


Abstract
Cooperative binding of ATP and RNA to DEAD-box helicases induces the closed conformation of their helicase core, with extensive interactions across the domain interface. The bound RNA is bent, and its distortion may constitute the first step towards RNA unwinding. To dissect the role of the conformational change in the helicase core for RNA unwinding, we characterized the RNA-stimulated ATPase activity, RNA unwinding and the propensity to form the closed conformer for mutants of the DEAD box helicase YxiN. The ATPase-deficient K52Q mutant forms a closed conformer upon binding of ATP and RNA, but is deficient in RNA unwinding. A mutation in motif III slows down the catalytic cycle, but neither affects the propensity for the closed conformer nor its global conformation. Hence, the closure of the cleft in the helicase core is necessary but not sufficient for RNA unwinding. In contrast, the G303A mutation in motif V prevents a complete closure of the inter-domain cleft, affecting ATP binding and hydrolysis and is detrimental to unwinding. Possibly, the K52Q and motif III mutants still introduce a kink into the backbone of bound RNA, whereas G303A fails to kink the RNA substrate.
INTRODUCTION
Members of the DEAD-box family of RNA helicases participate in all processes involving structural changes of RNA. Their common structure is a helicase core composed of two flexibly linked RecA domains (Figure 1). In many helicases, the core is flanked by additional domains that mediate interactions with other proteins, or provide high affinity- and/or high specificity binding sites for RNA substrates. Thus, these flanking domains define the specific function of a particular helicase, whereas the helicase core is a degenerate nucleotide-dependent switch with affinities for nucleic acids regulated by the nucleotide state. The core region carries all conserved signature motifs in a similar 3D arrangement in different helicases (1), pointing to similar contributions of the conserved motifs to DEAD box helicase activities. A conserved phenylalanine, the Q-motif, motifs I, Ia, Ib, II and III are located in the N-terminal RecA-like domain, and motifs IV, V and VI are in the C-terminal domain (1). In extensive mutational analyses of a number of DEAD box helicases, and in structural studies (2–6), ATP binding and hydrolysis were assigned to motifs I (Walker A motif, AxTGxGKT) and II (Walker B motif, DEAD), with contributions from motif VI (HRIGRTGR). The conserved glutamine in the Q-motif determines the specificity for adenine nucleotides (7). The motifs Ia (PTRELA) and Ib (TPGR), and IV, V (ARGID) and VI contribute to RNA binding (1,2,5,6). Most importantly, coupling of ATPase and unwinding activities has been ascribed to motif III [SAT (8,9)]. Altogether, these studies could confirm similar roles for the conserved signature motifs in many DEAD box helicases, but revealed complex contributions of various motifs to ATP binding and hydrolysis, to RNA binding and to unwinding. Structural studies demonstrated that extensive interactions between conserved motifs within the N- and C-terminal RecA-like domain most likely relay the nucleotide state to the RNA-binding site and thus contribute to RNA unwinding. The crystal structures of the DEAD box helicases Vasa [Drosophila (2)], eIF4A-III [human (3,4)] and Ddx19 ([human (5)] or Dbp5 [yeast (6)] in complex with ADPNP and a single-stranded RNA capture the closed conformation of DEAD box helicases and reveal an extensive interaction network between the conserved motifs from both RecA-like domains across the domain interface. ADPNP is bound in the cleft between the domains, and RNA is bound to a bipartite RNA-binding site formed by both RecA-like domains (Figure 1). The simultaneous assembly of the ATPase site and the RNA-binding site in the closed conformer rationalizes the RNA-stimulated ATPase activity and the cooperativity between ATP and RNA binding. The single-stranded RNA bound to Vasa and Ddx19 is kinked, and it has been suggested that this kink destabilizes an RNA duplex as a first step towards unwinding (2,5).
Figure 1.

DEAD box helicase motifs and position of mutations in motifs I, III and V. The homology model for the YxiN helicase core was constructed with Geno3D (23) using Vasa (PDB-ID 2db3) as a template. Mutations were introduced into motifs I (green, K52Q), motif III (orange, S182A/T184A) and motif V (blue, G303A). ADPNP (cyan) and RNA (yellow) are depicted in stick representation as they are bound to Vasa. The lower panel illustrates the context of the conserved helicase motifs. The positions for fluorophore attachment are indicated in red.
YxiN is a DEAD box helicase from Bacillus subtilis that specifically binds to hairpin 92 in the 23S ribosomal RNA via its C-terminal domain. RNA binding stimulates the intrinsic ATPase activity by three orders of magnitude (10–12). YxiN is in an open conformation with a large inter-domain cleft in the absence of RNA or nucleotide (12,13). In response to cooperative binding of the non-hydrolyzable ATP-analog ADPNP and RNA, YxiN undergoes a conformational change in its helicase core, leading to a closed conformation similar to Vasa and eIF4A-III (12). To investigate the relation of this conformational change to RNA unwinding, we used YxiN variants carrying a mutation in the Walker A motif (motif I, YxiN_K52Q, Figure 1), a mutation in the SAT motif (motif III, S182A/T184A, YxiN_AAA), or a mutation in motif V (YxiN_G303A). The lysine in motif I contacts the β- and γ-phosphates of ATP, and mutations have been reported to diminish ATPase activity in various DEAD box helicases (8,14). The role of ATP binding for DEAD box helicases has previously been addressed using the non-hydrolyzable ATP analog ADPNP, but the concern has been raised that the ADPNP bound state is an off-pathway intermediate (15). The K52Q mutant is thus ideally suited to study the ATP state of DEAD box helicases in more detail without relying on non-hydrolyzable ATP analogs. The two other mutants are examples of so-called ‘uncoupled’ mutants: motif III has been implicated in coupling ATP hydrolysis to RNA unwinding. Mutations do not interfere with ATP hydrolysis or RNA binding, but abolish or reduce helicase activity (8,16). Motif III is the only conserved motif that does not interact with ADPNP or RNA in the Vasa structure (2). In contrast, motif V is the only motif that interacts with both ADPNP and RNA (2). Similar to mutations in motif III, mutation of the conserved glycine in motif V leads to a DEAD box protein that hydrolyzes ATP and binds RNA but does not show unwinding activity (17). Thus, these mutants should be ideally suited to study coupling effects. To dissect the relation between the conformational change in the helicase core and RNA unwinding, we compared the RNA-dependent ATPase activity, the propensity to undergo the conformational change, and RNA unwinding activity for the three YxiN constructs with mutations in motif I, III, or V that are impaired at different points of the catalytic cycle.
MATERIAL AND METHODS
Construction and purification of YxiN mutants
Site-directed mutagenesis was performed according to the Quikchange protocol (Stratagene). YxiN wild-type and mutants were purified as described (11,12).
Steady-state ATPase activity
Adenine nucleotides were purchased from Pharma Waldhof or JenaBioscience. Steady-state ATP hydrolysis was monitored in a coupled enzymatic assay at 37°C via the decrease in A340 due to oxidation of NADH to NAD+ (18) as described (10–12). Assay conditions were 50 mM HEPES/KOH, pH 7.5, 175 mM KCl, 10 mM MgCl2, 100 µM DTT, 200 µM NADH, 1 mM PEP, 13 µg ml−1 LDH, 23 µg ml−1 PK and 10 nM YxiN wild-type, YxiN′, 30 nM YxiN_AAA, YxiN′_AAA, 50 nM YxiN_G303A, YxiN′_G303A, 1 μM YxiN_K52Q or 200 nM YxiN′_K52Q. Initial reaction velocities were calculated from the absorbance change ΔA340/Δt using the extinction coefficient ε340, NADH = 6300 M−1 cm−1. Data from ATP-dependent experiments were analyzed using the standard Michaelis–Menten model. Data from RNA-dependent experiments were analyzed using the explicit binding equation
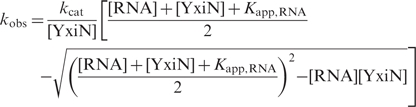 |
1 |
kobs is the observed rate constant at a certain RNA concentration, kcat is the turnover number and Kapp,RNA is the apparent dissociation constant for RNA. For all constructs except YxiN_AAA, Kapp,RNA values from the standard Michaelis–Menten or the explicit binding model were in good agreement. For YxiN_AAA and YxiN′_AAA, data were not well-described by the explicit binding model, and thus were analyzed using the standard Michaelis-Menten model (Figure 2a, Table 1). All experiments were repeated at least three times.
Figure 2.

Steady-state ATPase activity and RNA unwinding. (a) ATP hydrolysis rates as a function of RNA concentration in the presence of 5 mM ATP. Wild-type YxiN (squares) is a Michaelis–Menten enzyme with a kcat of 2.4 s−1 and a Kapp,RNA of 7 nM. YxiN′ (open squares) has wild-type like properties. YxiN_AAA (circles), YxiN′_AAA (open circles), YxiN_G303A (triangles), YxiN′_G303A (open triangles). YxiN_K52Q hydrolysis was within background (stars). Data for YxiN_AAA and YxiN′_AAA could not be described using the explicit binding model and were thus described using the standard Michaelis–Menten model (see Methods section). All experiments were performed at least in triplicate. Kapp,RNA and kcat values are summarized in Table 1. (b) ATP hydrolysis rates as a function of ATP concentration for YxiN_wild-type (squares), YxiN_AAA (circles) and YxiN_G303A (triangles) in the presence of RNA (260 nM, 55 nM, and 603 nM, respectively). Experiments were performed in triplicate. Michaelis–Menten parameters are summarized in Table 2. (c) Unwinding of the 32/9mer minimal RNA substrate by mutants in wildtype background. YxiN_AAA unwinds RNA to the same extent as wild-type YxiN, but much more slowly. YxiN_K52Q and YxiN_G303A are unwinding-deficient. None of the proteins unwinds RNA in the absence of ATP. Numbers below the lanes indicate the fraction of single strand. (d) RNA unwinding by mutants in YxiN′ background. The effect on RNA unwinding is identical to the results in (c) where the mutations were introduced into wild-type YxiN. All depicted data are representative results and have been reproduced at least twice.
Table 1.
Parameters for steady-state ATP hydrolysis by YxiN wild-type and mutants
| kcat (s−1) | Kapp,RNA (nM) | |
|---|---|---|
| YxiN_wild-type | 2.4 (±0.3) | 7 (±2) |
| YxiN_K52Q | n.d. | n.d. |
| YxiN_AAA | 1.36 (±0.03)a | 15 (±1)a |
| YxiN_G303A | 0.55 (±0.03) | 9 (±4) |
| YxiN′ | 2.6 (±0.1) | 13 (±4) |
| YxiN′_K52Q | n.d. | n.d. |
| YxiN′_AAA | 1.1 (±0.2)a | 20 (±3)a |
| YxiN′_G303A | 0.55 (±0.02) | 18 (±4) |
YxiN′ denotes the FRET construct YxiN_C61/267A_A115/S229C. Standard deviations were calculated from individual parameters of at least three independent experiments (Figure 2a).
aParameters were obtained using the standard Michaelis–Menten model to describe the data (see ‘Methods’ section).
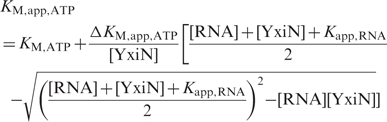
To determine coupling energies for RNA and ATP binding, KM,app,ATP values were determined in ATP-dependent experiments at four different RNA concentrations. KM,app,ATP values were plotted as a function of the RNA concentration, and described using
 |
2 |
Kapp,RNA is the apparent dissociation constant for RNA. Kapp,RNA values from the fit were in reasonable agreement with the values determined directly (Figure 2a). KM,ATP denotes the KM value in the absence of RNA, and the KM,app,ATP at saturating RNA concentration is KM,ATP + ΔKM,app,ATP. These KM values were converted to ΔG values; their difference is the coupling energy.
RNA substrates
The 153mer RNA substrate comprising nucleotides 2483–2635 of the B. subtilis 23S rRNA was generated by T7 polymerase in vitro transcription as described (11).
The 32/9mer minimal RNA substrate comprising hairpin 92 of the 23S rRNA was constructed by annealing a synthetic 32mer and a synthetic 9mer as described (11,12). Unwinding assays were performed with 5 μM RNA and 10 μM YxiN in 50 mM HEPES, pH 7.2, 150 mM KCl, 5% glycerol, 0.1 mM DTT, 0.1 mg/ml BSA, 5 mM MgCl2 at 25°C and products were analyzed by native polyacrylamide gel electrophoresis as described (11,12). All experiments were reproduced at least twice.
Fluorescent labeling and smFRET experiments
Fluorescent labeling of cysteines was performed in 50 mM Tris–HCl, pH 7.5, 500 mM NaCl, 0.5 mM TCEP at a protein concentration of ∼50 μM with a 3-fold molar excess of Alexa488-maleimide (A488, donor) and a 4-fold molar excess of Alexa546-maleimide (A546, acceptor) for 1 h at 25°C. The reaction was stopped by adding 1 mM BME, and free dye was removed by size exclusion chromatography on Bio-Rad Micro Bio-Spin Columns. Labeling efficiencies were determined from absorbance ratios at 493 nm (A488, corrected for A546 contributions) or 554 nm (A546) and 280 nm (protein, corrected for dye contributions).
Single molecule FRET experiments were performed using a home-built confocal microscope as described (12). Only fluorescence bursts above a threshold of 100 photons were considered in the analysis. Measured background-corrected fluorescence intensities were corrected for crosstalk (α donor crosstalk in acceptor channel, β acceptor crosstalk in donor channel), different quantum yields and detection efficiencies of donor and acceptor fluorescence (γ), and direct excitation of the acceptor (δ), and converted into FRET efficiencies as described (12). Measurements were performed at room temperature (25°C) in 50 mM Tris–HCl, pH 7.5, 150 mM NaCl, 5 mM MgCl2 with 40 pM fluorescently labeled protein (concentration of donor fluorophore), 5 mM nucleotide and 200 nM 153mer RNA. For conversion of FRET efficiencies into distances, previously determined Förster distances were used (12).
RESULTS AND DISCUSSION
Motif mutations affect RNA stimulated ATPase activity and RNA unwinding differently
To test the effect of mutations in the conserved motifs on the RNA-stimulated ATP hydrolysis, the steady-state ATPase activity was determined for wild-type YxiN and the mutants in the presence of a 153mer derived from 23S ribosomal RNA (Figure 2a, Table 1). YxiN is a Michaelis–Menten enzyme with a kcat of 2.4 s−1 and the apparent Kapp,RNA value for this RNA of 7 nM. The KM,app,ATP value for ATP in the presence of RNA is 1.0 mM (Figure 2b, Table 2). ATP hydrolysis by the motif I mutant YxiN_K52Q was close to the background hydrolysis under the experimental conditions, confirming that it is ATPase-deficient. The motif III double mutant YxiN_AAA exhibits a reduced kcat of 1.4 s−1, demonstrating that the rate-limiting step in ATP hydrolysis is decelerated. For the Escherichia coli homolog of YxiN, DbpA, it has been shown previously that ATP hydrolysis and phosphate release are rate-limiting in the RNA-stimulated ATPase reaction (15). Consequently, the apparent Kapp,RNA for RNA reflects binding to the nucleotide-states populated under steady-state conditions, which for DbpA are the ATP- and ADP-Pi states. The Kapp,RNA value of YxiN_AAA is slightly increased compared to wild-type (15 nM). Assuming similar steady-state populations for YxiN as for its E. coli homolog, this suggests that the ATP and/or ADP-Pi state bind RNA with similar affinities compared to wild-type YxiN. The KM,app,ATP of YxiN_AAA for ATP is slightly reduced compared to wild-type to 0.6 mM, indicative of a minor increase in ATP affinity. The mutation G303A in motif V drastically reduced the ATPase rate to kcat = 0.55 s−1 and the KM,app,ATP value is increased ∼2-fold compared to wild-type YxiN. Thus, ATP binding and the ATP hydrolysis rate are affected when the conserved glycine is replaced by alanine. In contrast, the Kapp,RNA value (9 nM) is similar to wild-type YxiN.
Table 2.
Michaelis–Menten parameters for steady-state ATP hydrolysis by YxiN wild-type and mutants: KM,app,ATP values for ATP in the presence of 153mer RNA (see Figure 2b)
| KM,app,ATP (mM) | |
|---|---|
| YxiN_wild-type | 1.0 (±0.4) |
| YxiN_AAA | 0.6 (±0.1) |
| YxiN_G303A | 1.7 (±0.2) |
Standard deviations were calculated from individual KM,app,ATP values determined in at least three independent experiments (Figure 2b).
A hallmark feature of DEAD box helicases is the cooperative binding of ATP and RNA substrate (19). Experimentally, cooperativity can be assessed by determining nucleotide affinities in the absence and presence of RNA substrate (12), or by determining RNA affinities in the absence and presence of nucleotide. However, RNA binding to YxiN is dominated by the high-affinity interaction of hairpin 92 in ribosomal RNA with the RNA-recognition motif in the C-terminal domain, and this interaction will mask nucleotide-dependent RNA affinity changes of the helicase core in titration experiments. In the reciprocal titration experiment of nucleotide with YxiN, low nucleotide affinities require high protein concentrations exceeding 100 μM. Solubility problems of the mutants used here precluded such a determination of Kd values, but when the analysis was restricted to data for protein concentrations below 80 μM, relative affinities could be deduced from initial slopes of the titration curves of mantADPNP with YxiN (data not shown). These data point towards reduced ADPNP affinities of YxiN_G303A and YxiN_K52Q compared to wild-type. YxiN_AAA appears to bind ADPNP with similar affinity as wild-type, but the analysis had to be restricted to protein concentrations below 10 μM. In addition, we therefore determined apparent KM,app,ATP values for ATP at different RNA concentrations in steady-state ATPase assays as a measure for ATP affinity (Table 2, data not shown). For wild-type YxiN, the KM,app,ATP value for ATP is markedly reduced with increasing RNA concentrations as a result of thermodynamic coupling. From the extrapolated KM,app,ATP at RNA saturation (0.5 mM) and the extrapolated KM,ATP in the absence of RNA (9 mM, data not shown), a coupling energy of 7.5 kJ/mol (1.8 kcal/mol) can be calculated (see ‘Methods’ section), consistent with the coupling energy for the same RNA substrate in the E. coli homolog DbpA [1.3 kcal/mol, (19)]. YxiN_G303A and YxiN_AAA show less cooperativity between ATP and RNA binding. The coupling energies are reduced to ∼3-4 kJ/mol (∼0.7–1.0 kcal/mol), indicating a substantial loss in thermodynamic coupling in both mutants.
To address the effect of the mutations on coupling of the ATPase activity to RNA unwinding, helicase assays were performed. As unwinding of the helix adjacent to hairpin 92 is reversible in the 153mer RNA substrate and cannot be detected experimentally, a bipartite 32/9mer subfragment of the 153mer was used as the minimal YxiN substrate (Figure 2c). Due to the limited thermodynamic stability of the 9 bp double helix, these experiments have to be performed at 25°C. It should be noted that unwinding assays are performed under single turnover conditions with respect to RNA, but provide multiple turnover conditions with respect to ATP hydrolysis. As a consequence, it cannot be excluded that YxiN undergoes multiple ATPase cycles while it remains bound to the RNA via its C-terminal domain.
Unwinding of this 32/9mer by wild-type YxiN is complete within three minutes and requires ATP (11,12) (Figure 2c). YxiN_AAA unwinds the RNA substrate to a similar extent as the wild-type protein, but displaces the 9mer more slowly. This reduced unwinding velocity might be a consequence of the reduced kcat for ATP hydrolysis, or of the less efficient coupling between RNA and ATP binding. In contrast, neither for YxiN_K52Q nor for YxiN_G303A was helicase activity observed, indicating that both mutants cannot complete the catalytic cycle. This is expected for YxiN_K52Q due to its deficiency in ATP hydrolysis. YxiN_G303A, however, still shows RNA-stimulated ATP hydrolysis, suggesting that the functional interaction between motifs from both RecA-like domains is maintained. The lack of unwinding activity indicates that the catalytic cycle is impaired at a different stage for YxiN_G303A.
Closure of the inter-domain cleft in the helicase core
We next probed whether the YxiN mutants were still able to undergo the conformational change in the helicase core upon binding of RNA and ATP in single molecule FRET (smFRET) experiments. For these measurements, YxiN mutants were used in which the two solvent-accessible cysteines C61 and C267 were replaced by alanines, and two cysteines were introduced at positions 115 and 229 in each RecA domain in the helicase core (YxiN_C61/267A_A115/S229C, abbreviated as YxiN′) for fluorescent labeling. In a previous smFRET study we have used a different construct with the intrinsic cysteines replaced by serines (12), which displayed markedly reduced RNA-stimulated ATPase activities compared to wild-type. In contrast, YxiN′ exhibits similar RNA-stimulated ATPase and RNA unwinding activities as wild-type YxiN, and thus constitutes an improved YxiN construct for smFRET experiments (Supplementary Figure 1). The mutations in the conserved motifs were therefore introduced into YxiN′. These mutants displayed similar kcat and Kapp,RNA values in steady-state ATPase assays (Figure 2a, Table 1) and identical RNA unwinding activities as the corresponding motif mutants in wild-type background (Figure 2d).
SmFRET experiments were performed on freely diffusing donor/acceptor labeled YxiN′ in a confocal microscope. FRET histograms (Figure 3) show a FRET efficiency of ∼0.38 for all constructs in the absence of ligands, suggesting that the conformations of the mutants are similar. Upon addition of 153mer RNA substrate and the non-hydrolyzable ATP analog ADPNP, the FRET efficiency in the wild-type protein shifted to a value of ∼0.7. Using the combined results from FRET experiments with five constructs carrying donor and acceptor fluorophores in different positions on each side of the inter-domain cleft, we have previously established that the increase in FRET efficiency between dyes at positions 115 and 229 reflects a closure of the inter-domain cleft in response to RNA and ATP binding (12). The FRET efficiency remained at 0.38, and the protein retained the open conformation when only RNA or nucleotide was present (data not shown), in agreement with our earlier observations (12). In the presence of ATP and RNA, two populations were observed in the FRET histogram (Figure 3), with FRET efficiencies of 0.38 (open conformation) and 0.70 (closed conformation). Under these conditions, YxiN can hydrolyze ATP and complete catalytic cycles while in the confocal volume, and is thus captured in different conformations at different points in its catalytic cycle. In contrast, it is trapped in the closed pre-hydrolysis conformation in the presence of the non-hydrolyzable ADPNP.
Figure 3.

Conformational changes in the helicase core: smFRET experiments. FRET histograms of YxiN′, YxiN′_K52Q, YxiN′_AAA and YxiN′_G303A in the absence of ligands, and in the presence of 153mer RNA and ADPNP or ATP. The broken line indicates the FRET efficiency for the (completely) closed conformer of YxiN. YxiN′_K52Q and YxiN′_AAA have a similar propensity to form the closed conformer as wild-type YxiN, and the FRET efficiencies for the closed conformer are identical. YxiN′_G303A does not populate the closed conformer to the same extent, and the slightly reduced FRET efficiency of the closed conformer indicates that the cleft is wider compared to the closed conformation of wild-type YxiN.
The motif I mutant YxiN_K52Q is in the open conformation in the absence of substrate as well as in the presence of ATP, ADPNP or RNA alone. In the presence of RNA and ADPNP, or RNA and ATP, the FRET efficiency increased to 0.70 (Figure 3), characteristic of the closed conformation. The FRET histograms for the ADPNP- and ATP-states of YxiN_K52Q are similar, and closely resemble the histogram for wild-type YxiN in the RNA- and ADPNP-bound form. These data confirm that the global conformation of YxiN in the ADPNP-bound state is indeed similar to the ATP-state, in contrast to previous suggestions (15,20,21). Importantly, they provide strong evidence that ATP binding, not a subsequent step, populates the closed helicase conformer with high RNA affinity. Furthermore, the observation that YxiN_K52Q adopts a closed conformation in the RNA- and ATP-bound state, but is unwinding-deficient, demonstrates that the closure of the inter-domain cleft in the helicase core is not sufficient to promote RNA unwinding. This conclusion is consistent with the observation that ADPNP promotes closure of the inter-domain cleft in the helicase core, but does not support RNA unwinding in YxiN (12,22) and other DEAD-box helicases (21).
The motif III mutant YxiN_AAA showed similar FRET histograms to wild-type YxiN in the absence of ligands, and in the presence of RNA and ATP, or RNA and ADPNP (Figure 3), indicating that the SAT motif is not required for the conformational change to occur. Strikingly, the histograms for wild-type YxiN and YxiN_AAA in the presence of RNA and ATP are similar, indicating similar equilibrium populations of open and closed states despite the different rates of ATP hydrolysis. These similar populations suggest that opening and closing rates are equally affected in the YxiN_AAA mutant. While the global conformation of YxiN_AAA appears to be similar to wild-type YxiN, the reduced kcat points to local structural differences that lead to non-optimal alignment of residues involved in ATP hydrolysis, and the reduced thermodynamic coupling indicates differences in inter-domain communication.
Finally, the motif V mutant G303A showed a unique behavior in the presence of RNA and ADPNP, or RNA and ATP, with a very broad distribution of FRET efficiencies (Figure 3). The distribution can be described by two underlying populations, one with a mean FRET efficiency of around ∼0.37 for the open state, and a second state with a mean FRET efficiency of ∼0.65 resembling the closed conformation. The difference between FRET efficiencies of 0.65 for YxiN_G303A and 0.70 as observed for the closed conformations of wild-type YxiN, YxiN_K52Q and YxiN_AAA is significant and was reproduced in multiple independent experiments. The observed difference in the FRET efficiencies translates into a difference in donor/acceptor distance of 0.2 nm, and points towards a slightly wider cleft between the RecA domains in the closed conformation than in the other constructs. The incomplete closure of the cleft is not unexpected as the replacement of the highly conserved glycine by an alanine leads to steric hindrance at the inter-domain interface, as is evident from the Vasa, eIF4A-III and Ddx19 structures (2–6). Such an incomplete cleft-closure would lead to a misassembled catalytic site for ATP hydrolysis, consistent with the significantly reduced ATPase rate of YxiN_G303A. Similarly, the slightly increased Kapp,RNA value for RNA suggests that the two RNA-interaction sites on the RecA domains in YxiN_G303A may not be optimally aligned to form the bipartite RNA-binding site, and the partial loss in thermodynamic coupling of ATP and RNA binding indicates differences in inter-domain communication, again consistent with a different, more open conformation of the closed helicase core.
The model for RNA unwinding that has been proposed based on the Vasa structure predicts that uncoupled mutants still adopt a closed conformation of the helicase core, but do not close completely, and thus may not introduce a kink into the RNA substrate (2). Assuming that the formation of a completely closed conformer is linked to this RNA distortion, our results suggest that YxiN_K52Q and YxiN_AAA that form similar closed conformers as wild-type YxiN should kink the RNA (Figure 4), whereas YxiN_G303A does not. Similar to wild-type YxiN in the presence of ADPNP, YxiN_K52Q is unwinding-deficient because a critical step subsequent to ATP binding and formation of the closed conformer is blocked. YxiN_AAA unwinds RNA more slowly than wild-type because ATP hydrolysis (or product release) is decelerated. YxiN_G303A, in contrast, binds ATP and RNA, but does not adopt a completely closed conformation (Figure 4), and hence might fail to distort the bound RNA. Such a lack of initial distortion would thus prevent the efficient coupling of ATP hydrolysis to RNA unwinding. In this scenario, YxiN_G303A constitutes an uncoupled mutant as proposed by the Vasa unwinding model. Future experiments that directly address the conformation of RNA, such as the structure determination of double-stranded RNA bound to the closed helicase, could shed light on RNA distortions and their origin.
Figure 4.

Effect of mutations on the RNA helicase catalytic cycle. Possible model for RNA unwinding consistent with the data presented in this work. The left branch represents a productive unwinding cycle (interrupted for YxiN_K52Q), the right branch depicts the uncoupled cycle for YxiN_G303A. YxiN is in an open conformation in the absence of ligands (1). Binding of ATP (triangle) and RNA (line-pair) induces a closure of the inter-domain cleft in the helicase core, which introduces a kink into the bound RNA and unwinds the terminal base-pairs (2). YxiN_G303A does not close completely, and possibly does not bend the bound RNA to the same extent (2′). ATP is hydrolyzed, and product release is coupled to dissociation of the unwound RNA (3,4). YxiN_K52Q cannot undergo hydrolysis, and the initial kink is not sufficient for RNA unwinding, rendering YxiN_K52Q unwinding-deficient. The RNA bound to YxiN_G303A dissociates in a double-stranded form upon ADP or phosphate release (3′–4′). The catalytic cycle for YxiN_AAA is similar to wild-type YxiN, but individual steps and thus the overall unwinding reaction are decelerated. Future experiments need to focus on the conformation of bound RNA, and on the identity of the second critical step for RNA unwinding after formation of the closed conformer.
CONCLUSIONS
We have shown here that three mutants of the DEAD box helicase YxiN with mutations in motifs I, III and V that are unwinding-deficient or show reduced unwinding activity still adopt a closed conformation in the presence of RNA and ATP. The conformation of the ATPase-deficient YxiN_K52Q/ATP/RNA complex resembles the corresponding ADPNP state of wild-type YxiN, demonstrating that binding of nucleotide and RNA is sufficient to stabilize the closed helicase conformer. Furthermore, our previous assignment of the closed conformation as the pre-hydrolysis state is independently confirmed. Mutation of the SAT motif affects RNA unwinding by YxiN, but does not completely decouple ATP hydrolysis and RNA unwinding. YxiN_G303A adopts a less compact closed conformation, which is most likely caused by steric hindrance due to the alanine side-chain. Our results show that mutations can affect kcat, either by removal of the catalytic amino acid (YxiN_K52Q), or by affecting the alignment of catalytic residues at the interface of the RecA domains (YxiN_AAA and YxiN_G303A). As the ATPase activity provides a timing function for the catalytic cycle, RNA unwinding is similarly affected. Additionally, mutations can increase Kapp,RNA and KM,app,ATP, indicative of weaker interactions with RNA and ATP due to a misalignment of the bipartite RNA-binding site and the ATPase site, and decrease the thermodynamic coupling between interactions with ATP and RNA. Consequently, subtle changes in the inter-domain communication can affect RNA helicase activity in a complex manner. The two mutants studied here represent different degrees of misalignment and of decoupling: In YxiN_AAA, the global conformation of the helicase core and thus RNA binding are not affected, but local conformational differences lead to a decreased kcat. Altogether, this mutant can still unwind RNA, but the reaction occurs more slowly. In YxiN_G303A, the global conformation is affected, leading to a decrease in ATPase activity and a severe drop in ATP and RNA affinities. The combination of these effects is detrimental for unwinding activity. It has previously been suggested that the introduction of a kink into the RNA upon formation of the closed conformer initiates unwinding (2), and it may be speculated that YxiN_K52Q and YxiN_AAA still kink their RNA substrate, whereas YxiN_G303A may not kink the RNA (or not to the same extent). Consistent with the observation that ADPNP does not support unwinding (12,22), we postulate that ATP hydrolysis or product release is coupled to a subsequent final step of unwinding. This critical step is absent in YxiN_K52Q, and significantly slowed down in YxiN_AAA. To identify the final step that leads to RNA unwinding, future studies will have to address the conformation of RNA bound to the helicase directly.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
VolkswagenStiftung and the Swiss National Science Foundation. The Open Access publication charge for this paper has been waived by Oxford University Press.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Ines Hertel for excellent technical assistance.
REFERENCES
- 1.Cordin O, Banroques J, Tanner NK, Linder P. The DEAD-box protein family of RNA helicases. Gene. 2006;367:17–37. doi: 10.1016/j.gene.2005.10.019. [DOI] [PubMed] [Google Scholar]
- 2.Sengoku T, Nureki O, Nakamura A, Kobayashi S, Yokoyama S. Structural basis for RNA unwinding by the DEAD-box protein Drosophila Vasa. Cell. 2006;125:287–300. doi: 10.1016/j.cell.2006.01.054. [DOI] [PubMed] [Google Scholar]
- 3.Andersen CB, Ballut L, Johansen JS, Chamieh H, Nielsen KH, Oliveira CL, Pedersen JS, Seraphin B, Le Hir H, Andersen GR. Structure of the exon junction core complex with a trapped DEAD-box ATPase bound to RNA. Science. 2006;313:1968–1972. doi: 10.1126/science.1131981. [DOI] [PubMed] [Google Scholar]
- 4.Bono F, Ebert J, Lorentzen E, Conti E. The crystal structure of the exon junction complex reveals how it maintains a stable grip on mRNA. Cell. 2006;126:713–725. doi: 10.1016/j.cell.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 5.Collins R, Karlberg T, Lehtio L, Schutz P, van den Berg S, Dahlgren LG, Hammarstrom M, Weigelt J, Schuler H. The DExD/H-box RNA helicase DDX19 is regulated by an alpha -helical switch. J. Biol. Chem. 2009;284:10296–10300. doi: 10.1074/jbc.C900018200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.von Moeller H, Basquin C, Conti E. The mRNA export protein DBP5 binds RNA and the cytoplasmic nucleoporin NUP214 in a mutually exclusive manner. Nat. Struct. Mol. Biol. 2009;16:247–254. doi: 10.1038/nsmb.1561. [DOI] [PubMed] [Google Scholar]
- 7.Tanner NK, Cordin O, Banroques J, Doere M, Linder P. The Q motif: a newly identified motif in DEAD box helicases may regulate ATP binding and hydrolysis. Mol. Cell. 2003;11:127–138. doi: 10.1016/s1097-2765(03)00006-6. [DOI] [PubMed] [Google Scholar]
- 8.Pause A, Sonenberg N. Mutational analysis of a DEAD box RNA helicase: the mammalian translation initiation factor eIF-4A. EMBO J. 1992;11:2643–2654. doi: 10.1002/j.1460-2075.1992.tb05330.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pause A, Methot N, Sonenberg N. The HRIGRXXR region of the DEAD box RNA helicase eukaryotic translation initiation factor 4A is required for RNA binding and ATP hydrolysis. Mol. Cell Biol. 1993;13:6789–6798. doi: 10.1128/mcb.13.11.6789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kossen K, Uhlenbeck OC. Cloning and biochemical characterization of Bacillus subtilis YxiN, a DEAD protein specifically activated by 23S rRNA: delineation of a novel sub-family of bacterial DEAD proteins. Nucleic Acids Res. 1999;27:3811–3820. doi: 10.1093/nar/27.19.3811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Karow AR, Theissen B, Klostermeier D. Authentic interdomain communication in an RNA helicase reconstituted by expressed protein ligation of two helicase domains. FEBS J. 2007;274:463–473. doi: 10.1111/j.1742-4658.2006.05593.x. [DOI] [PubMed] [Google Scholar]
- 12.Theissen B, Karow AR, Kohler J, Gubaev A, Klostermeier D. Cooperative binding of ATP and RNA induces a closed conformation in a DEAD box RNA helicase. Proc. Natl Acad. Sci. USA. 2008;105:548–553. doi: 10.1073/pnas.0705488105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang S, Overgaard MT, Hu Y, McKay DB. The Bacillus subtilis RNA Helicase YxiN is Distended in Solution. Biophys. J. 2007;94:L01–L03. doi: 10.1529/biophysj.107.120709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rozen F, Pelletier J, Trachsel H, Sonenberg N. A lysine substitution in the ATP-binding site of eucaryotic initiation factor 4A abrogates nucleotide-binding activity. Mol. Cell Biol. 1989;9:4061–4063. doi: 10.1128/mcb.9.9.4061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Henn A, Cao W, Hackney DD, De La Cruz EM. The ATPase cycle mechanism of the DEAD-box rRNA helicase, DbpA. J. Mol. Biol. 2008;377:193–205. doi: 10.1016/j.jmb.2007.12.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rocak S, Emery B, Tanner NK, Linder P. Characterization of the ATPase and unwinding activities of the yeast DEAD-box protein Has1p and the analysis of the roles of the conserved motifs. Nucleic Acids Res. 2005;33:999–1009. doi: 10.1093/nar/gki244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cheng Z, Coller J, Parker R, Song H. Crystal structure and functional analysis of DEAD-box protein Dhh1p. RNA. 2005;11:1258–1270. doi: 10.1261/rna.2920905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Adam H. Methoden der enzymatischen Analyse. H.U.(Hrsg.), Verlag Chemie, Weinheim: Bergmeyer; 1962. pp. 573–577. [Google Scholar]
- 19.Polach KJ, Uhlenbeck OC. Cooperative binding of ATP and RNA substrates to the DEAD/H protein DbpA. Biochemistry. 2002;41:3693–3702. doi: 10.1021/bi012062n. [DOI] [PubMed] [Google Scholar]
- 20.Chen Y, Potratz JP, Tijerina P, Del Campo M, Lambowitz AM, Russell R. DEAD-box proteins can completely separate an RNA duplex using a single ATP. Proc. Natl Acad. Sci. USA. 2008;105:20203–20208. doi: 10.1073/pnas.0811075106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu F, Putnam A, Jankowsky E. ATP hydrolysis is required for DEAD-box protein recycling but not for duplex unwinding. Proc. Natl Acad. Sci. USA. 2008;51:20209–20214. doi: 10.1073/pnas.0811115106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kossen K, Karginov FV, Uhlenbeck OC. The carboxy-terminal domain of the DExDH protein YxiN is sufficient to confer specificity for 23S rRNA. J. Mol. Biol. 2002;324:625–636. doi: 10.1016/s0022-2836(02)01140-3. [DOI] [PubMed] [Google Scholar]
- 23.Combet C, Jambon M, Deleage G, Geourjon C. Geno3D: automatic comparative molecular modelling of protein. Bioinformatics. 2002;18:213–214. doi: 10.1093/bioinformatics/18.1.213. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.