models of time from amnestic mild cognitive impairment to Alzheimer disease
DISCUSSION
This study presents associations with future clinical change of 2 core disease indicators in AD: MRI and CSF. We showed that both baseline MRI and CSF are independently associated with future cognitive decline. When average CDR-SB over time by biomarker quartiles was compared within each diagnostic group, MRI was more predictive of decline on the CDR-SB than CSF using likelihood ratio tests. This can also be observed in figure 1, where there is an ordered but no clear separation of cognitive decline by 25th, 50th, and 75th percentiles of t-tau and Aβ1–42. This relationship still holds after adjusting for baseline cognitive performance, supporting the idea that biomarkers provide additional information to clinical assessments. One practical implication of these findings is that either structural MRI by itself or in combination with CSF t-tau/Aβ1–42 levels could be used to identify subjects with MCI or AD at higher risk to decline more rapidly than others. This predictive power could be employed in certain clinical trial designs, such as phase II studies, where small numbers of subjects are studied to gain a preliminary sense of efficacy and could also be used in subset analyses of more rapid progressors in larger phase III studies.
In our time-to-event analysis, both STAND and log(t-tau/Aβ1–42) were highly significant predictors of time to conversion of aMCI to AD. Our results are in concordance with most of the existing CSF and MRI literature, which has shown that MRI biomarkers such as hippocampal, entorhinal, and ventricular volumes and brain atrophy rates20–27 and CSF biomarker, t-tau/Aβ1–42,5,6,28,29 are significant predictors of future cognitive decline. Our results suggest that structural brain changes provide slightly better information than CSF to predict future clinical course of disease in subjects who meet criteria for MCI at baseline.
This is in contradiction with an earlier CSF-MRI study that found MRI biomarkers were not better predictors of future conversion compared to CSF biomarkers with a follow-up of 19 months.13 The authors13 used visual grading of MRI scans for assessing medial temporal atrophy, in contrast to the automated analysis used here, which could account for the difference in results. It has been shown that the computerized scoring of MRI scans for neurodegenerative atrophy is more reliable than visual scoring of MRI scans30 and there is a better correlation between Braak stage and STAND scores (r = 0.62) than hippocampal volume measurements (r = 0.4) in subjects who have undergone antemortem MRI scans and then come to autopsy,11 supporting the use of STAND scores here.
MRI, Aβ1–42, and t-tau reflect different aspects of AD pathology. Low CSF Aβ1–42 is a marker of fibrillary amyloid deposition in plaques. Near complete concordance is present between individuals with positive Pittsburgh Compound B (PIB)–PET scans and those with low CSF Aβ1–42.31 Although correlations with Aβ1–42 were present in our study, well accepted reasons exist to explain why Aβ1–42 might not correlate highly with clinical indices of disease intensity, where intensity is defined as rate of change. Amyloid deposition is regarded to be an early event that occurs prior to clinical symptoms, and therefore CSF Aβ1–42 is not a good leading indicator of near term cognitive decline.32
Increased CSF t-tau is a marker of neuronal injury which correlates well with NFT stage and NFT load.33,34 Atrophy on structural MRI also correlates with Braak NFT stage and NFT load35–38 but the most proximate histologic correlate of MRI volume loss is loss of neurons and synapses.8,9 It may at first be surprising to find that correlations with clinical disease progression are slightly stronger for MRI vs t-tau given that CSF t-tau is usually regarded as direct marker of neuronal injury. A possible explanation for the better correlation between MRI and cognitive/functional performance than that with t-tau is simply that MRI may be a more stable indicator of neuronal injury. Brain volume quantification with MRI has nothing analogous to daily turnover of a soluble protein with inevitable diurnal variation.39 Lower physiologic variation in brain volume may translate into stronger correlations between MRI and clinical indices of disease progression over many subjects. Another possible explanation is that MRI measures at a fixed point in time reflect cumulative damage while t-tau reflects recent or transient damage; e.g., t-tau levels become elevated immediately after acute brain injury.
There are some limitations to the study. First, the ADNI cohort is not generalizable to the general population. The recruitment mechanisms were those used for clinical trials in AD and included memory clinics, patient registries, public media campaigns, and other forms of public advertisements. Second, although numbers of subjects were relatively large, the period of clinical follow-up was relatively short, with median follow-up times of 2.0 years in CN, 1.5 years in aMCI, and 1.0 years in AD. Thus, conclusions about the relationship between baseline MRI and CSF to future clinical course pertain only to relatively short-term clinical outcomes.
Our main goal of this study was to better understand and compare the effect of CSF and global structural atrophy levels on risk of progression from aMCI to dementia. However, the findings provide some possibly useful information for a prospective clinical trial examining time from aMCI to dementia. To increase event rates, enrollment could be restricted to higher-risk subjects who have CSF or STAND scores above or below a certain value, although this may come at the risk of reducing generalizability. Alternatively, these measures could be used as stratification factors so that the study arms have not only similar demographic profiles but similar subclinical CSF and MRI profiles.
AUTHOR CONTRIBUTIONS
Statistical analysis was conducted by Heather J. Wiste, BA, and Stephen D. Weigand, MS.
ACKNOWLEDGMENT
Data used in the preparation of this article were obtained from the Alzheimer's Disease Neuroimaging Initiative (ADNI) database (www.loni.ucla.edu\ADNI). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report.
DISCLOSURE
Dr. Vemuri receives support from the Robert H. Smith Family Foundation Research Fellowship and NIH [R01-AG11378]. Heather Wiste reports no disclosures. Stephen Weigand reports no disclosures. Dr. Shaw receives support from the NIH [U01-AG024904]. Dr. Trojanowski receives support from the NIH [U01-AG024904]. Dr. Weiner receives support from the NIH [U01-AG024904, P01-AG19724, R01 AG10897, and P41 RR023953]. Dr. Knopman serves as an Associate Editor of Neurology®; served on a data safety monitoring board for Sanofi Aventis; and is an investigator in a clinical trial sponsored by Elan Pharmaceuticals and by Forest Laboratories. Dr. Petersen has served as a consultant to GE Healthcare; has served on a data safety monitoring board for Elan and Wyeth Pharmaceuticals; and receives research support from the NIH [P50-AG16574 and U01-AG06786]. Dr. Jack is an investigator in clinical trials sponsored by Pfizer; serves as a consultant for Elan Pharmaceuticals; and receives research support from the NIH [R01-AG11378] and the Alexander Family Alzheimer's Disease Research Professorship of the Mayo Foundation.
Supplementary Material
Received December 5, 2008. Accepted in final form May 7, 2009.
Address correspondence and reprint requests to Dr. Clifford R. Jack, Jr., Mayo Clinic and Foundation, 200 First Street SW, Rochester, MN 55905 jack.clifford@mayo.edu
Supplemental data at www.neurology.org
See page 287
*Investigators of The Alzheimer's Disease Neuroimaging Initiative are listed at www.loni.ucla.edu\ADNI\Collaboration\ADNI_Manuscript_Citations.pdf.
Supported by NIH grant AG11378; Robert H. Smith Family Foundation Research Fellowship; the Alexander Family AD Research Professorship of the Mayo Foundation; and U.S.A. and Opus building NIH grant C06 RR018898.
Disclosure: Author disclosures are provided at the end of the article.
REFERENCES
- 1.Price JL, Ko AI, Wade MJ, Tsou SK, McKeel DW, Morris JC. Neuron number in the entorhinal cortex and CA1 in preclinical Alzheimer disease. Arch Neurol 2001;58:1395–1402. [DOI] [PubMed] [Google Scholar]
- 2.Andreasen N, Gottfries J, Vanmechelen E, et al. Evaluation of CSF biomarkers for axonal and neuronal degeneration, gliosis, and beta-amyloid metabolism in Alzheimer's disease. J Neurol Neurosurg Psychiatry 2001;71:557–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tapiola T, Overmyer M, Lehtovirta M, et al. The level of cerebrospinal fluid tau correlates with neurofibrillary tangles in Alzheimer's disease. Neuroreport 1997;8:3961–3963. [DOI] [PubMed] [Google Scholar]
- 4.Strozyk D, Blennow K, White LR, Launer LJ. CSF Abeta 42 levels correlate with amyloid-neuropathology in a population-based autopsy study. Neurology 2003;60:652–656. [DOI] [PubMed] [Google Scholar]
- 5.Fagan AM, Roe CM, Xiong C, Mintun MA, Morris JC, Holtzman DM. Cerebrospinal fluid tau/beta-amyloid ratio as a prediction of cognitive decline in nondemented older adults. Arch Neurol 2007;64:343–349. [DOI] [PubMed] [Google Scholar]
- 6.Li G, Sokal I, Quinn JF, et al. CSF tau/Abeta42 ratio for increased risk of mild cognitive impairment: a follow-up study. Neurology 2007;69:631–639. [DOI] [PubMed] [Google Scholar]
- 7.Blennow K, Hampel H. CSF markers for incipient Alzheimer's disease. Lancet Neurol 2003;2:605–613. [DOI] [PubMed] [Google Scholar]
- 8.Bobinski M, de Leon MJ, Wegiel J, et al. The histological validation of post mortem magnetic resonance imaging-determined hippocampal volume in Alzheimer's disease. Neuroscience 2000;95:721–725. [DOI] [PubMed] [Google Scholar]
- 9.Zarow C, Vinters HV, Ellis WG, et al. Correlates of hippocampal neuron number in Alzheimer's disease and ischemic vascular dementia. Ann Neurol 2005;57:896–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vemuri P, Gunter JL, Senjem ML, et al. Alzheimer's disease diagnosis in individual subjects using structural MR images: validation studies. Neuroimage 2008;39:1186–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vemuri P, Whitwell JL, Kantarci K, et al. Antemortem MRI based STructural Abnormality iNDex (STAND)-scores correlate with postmortem Braak neurofibrillary tangle stage. Neuroimage 2008;42:559–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.de Leon MJ, DeSanti S, Zinkowski R, et al. MRI and CSF studies in the early diagnosis of Alzheimer's disease. J Intern Med 2004;256:205–223. [DOI] [PubMed] [Google Scholar]
- 13.Bouwman FH, Schoonenboom SN, van der Flier WM, et al. CSF biomarkers and medial temporal lobe atrophy predict dementia in mild cognitive impairment. Neurobiol Aging 2007;28:1070–1074. [DOI] [PubMed] [Google Scholar]
- 14.Wahlund LO, Blennow K. Cerebrospinal fluid biomarkers for disease stage and intensity in cognitively impaired patients. Neurosci Lett 2003;339:99–102. [DOI] [PubMed] [Google Scholar]
- 15.Jack CR Jr, Bernstein MA, Fox NC, et al. The Alzheimer's Disease Neuroimaging Initiative (ADNI): MRI methods. J Magn Reson Imaging 2008;27:685–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”: a practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975;12:189–198. [DOI] [PubMed] [Google Scholar]
- 17.Hughes CP, Berg L, Danziger WL, Coben LA, Martin RL. A new clinical scale for the staging of dementia. Br J Psychiatry 1982;140:566–572. [DOI] [PubMed] [Google Scholar]
- 18.Pinheiro JC, Bates DM. Mixed-effects Models in S and S-PLUS. New York: Springer; 2004. [Google Scholar]
- 19.Therneau T, Grambsch P. Modeling Survival Data: Extending the Cox Model (Statistics for Biology and Health). New York: Springer; 2001. [Google Scholar]
- 20.Convit A, de Asis J, de Leon MJ, Tarshish CY, De Santi S, Rusinek H. Atrophy of the medial occipitotemporal, inferior, and middle temporal gyri in non-demented elderly predict decline to Alzheimer's disease. Neurobiol Aging 2000;21:19–26. [DOI] [PubMed] [Google Scholar]
- 21.Csernansky JG, Wang L, Swank J, et al. Preclinical detection of Alzheimer's disease: hippocampal shape and volume predict dementia onset in the elderly. Neuroimage 2005;25:783–792. [DOI] [PubMed] [Google Scholar]
- 22.Devanand DP, Pradhaban G, Liu X, et al. Hippocampal and entorhinal atrophy in mild cognitive impairment: prediction of Alzheimer disease. Neurology 2007;68:828–836. [DOI] [PubMed] [Google Scholar]
- 23.Fleisher AS, Sun S, Taylor C, et al. Volumetric MRI vs clinical predictors of Alzheimer disease in mild cognitive impairment. Neurology 2008;70:191–199. [DOI] [PubMed] [Google Scholar]
- 24.Fox NC, Scahill RI, Crum WR, Rossor MN. Correlation between rates of brain atrophy and cognitive decline in AD. Neurology 1999;52:1687–1689. [DOI] [PubMed] [Google Scholar]
- 25.Jack CR Jr, Petersen RC, Xu YC, et al. Prediction of AD with MRI-based hippocampal volume in mild cognitive impairment. Neurology 1999;52:1397–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jack CR Jr, Shiung MM, Weigand SD, et al. Brain atrophy rates predict subsequent clinical conversion in normal elderly and amnestic MCI. Neurology 2005;65:1227–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stoub TR, Bulgakova M, Leurgans S, et al. MRI predictors of risk of incident Alzheimer disease: a longitudinal study. Neurology 2005;64:1520–1524. [DOI] [PubMed] [Google Scholar]
- 28.Hansson O, Zetterberg H, Buchhave P, Londos E, Blennow K, Minthon L. Association between CSF biomarkers and incipient Alzheimer's disease in patients with mild cognitive impairment: a follow-up study. Lancet Neurol 2006;5:228–234. [DOI] [PubMed] [Google Scholar]
- 29.Schoonenboom NS, van der Flier WM, Blankenstein MA, et al. CSF and MRI markers independently contribute to the diagnosis of Alzheimer's disease. Neurobiol Aging 2008;29:669–675. [DOI] [PubMed] [Google Scholar]
- 30.Kloppel S, Stonnington CM, Barnes J, et al. Accuracy of dementia diagnosis: a direct comparison between radiologists and a computerized method. Brain 2008;131(Pt 11):2969–2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fagan AM, Mintun MA, Mach RH, et al. Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Abeta42 in humans. Ann Neurol 2006;59:512–519. [DOI] [PubMed] [Google Scholar]
- 32.Ingelsson M, Fukumoto H, Newell KL, et al. Early Abeta accumulation and progressive synaptic loss, gliosis, and tangle formation in AD brain. Neurology 2004;62:925–931. [DOI] [PubMed] [Google Scholar]
- 33.Buerger K, Ewers M, Pirttila T, et al. CSF phosphorylated tau protein correlates with neocortical neurofibrillary pathology in Alzheimer's disease. Brain 2006;129:3035–3041. [DOI] [PubMed] [Google Scholar]
- 34.Clark CM, Xie S, Chittams J, et al. Cerebrospinal fluid tau and beta-amyloid: how well do these biomarkers reflect autopsy-confirmed dementia diagnoses? Arch Neurol 2003;60:1696–1702. [DOI] [PubMed] [Google Scholar]
- 35.Csernansky JG, Hamstra J, Wang L, et al. Correlations between antemortem hippocampal volume and postmortem neuropathology in AD subjects. Alzheimer Dis Assoc Disord 2004;18:190–195. [PubMed] [Google Scholar]
- 36.Gosche KM, Mortimer JA, Smith CD, Markesbery WR, Snowdon DA. Hippocampal volume as an index of Alzheimer neuropathology: findings from the Nun Study. Neurology 2002;58:1476–1482. [DOI] [PubMed] [Google Scholar]
- 37.Jack CR Jr, Dickson DW, Parisi JE, et al. Antemortem MRI findings correlate with hippocampal neuropathology in typical aging and dementia. Neurology 2002;58:750–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Silbert LC, Quinn JF, Moore MM, et al. Changes in premorbid brain volume predict Alzheimer's disease pathology. Neurology 2003;61:487–492. [DOI] [PubMed] [Google Scholar]
- 39.de Leon MJ, Segal S, Tarshish CY, et al. Longitudinal cerebrospinal fluid tau load increases in mild cognitive impairment. Neurosci Lett 2002;333:183–186. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.