

Abstract
We examined the role of phosphoinositide 3-kinase (PI3K) in integrin-mediated eosinophil adhesion. Δp85, a dominant-negative form of the class IA PI3K adaptor subunit, was fused to an HIV-TAT protein transduction domain (TAT-Δp85). Recombinant TAT-Δp85 inhibited interleukin (IL)-5–stimulated phosphorylation of protein kinase B, a downstream target of PI3K. β2-Integrin–dependent adhesion caused by IL-5 to the plated intracellular adhesion molecule-1 surrogate, bovine serum albumin, was inhibited by TAT-Δp85 in a concentration-dependent manner. Similarly, two PI3K inhibitors, wortmannin and LY294002, blocked eosinophil adhesion to plated bovine serum albumin. By contrast, β1-integrin–mediated eosinophil adhesion to vascular cell adhesion moelcule-1 was not blocked by TAT-Δp85, wortmannin, or LY294002. Rottlerin, a protein kinase C (PKC)-δ inhibitor, also blocked β2-integrin adhesion of eosinophils caused by IL-5, whereas β1 adhesion to vascular cell adhesion molecule-1 was not affected. IL-5 caused translocation of PKCδ from the cytosol to cell membrane; inhibition of PI3K by wortmannin blocked translocation of PKCδ. Western blot analysis demonstrated that extracellular signal–regulated kinase phosphorylation, a critical intermediary in adhesion elicited by IL-5, was blocked by inhibition of either PI3K or PKC-δ. These data suggest that extracellular signal–regulated kinase–mediated adhesion of β2-integrin caused by IL-5 is mediated in human eosinophils by a class IA PI3K through activation of a PKCδ pathway.
Keywords: phosphoinositide 3-kinase, eosinophil, adhesion
Phosphorylation by extracellular signal–regulated kinase (ERK) activates group IVa cytosolic phospholipase A2 (cPLA2) to cause β2-integrin–mediated adhesion of eosinophils to the endothelial counterligand, intercellular adhesion molecule-1 (ICAM-1). Activation of cPLA2 results in the hydrolysis of phosphatidylcholine to produce platelet-activating factor (PAF), which has been shown recently to be a necessary step for β2-integrin adhesion in granulocytes (1, 2). However, the signal transduction pathways leading to phosphorylation of ERK, a requisite step in the phosphorylation of cPLA2 and eosinophil adhesion, have not been well elucidated.
Phosphoinositide 3-kinase (PI3K) has been implicated in a wide variety of cellular functions, including mitogenesis, cell adhesion and motility, inflammatory mediator secretion, and protection from apoptosis (3, 4). The isoforms of PI3K are divided into three main classes. The class IA PI3K is activated by tyrosine kinases and consists of a heterodimer composed of a 110-kD (p110α, β, δ) catalytic subunit and an adaptor protein (p85α, p85β, p55α, p55γ, p50α) (5). Class IB enzymes contain a unique catalytic subunit (p110γ) associated with a regulatory subunit p101 and signal downstream of heterotrimeric G-protein–coupled receptors. Class II PI3K is characterized by the presence of a C2 domain, and a class III PI3K uses only phosphatidylinositol as a substrate. The function of each of the classes of PI3K-isoform has not been well elucidated due to the lack of isoform-selective inhibitors.
PI3K has been reported to participate in the early events leading to both activation and inactivation of ERK, depending on cell type and stimulus (6–8). PI3K regulates cell functions through activation of several downstream molecules, including Akt/protein kinase B, phosphoinositide-dependent kinase-1, and protein kinase C (PKC) (4). Among them, PKC has been found to activate ERK (9).
We previously have shown that ERK phosphorylation caused by PI3K mediates adhesion of eosinophils to ICAM-1 (2). However, the mechanism by which PI3K regulates ERK-mediated eosinophil adhesion has not been determined. Accordingly, the specific regulatory isoform(s) of PI3K that causes ERK-mediated adhesion also has not been elucidated. The objective of this investigation was to elucidate further the signaling pathways regulating integrin-mediated adhesion of eosinophils in interleukin (IL)-5–stimulated cells and to determine the potential role of PI3K. We also sought to determine the specific isoforms of PI3K that likely participate in the regulation of β2-integrin adhesion. We found that eosinophil adhesion by β2-integrin to ICAM-1 surrogate is regulated by PKC-δ through the activation of class IA PI3K. This regulation is selective for β2-integrin, as we also found that activation of PKCδ through PI3K does not cause β1-integrin adhesion of eosinophils to vascular adhesion molecule-1 (VCAM-1).
MATERIALS AND METHODS
Materials
Eosinophil isolation materials were obtained from Miltenyi Biotec (Sunnyvale, CA). Bovine serum albumin (BSA) fraction V was purchased from Sigma-Aldrich (St. Louis, MO). Wortmannin, LY294002, rottlerin, PKB inhibitor, and rapamycin were purchased from EMD Biosciences (San Diego, CA). BL21 Escherichia coli was obtained from Novagen (Madison, WI). Ni-NTA columns and Penta-His Ab were purchased from Qiagen (Valencia, CA). ERK1/2 antibody, Ser473 phosphorylation-specific and nonspecific PKB antibodies were purchased from Cell Signaling Technology (Beverly, MA). Polyclonal anti-p85 subunit of PI3K was purchased from Upstate Biotechnology (Lake Placid, NY). Polyclonal rabbit anti-PKC δ (sc-937) was obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Phosphorylation-specific ERK1/2 polyclonal antibody was purchased from Promega (Madison, WI). FITC-labeled goat anti-rabbit Ig was purchased from Becton-Dickinson (Mountain View, CA). Donkey anti-rabbit Ig and goat anti-mouse Ig conjugated with horseradish peroxidase and PD-10 columns were purchased from Amersham (Arlington Heights, IL). Polystyrene 96-well microplates were purchased from Costar (Cambridge, MA). Δp85 cDNA was a gift from Dr. M. Kasuga (Kobe University Graduate School of Medicine, Kobe, Japan). PTAT plasmid was a gift from Dr. S. Dowdy (University of California San Diego, La Jolla, CA).
Generation of TAT Fusion Protein
A detailed method for the generation of dominant-negative TAT-Δp85 and TAT-GFP (Figure 1) has been described previously (10). Briefly, a cDNA fragment encoding dominant-negative p85 was amplified by PCR from the Δp85 cDNA with the deletion of 35 amino acids from residues 478–513 with the insertion of two amino acids in pGEX. The PCR product was inserted into a pCRII TOPO vector. The plasmid was digested with AgeI/EcoRI and ligated into an AgeI/EcoRI digested pTAT vector using T4 ligase.
Figure 1.

Effect of TAT-Δp85 on IL-5–induced phosphorylation of PKB, a downstream target of PI3K. (A) Structure of TAT fusion protein used in this study. Six His residues and the 11–amino acid TAT peptide precede the N-terminal of the Δp85. The 11 amino acids of TAT are the protein transduction domain. (B) Time-dependent effect of TAT-Δp85 transduction on PI3K activation. Eosinophils were preincubated with 600 nM TAT-Δp85 at 37°C for indicated times and then stimulated with 10 ng/ml IL-5 for 10 min, and cell lysates were separated by SDS-PAGE and probed with anti-phosphorylated PKB antibody (upper panel, PKB-phos) and with antibody for total PKB (lower panel, PKB-total) to demonstrate equal loading in all lanes. (C) Concentration-dependent effect of TAT-Δp85 transduction on PI3K activation. Eosinophils were preincubated with various concentrations of TAT-Δp85 for 15 min and then stimulated with 10 ng/ml IL-5 for 10 min. PKB phosphorylation and expression were measured as previously described.
Purification of TAT fusion proteins was performed using a method modified from Myou and coworkers (11). Briefly, TAT-Δp85 was purified by sonication of high expressing BL21 E. coli in 10 ml buffer Z (8 M urea, 20 mM HEPES, pH 8.0, 100 mM NaCl). Cellular lysates were resolved by centrifugation, loaded onto 5-ml Ni-NTA columns in buffer Z, washed, and eluted with 250 mM of imidazole in phosphate-buffered saline. Imidazole was removed from the resultant protein solution using PD-10 column. Each fusion protein was flash-frozen at −80°C.
Isolation of Human Peripheral Blood Eosinophils
Eosinophils were isolated from mildly atopic human volunteers by a method modified from Hansel and colleagues (12). The protocol was approved by the University of Chicago Institutional Review Board, and informed written consent was obtained from all volunteers in this study. Briefly, percoll centrifugation (density 1.089 g/ml) was employed to isolate granulocytes and was followed by hypotonic lysis of RBC, and finally, immunomagnetic depletion of neutrophils by the magnetic cell separation system using anti-CD16–coated MACS particles. Eosinophil purity of ⩾ 98% was routinely obtained, as assessed by Wright-Giemsa staining. Cells were kept on ice until use.
Eosinophil Adhesion
Eosinophil adhesion was assessed as residual eosinophil peroxidase (EPO) activity of adherent cells (1, 13). We have validated previously an in vitro adhesion assay for which plated BSA serves as a surrogate for ICAM-1 (13). This method has been established to replicate specifically all components of β2-integrin adhesion; β2-integrin adhesion also is blocked selectively in identical concentration-related fashion for eosinophils plated on BSA as for plated ICAM-1 using mAb directed against CD11b and CD18, the common β-chain for β2-integrin. Briefly, 100 μl of 10 μg/ml BSA or 50 μl of VCAM-1 (5 μg/ml) was added to flat-bottom 96-well microtiter plates and incubated at 4°C overnight. BSA or VCAM-1 was decanted, and 200 μl/well heat-inactivated 10% fetal bovine serum in phosphate-buffered saline was added to coated wells. After 60 min incubation at 37°C, the wells were decanted and washed with Hanks' balanced salt solution (HBSS) before the addition of eosinophils. Cells (104/100 μl HBSS/0.1% gelatin) were preincubated with various concentration of wortmannin, LY294002, TAT-Δp85, or rottlerin for 20 min at 37°C. Cells then were added to each well of BSA- or VCAM-1–coated microplates with 10 ng/ml of IL-5, and were allowed to settle for 10 min on ice. Plates were rapidly warmed to 37°C and incubated for 30 min. After washing with HBSS, 100 μl of HBSS/0.1% gelatin was added to the reaction wells, and serial dilutions of the original cell suspension were added to the empty wells to generate a standard curve. A total of 100 μl of EPO substrate (1 mM H2O2, 1 mM o-phenylenediamine, and 0.1% Triton X-100 in Tris buffer, pH 8.0) was then added to the wells. After a 30-min incubation at room temperature, 50 μl of 4 N H2SO4 was added to stop the reaction. Absorbance was measured at 490 nM in a microplate reader (Thermomax; Molecular Devices, Menlo Park, CA). All assays were performed in duplicate. Data storage and analysis were facilitated by the use of computer software interfaced with the reader (Softmax; Molecular Devices). The detection of EPO by this assay was linear between concentrations of 1.0 × 103 and 1.5 × 104 cells/well, as determined by a standard curve.
Immunoblot Analysis of ERK1/2 and PKB
Eosinophils (106/group) were preincubated with 100 nM wortmannin or 10 μM rottlerin for 20 min, or TAT-Δp85 for 15 min, and then stimulated with 10 ng/ml of IL-5 for 10 min at 37°C. The reaction was stopped by centrifugation at 11,000 × g for 2 min. The pellets were lysed by sonication in 40 μl of boiling buffer (50 mM Tris, pH 7.4, 1% SDS, 2 mM EGTA, 2 mM EDTA). The supernatants then were mixed with 8 μl of 6× sample buffer and boiled for 5 min. The samples were collected and saved at −70°C. Samples were subjected to SDS-PAGE, using 10% acrylamide gels under reducing condition (15 mA/gel). Electrotransfer of proteins from the gels to polyvinylidene fluoride membrane was achieved using a semidry system (400 mA, 60 min). The membrane was blocked with 1% BSA for 60 min and then incubated with 1/5,000 anti-phosphorylation–specific ERK1/2, 1/1,000 anti-ERK1/2, 1/1,000 anti-Ser 473 phosphorylation–specific PKB, or 1/1,000 PKB antibody diluted in Tris-buffer saline plus tween-20 overnight. The membranes then were washed three times for 20 min with TBST. Donkey anti-rabbit IgG conjugated with horseradish peroxidase was diluted 1/3,000 and incubated with polyvinylidene fluoride membrane for 60 min. The membrane was again washed three times and assayed by an enhanced chemiluminescence system from Amersham.
Immunoblotting of Cytosolic and Membrane Fractions of Eosinophils
Eosinophils (2 × 105/group) were pretreated with 100 nM of wortmannin, a PI3K inhibitor, for 20 min or 600 nM TAT-Δp85, a dominant-negative class IA PI3K inhibitor, for 15 min. The reaction was stopped by centrifugation at 11,000 × g for 2 min. The pellets were lysed in 50 μl lysis buffer and incubated on ice for 20 min, followed by the centrifugation at 11,000 × g for 2 min. Then the supernatant was centrifuged at 100,000 × g for 60 min. The supernatant (cytosolic fraction) was removed, and the pellet (membrane fraction) was resuspended in 50 μl of boiling lysis buffer. Both fractions were mixed with 10 μl of loading buffer and boiled for 5 min. The fractions were separated on 7.5% SDS-PAGE and transferred to membrane by semidry transfer. After incubation with polyclonal anti–PKC-δ antibody, blots were amplified and visualized using anti-rabbit IgG and enhanced chemiluminescence.
Coimmunoprecipitation Assay
To assess the physical association between PI3K and PKC-δ, aliquots of eosinophils (5 × 106/group) were stimulated with or without IL-5 for 10 min, and the reactions were stopped by centrifugation at 11,000 × g for 2 min. The pellets were lysed in 500 μl lysis buffer (20 mM Tris-HCl [pH 7.4], 30 mM Na4P2O7, 50 mM NaF, 40 mM NaCl, 5 mM EDTA, 1% Nonidet P-40, 10 μg/ml leupeptin, 5 μg/ml aprotinin, 1 mM PMSF, 2 mM Na3VO4, and 0.5% deoxycholic acid) and incubated on ice for 20 min. The lysates were centrifuged at 11,000 × g for 2 min, and 5 μl of polyclonal anti-p85 subunit of PI3K Ab was added to the supernatant and incubated with rocking at 4°C overnight. The lysates were further incubated with protein A/G agarose beads for 45 min and washed four times with lysis buffer, and the pellets were resuspended in 40 μl of loading buffer and boiled for 5 min. The immunoprecipitated proteins were separated on 7.5% SDS-PAGE and transferred to membrane by semidry transfer. After incubation with 1:400 polyclonal anti–PKC-δ, blots were amplified and visualized using anti-rabbit IgG and enhanced chemiluminescence. The membrane was stripped, and reprobed with 1:5,000 anti-p85 antibody to verify the equal precipitation among samples.
Statistical Analysis
All measurements were expressed as mean ± SEM. Variation between two groups was tested using Student's t test. Variation between more than two groups was tested using ANOVA followed by Fisher's protected least significant difference. A value of P < 0.05 was accepted as statistically significant.
RESULTS
Efficacy of TAT-Δp85 on the Inhibition of PI3K Activation
To access the efficacy of TAT-Δp85 protein transduction, time- and concentration-curves first were generated. PI3K activation was measured as a function of the phosphorylation of its downstream substrate, PKB. Eosinophils were incubated with 600nM of dominant-negative TAT-Δp85 for ⩽ 15 min, and IL-5–induced PKB phosphorylation was analyzed by Western blot (Figure 1B). TAT-Δp85 inhibited PKB phosphorylation caused by IL-5 in a time-dependent fashion. PKB phosphorylation decreased after 2 min incubation with TAT-Δp85 and remained effective in inhibiting PI3K activation for ⩾ 15 min. Consequently, a 15-min incubation time was used in the subsequent experiments. TAT-Δp85 also caused concentration-dependent blockade of PKB phosphorylation elicited by IL-5. PKB phosphorylation was reduced at 300 nM and blocked completely with 600–900 nM TAT-Δp85 (Figure 1C). By contrast, 900 nM TAT-GFP control (TAT-fused to green fluorescent protein) had no effect on PKB phosphorylation caused by IL-5. Consequently, 600 nM of TAT-Δp85 was used in the subsequent experiments. These results demonstrated that TAT-Δp85 functionally suppressed PKB phosphorylation after transduction into eosinophils using HIV-TAT protein.
Effects of PI3K Inhibition on Adhesion of Eosinophils to Plated BSA Caused by IL-5
Using the ICAM-1 assay with BSA as a fully validated replicate surrogate (see Materials and Methods), two PI3K pharmacologic inhibitors (wortmannin and LY294002) and TAT-Δp85, which inhibits class IA PI3K, were used to determine whether PI3K was involved in eosinophil adhesion. Eosinophil adhesion caused by IL-5 was blocked by (1) wortmannin, (2) LY294002, or (3) dominant-negative TAT-Δp85 in a concentration-dependent manner (Figure 2A). IL-5–stimulated adhesion decreased from 32.5 ± 3.5% to 12.5 ± 4.8% with 100 nM wortmannin (P < 0.05), from 23.0 ± 5.1% to 7.9 ± 3.2% with 900 nM TAT-Δp85 (P < 0.01), and from 27.3 ± 4.3% to 15.4 ± 3.6% with 50 μM Ly294002 (P < 0.01). Specificity of blockade with TAT-protein has been established in a previous report (10, 11). Accordingly, preincubation of eosinophils with ⩽ 1 μM TAT-GFP has been shown previously to have no inhibitory effect on eosinophil adhesion (11). In this study, TAT-Δp85 at the concentration used had not blocked β2-integrin upregulation caused by IL-5. We also have reported previously that IL-5–induced upregulation of this integrin was not blocked by TAT–dominant-negative Ras (11). In contrast to β2-integrin adhesion, which requires integrin activation, β1-integrin (VLA-4) adheres to VCAM-1 in its constitutive state (1). Eosinophil adhesion was 17.1 ± 1.5% for VCAM-1–coated wells versus 8.4 ± 0.5% for buffer-coated control wells (P < 0.01). Neither wortmannin, LY294002, nor TAT-Δp85 had any inhibitory effect on eosinophil adhesion to plated VCAM-1 (Figure 2B).
Figure 2.
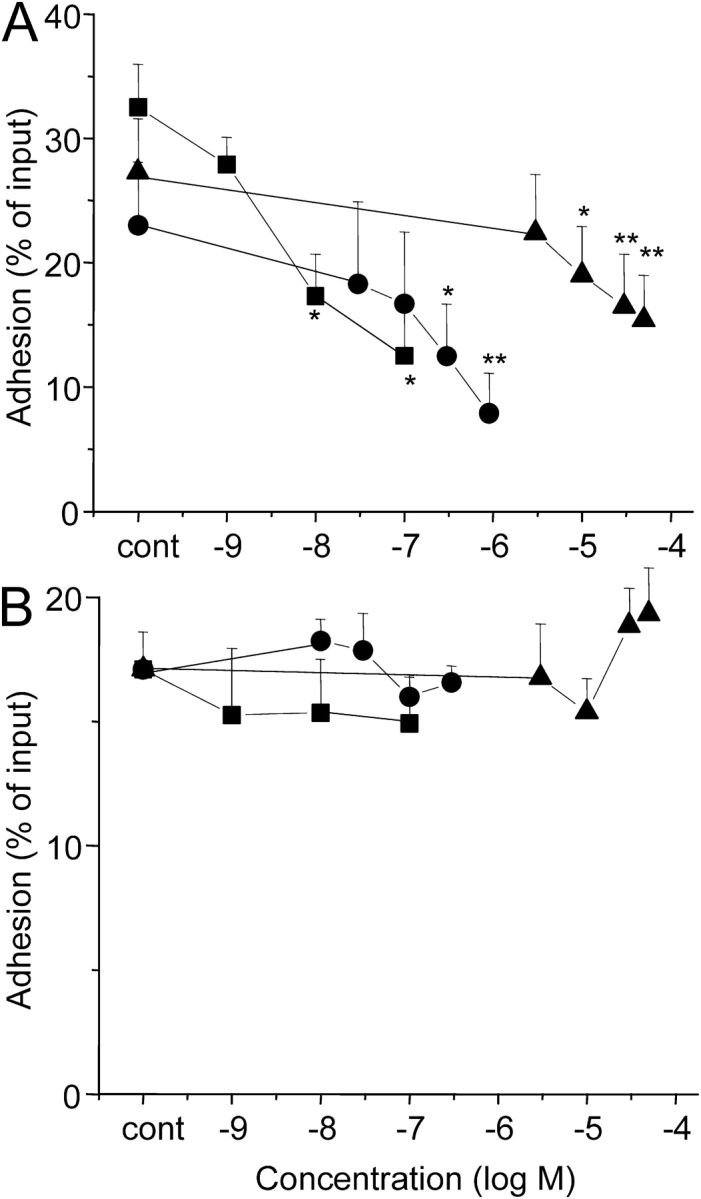
Effect of the PI3K inhibitors on integrin-mediated eosinophil adhesion. (A) Effect of PI3K inhibition on IL-5–induced β2-integrin–mediated adhesion to plated BSA. Eosinophils were preincubated with the specified concentrations of wortmannin (squares) or LY294002 (triangles) for 30 min, or dominant-negative TAT-Δp85 (circles) for 15 min at 37°C, followed by addition of IL-5 and then further incubated for 30 min at 37°C. The adherence of eosinophils was assessed in the presence of inhibitors as detailed in MATERIALS AND METHODS. Each point represents the mean ± SEM. *P < 0.05 and **P < 0.01 compared with each control, n = 5. (B) Effect of PI3K inhibition on β1-integrin–mediated eosinophil adhesion to plated VCAM-1. Eosinophils were preincubated with the specified concentrations of wortmannin or LY294002 for 30 min, or dominant-negative TAT-Δp85 for 15 min at 37°C, and then were adhered to VCAM-1–coated wells for 30 min at 37°C. The adherence of eosinophils was measured as above (n = 5).
Effects of PKCδ Inhibition on Adhesion of Eosinophils to Plated BSA Caused by IL-5
We next investigated which downstream signal transduction molecules of PI3K might be involved in IL-5–induced eosinophil adhesion. PKB, p70 S6 kinase, and PKC-δ have been reported to be the downstream targets for PI3K (14–16). Neither PKB inhibitor, nor S6 kinase inhibition by rapamycin, blocked eosinophil adhesion to plated BSA caused by IL-5 (Figure 3A). By contrast, rottlerin, a PKC-δ inhibitor, completely blocked IL-5–induced eosinophil adhesion to BSA in concentration-dependent manner (Figure 3B). Adhesion caused by IL-5 was ∼ 93% blocked by 10 μM rottlerin (P < 0.01). In contrast to β2-integrin adhesion, rottlerin had no inhibitory effect on eosinophil adhesion to plated VCAM-1 (Figure 3C).
Figure 3.

Effects of inhibition of PI3K downstream kinases on eosinophil adhesion. (A) Effect of the Akt/PKB inhibitor (squares) and rapamycin (circles) on IL-5–induced eosinophil adhesion to plated BSA. Eosinophils were preincubated with the specified concentrations of inhibitors for 30 min at 37°C, followed by addition of IL-5 and incubation for 30 min at 37°C. The adherence of eosinophils was assessed as detailed in MATERIALS AND METHODS in the presence of inhibitors (n = 4). (B) Effect of the PKC-δ inhibitor on IL-5–induced eosinophil adhesion to BSA. Eosinophils were preincubated with the specified concentrations of rottlerin for 20 min, followed by addition of IL-5 and incubation for 30 min at 37°C. The adherence of eosinophils was assessed as above (n = 5). (C) Effect of the PKC-δ on spontaneous eosinophil adhesion to VCAM-1. Eosinophils were preincubated with the specified concentrations of rottlerin for 20 min at 37°C, and incubated in VCAM-1–coated wells for 30 min at 37°C. The adherence of eosinophils was assessed as described in MATERIALS AND METHODS in the presence of inhibitors. Each point represents the mean ± SEM and compared with each control (n = 5). *P < 0.05 and **P < 0.01 compared with each control.
Effect of PI3K Inhibition on IL-5–Induced PKCδ Activation
In further studies, we examined whether PI3K inhibition suppressed PKC-δ activation caused by IL-5 in eosinophils. PKC activation was measured by its translocation from the cytosol to cell membrane. Western blot analysis showed the time-dependent translocation of PKC-δ (Figure 4A) from cytosol to membrane fractions induced by IL-5. Pretreatment of eosinophil with 100 nM of wortmannin or 600 nM of the dominant-negative TAT-Δp85 inhibited the translocation of PKC-δ to the membrane fractions (Figure 4B). These data suggested that IL-5–stimulated PKCδ activation was regulated by PI3K activation.
Figure 4.

Effect of PI3K inhibition on IL-5–induced PKC activation. (A) Time-dependent effect of IL-5 on the translocation of PKCδ to the cell membrane in eosinophils. Eosinophils (2 × 106/group) were stimulated with 10 ng/ml IL-5 for indicated time at 37°C. The cells were lysed and fractionated, and the cytosolic and membrane fractions were separated on 7.5% SDS-PAGE, followed by immunoblotting with polyclonal anti–PKC-δ antibody. (B) Effect of PI3K inhibition on IL-5–induced PKC translocation in human eosinophils. Eosinophils (2 × 106/group) were incubated with 100 nM wortmannin for 20 min, or 600 nM TAT-Δp85 for 15 min followed by addition of 10 ng/ml IL-5 for 10 min. The cell fractions were separated on 7.5% SDS-PAGE, followed by immunoblotting with polyclonal anti–PKC-δ antibody. The result shown is representative of three different experiments.
Coimmunoprecipitation of PI3K with PKC-δ
We further examined the physical association of PI3K and PKCδ in eosinophils. PKC-δ was present in the p85 subunit of PI3K immunoprecipitates with or without stimulation by IL-5 (Figure 5). Equal amount of p85 was precipitated in control and IL-5–stimulated cells. These results suggest that PKCδ is constitutively associated with class IA PI3K in human eosinophils.
Figure 5.

The physical association between PI3K and PKC-δ. Eosinophils (5 × 106/group) were stimulated with 10 ng/ml of IL-5 or buffer for 10 min at 37°C, and the cells were lysed and immunoprecipitated by anti-p85 subunit of PI3K antibody. The immunioprecipitated proteins were separated by SDS-PAGE and were probed by anti–PKC-δ (left panel) or anti-p85 (right panel) antibody after stripping. The results shown are representative of four different experiments.
Effect of Inhibition of PI3K and PKCδ on ERK1/2 Activation Caused by IL-5
In additional studies, we examined whether inhibition of PI3K or PKC-δ attenuated ERK1/2 activation caused by IL-5 in eosinophils (Figure 6). Eosinophils were pretreated with 100 nM of wortmannin for 30 min, 600 nM of TAT-Δp85 for 15 min, or 10 μM of rottlerin for 30 min, and then stimulated with 10 ng/ml IL-5 at 37°C for 10 min. Wortmannin (100 nM) and TAT-Δp85 (900 nM), in concentrations sufficient to block PI3K activity, attenuated ERK phosphorylation caused by IL-5 (Figure 6A). Rottlerin also attenuated ERK phosphorylation caused by IL-5 (Figure 6B). These data suggest that IL-5–stimulated ERK1/2 phosphorylation is elicited through a PI3K–PKC-δ pathway.
Figure 6.

Effects of inhibition of PI3K or PKCδ on IL-5–induced ERK1/2 activation. Eosinophils were incubated with 100 nM wortmannin and 10 μM rottlerin for 20 min, or 600 nM TAT-Δp85 for 15 min at 37°C, followed by addition of 10 ng/ml IL-5 for 10 min at 37°C. Eosinophils were lysed, and the lysates were loaded on 10% SDS-PAGE, followed by immunoblotting with anti-phosphorylation–specific ERK1/2 Ab (top panel) or anti-ERK1/2 Ab (bottom panel). The result shown is representative of four different experiments.
DISCUSSION
In this study, we found that class IA PI3K inhibition by dominant-negative TAT-Δp85 or pharmacologic inhibitors blocked (1) IL-5–stimulated translocation of PKC-δ from cytosol to membrane fractions, (2) ERK1/2 phosphorylation, and (3) β2-integrin–mediated eosinophil adhesion to plated ICAM-1 surrogate. We also found that the p85 adaptor unit of PI3K was physically associated with PKC-δ and that PKC-δ inhibition attenuated ERK phosphorylation and eosinophil adhesion to BSA caused by IL-5. By contrast, neither PI3K nor PKCδ inhibition suppressed eosinophil adhesion to VCAM-1. Accordingly, our data demonstrate that IL-5–induced eosinophil adhesion to plated ICAM-1 surrogate is regulated by class IA PI3K through PKC-δ and ERK, whereas eosinophil adhesion to VCAM-1 is not activated by PI3K.
We have reported previously that ERK phosphorylation, but not p38 or JNK phosphorylation, is essential for β2- but not β1-integrin adhesion of eosinophils (2). PI3K has been reported to participate in the early events leading to activation or inactivation of mitogen-activated protein kinase, depending on the cell type and stimulus (6–8). We have previously suggested a role of PI3K in ERK activation in eosinophil adhesion. However, the isoform and mechanism of action of PI3K has not been defined previously. For this study, we constructed a dominant-negative TAT-Δp85 to selectively inhibit class IA PI3K. Together with the pharmacologic inhibitors (wortmannin and LY294002) of PI3K, we found that selective inhibition of class IA PI3K blocked ERK phosphorylation and eosinophil adhesion to plated BSA in IL-5–stimulated eosinophils. It is interesting to note that all these PI3K inhibitors used in this study only partially attenuated eosinophil adhesion to BSA-coated plates at the concentrations that produced total PI3K inhibition. This suggests that a PI3K-independent pathway is involved in the regulation of β2-integrin adhesion caused by IL-5. We previously have found that Ras activation also regulates β2-integrin adhesion caused by IL-5, fMLP, or eotaxin (11). As both pathways appear to be involved in regulation of β2-integrin adhesion, it is not surprising that blockade of either one of these pathways is not fully efficacious in blocking adhesion. However, the complex interaction between Ras and PI3K in causing β2-integrin adhesion still requires further investigation.
We also examined the mechanism by which PI3K regulates eosinophil adhesion. A variety of downstream signal transduction molecules such as phosphoinositide-dependent kinase 1 (4), PKB (also termed Akt) (17), p70 S6 kinase (p70S6K) (18), and PKC (14, 16) are regulated by PI3K. PKB is a Ser/Thr kinase regulated by growth factors, cytokines, and G-protein–coupled receptors (17) and plays a role in a number of cellular processes, including cell proliferation, apoptosis, and gene transcription. In eosinophils, PKB is activated by fMLP, IL-5, and PAF (2, 19, 20). However, treatment with a PKB inhibitor caused no inhibitory effects on β2-integrin adhesion (Figure 3A). The PKB inhibitor used in this study was a phosphatidylinositol analog with an IC50 of 5.0 ± 1.7 μM for inhibition of PKB and IC50 values in the 1–10 μM range for cell growth inhibition (21). Hence, we used 10 μM PKB inhibitor to block PKB activity. Similarly, inhibition of S6K by rapamycin also had no effect on eosinophil adhesion caused by IL-5 (Figure 3A), while selective inhibition of PKCδ attenuated eosinophil adhesion (Figure 3B), suggesting that eosinophil adhesion caused by PI3K is mediated by PKC-δ.
There are currently 12 known isoforms of PKC; 8 PKC isoforms have been identified in human eosinophils (22). These include: PKC-α, -βI, -βII, -δ, -ε, -ζ, -ι, and -μ. PKC-δ is a member of the novel PKC subfamily and is a DAG-dependent, Ca2+-independent PKC isoform. PKC-δ activation can be controlled by PI3K (16). We demonstrate in this study that the activation of PKCδ was regulated by PI3K. We find that PI3K and PKC-δ are physically associated in human eosinophils (Figure 5), and PI3K inhibition blocks the translocation of PKC-δ from cytosol to membrane fractions (Figures 4A and 4B). These results suggest that PI3K may mediate its effect through PKC-δ. These findings comport with a prior study, which reported that PI3K and PKC-δ are physically associated following IL-5 stimulation in an erythroleukemia cell line (15). Prior investigations also have demonstrated that PI3K-dependent PKC-δ activation plays a critical role in IL-5– and LTB4-induced superoxide production in human eosinophils (20), as well as in the TNF-α–caused anti-apoptotic signaling in adherent neutrophils (23).
In summary, we find that IL-5–induced β2-integrin–dependent eosinophil adhesion is inhibited by pretreatment with PI3K inhibitors as well as inhibitors of PKC-δ. We also find that PI3K is functionally associated with PKC-δ, and PI3K inhibition blocked PKC-δ translocation from cytosol to cell membranes. Finally, we find that ERK1/2 phosphorylation is inhibited by either inhibition of PI3K or PKC-δ. These results suggest that IL-5–stimulated β2-integrin adhesion is regulated by ERK1/2 phosphorylation through a PI3K-dependent PKC-δ activation. By contrast, we find that β1-integrin–mediated eosinophil adhesion to VCAM-1 is not dependent on either PI3K or PKCδ, demonstrating for the first time this markedly different mechanism for regulation of these highly homologous β1- and β2-integrins.
Acknowledgments
The authors thank Dr. Masato Kasuga (Kobe University Graduate School of Medicine) for providing pGEX vectors containing p85 cDNA, and Dr. Steven Dowdy (University of California San Diego, La Jolla, CA) for providing TAT expression vector.
This work was supported by NIAID AI52109 (X.Z.), by NHLBI HL-46368 (A.R.L.), and by the AstraZeneca Traveling Fellowship (M.S.). A.R.L. is the principal investigator in the University of Chicago GlaxoSmithKline Center of Excellence in Asthma.
Conflict of Interest Statement: M.S. is a fellow funded in part from an unrestricted educational grant from AstraZeneca; A.R.L. has received $3,000 in the past 3 years for a GlaxoSmithKline Advisory Board, $13,000/year for a Merck Advisory Board, $24,000/year for Broncus/Asthmatx Advisory Board, and $3,500 for a Pfizer Advisory Board; S.M. has no declared conflicts of interest; E.B. has no declared conflicts of interest; A.Y.M. has no declared conflicts of interest; J.L. has no declared conflicts of interest; A.T.L. has no declared conflicts of interest; N.M.M. has no declared conflicts of interest; and X.Z. has no declared conflicts of interest.
References
- 1.Zhu X, Munoz NM, Kim KP, Sano H, Cho W, Leff AR. Cytosolic phospholipase A2 activation is essential for beta 1 and beta 2 integrin-dependent adhesion of human eosinophils. J Immunol 1999;163:3423–3429. [PubMed] [Google Scholar]
- 2.Zhu X, Jacobs B, Boetticher E, Myou S, Meliton A, Sano H, Lambertino AT, Munoz NM, Leff AR. IL-5-induced integrin adhesion of human eosinophils caused by ERK1/2-mediated activation of cPLA2. J Leukoc Biol 2002;72:1046–1053. [PubMed] [Google Scholar]
- 3.Wymann MP, Pirola L. Structure and function of phosphoinositide 3-kinases. Biochim Biophys Acta 1998;1436:127–150. [DOI] [PubMed] [Google Scholar]
- 4.Cantley LC. The phosphoinositide 3-kinase pathway. Science 2002;296:1655–1657. [DOI] [PubMed] [Google Scholar]
- 5.Dhand R, Hara K, Hiles I, Bax B, Gout I, Panayotou G, Fry MJ, Yonezawa K, Kasuga M, Waterfield MD. PI 3-kinase: structural and functional analysis of intersubunit interactions. EMBO J 1994;13:511–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ferby IM, Waga I, Sakanaka C, Kume K, Shimizu T. Wortmannin inhibits mitogen-activated protein kinase activation induced by platelet-activating factor in guinea pig neutrophils. J Biol Chem 1994;269:30485–30488. [PubMed] [Google Scholar]
- 7.Gerhardt CC, Gros J, Strosberg AD, Issad T. Stimulation of the extracellular signal-regulated kinase 1/2 pathway by human beta-3 adrenergic receptor: new pharmacological profile and mechanism of activation. Mol Pharmacol 1999;55:255–262. [DOI] [PubMed] [Google Scholar]
- 8.Moelling K, Schad K, Bosse M, Zimmermann S, Schweneker M. Regulation of Raf-Akt Cross-talk. J Biol Chem 2002;277:31099–31106. [DOI] [PubMed] [Google Scholar]
- 9.Corbit KC, Soh JW, Yoshida K, Eves EM, Weinstein IB, Rosner MR. Different protein kinase C isoforms determine growth factor specificity in neuronal cells. Mol Cell Biol 2000;20:5392–5403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Myou S, Leff AR, Myo S, Boetticher E, Meliton AY, Lambertino AT, Liu J, Xu C, Munoz NM, Zhu X. Activation of group IV cytosolic phospholipase A2 in human eosinophils by phosphoinositide 3-kinase through a mitogen-activated protein kinase-independent pathway. J Immunol 2003;171:4399–4405. [DOI] [PubMed] [Google Scholar]
- 11.Myou S, Zhu X, Boetticher E, Myo S, Meliton A, Lambertino A, Munoz NM, Leff AR. Blockade of focal clustering and active conformation in beta 2-integrin-mediated adhesion of eosinophils to intercellular adhesion molecule-1 caused by transduction of HIV TAT-dominant negative Ras. J Immunol 2002;169:2670–2676. [DOI] [PubMed] [Google Scholar]
- 12.Hansel TT, De Vries IJ, Iff T, Rihs S, Wandzilak M, Betz S, Blaser K, Walker C. An improved immunomagnetic procedure for the isolation of highly purified human blood eosinophils. J Immunol Methods 1991;145:105–110. [DOI] [PubMed] [Google Scholar]
- 13.Zhu X, Subbaraman R, Sano H, Jacobs B, Sano A, Boetticher E, Munoz NM, Leff AR. A surrogate method for assessment of beta(2)-integrin-dependent adhesion of human eosinophils to ICAM-1. J Immunol Methods 2000;240:157–164. [DOI] [PubMed] [Google Scholar]
- 14.Chou MM, Hou W, Johnson J, Graham LK, Lee MH, Chen CS, Newton AC, Schaffhausen BS, Toker A. Regulation of protein kinase C zeta by PI 3-kinase and PDK-1. Curr Biol 1998;8:1069–1077. [DOI] [PubMed] [Google Scholar]
- 15.Ettinger SL, Lauener RW, Duronio V. Protein kinase C delta specifically associates with phosphatidylinositol 3-kinase following cytokine stimulation. J Biol Chem 1996;271:14514–14518. [DOI] [PubMed] [Google Scholar]
- 16.Le Good JA, Ziegler WH, Parekh DB, Alessi DR, Cohen P, Parker PJ. Protein kinase C isotypes controlled by phosphoinositide 3-kinase through the protein kinase PDK1. Science 1998;281:2042–2045. [DOI] [PubMed] [Google Scholar]
- 17.Coffer PJ, Jin J, Woodgett JR. Protein kinase B (c-Akt): a multifunctional mediator of phosphatidylinositol 3-kinase activation. Biochem J 1998;335:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Proud CG. p70 S6 kinase: an enigma with variations. Trends Biochem Sci 1996;21:181–185. [PubMed] [Google Scholar]
- 19.Coffer PJ, Schweizer RC, Dubois GR, Maikoe T, Lammers JW, Koenderman L. Analysis of signal transduction pathways in human eosinophils activated by chemoattractants and the T-helper 2-derived cytokines interleukin-4 and interleukin-5. Blood 1998;91:2547–2557. [PubMed] [Google Scholar]
- 20.Bankers-Fulbright JL, Kita H, Gleich GJ, O'Grady SM. Regulation of human eosinophil NADPH oxidase activity: a central role for PKCdelta. J Cell Physiol 2001;189:306–315. [DOI] [PubMed] [Google Scholar]
- 21.Hu Y, Qiao L, Wang S, Rong SB, Meuillet EJ, Berggren M, Gallegos A, Powis G, Kozikowski AP. 3-(Hydroxymethyl)-bearing phosphatidylinositol ether lipid analogues and carbonate surrogates block PI3-K, Akt, and cancer cell growth. J Med Chem 2000;43:3045–3051. [DOI] [PubMed] [Google Scholar]
- 22.Evans DJ, Lindsay MA, Webb BL, Kankaanranta H, Giembycz MA, O'Connor BJ, Barnes PJ. Expression and activation of protein kinase C-zeta in eosinophils after allergen challenge. Am J Physiol 1999;277:L233–L239. [DOI] [PubMed] [Google Scholar]
- 23.Kilpatrick LE, Lee JY, Haines KM, Campbell DE, Sullivan KE, Korchak HM. A role for PKC-delta and PI 3-kinase in TNF-alpha-mediated antiapoptotic signaling in the human neutrophil. Am J Physiol Cell Physiol 2002;283:C48–C57. [DOI] [PubMed] [Google Scholar]