

Abstract
The incidence of serious photochemical smog events is steadily growing in urban environments around the world. The electrophilic metabolites of 1-nitronaphthalene (1-NN), a common air pollutant in urban areas, have been shown to bind covalently to proteins. 1-NN specifically targets the airway epithelium, and the toxicity is synergized by prior long-term ozone exposure in rat. In this study we investigated the formation of 1-NN protein adducts in the rat airway epithelium in vivo and examined how prior long-term ozone exposure affects adduct formation. Eight adducted proteins, several involved in cellular antioxidant defense, were identified. The extent of adduction of each protein was calculated, and two proteins, peroxiredoxin 6 and biliverdin reductase, were adducted at high specific activities (0.36–0.70 and 1.0 nmol adduct/nmol protein). Furthermore, the N-terminal region of calreticulin, known as vasostatin, was adducted only in ozone-exposed animals. Although vasostatin was adducted at relatively low specific activity (0.01 nmol adduct/nmol protein), the adduction only in ozone-exposed animals makes it a candidate protein for elucidating the synergistic toxicity between ozone and 1-NN. These studies identified in vivo protein targets for reactive 1-NN metabolites that are potentially associated with the mechanism of 1-NN toxicity and the synergistic effects of ozone.
Keywords: 1-nitronaphthalene, calreticulin, ozone, protein adduct, proteomics
Lung disease is a leading cause of morbidity and mortality worldwide, and anthropogenic air pollutants are known to be a causative factor in urban areas (1). Polycyclic aromatic hydrocarbons (PAHs) are abundant pollutants in vehicle emissions and can have serious effects on human health due to their mutagenicity. The nitrated derivatives of PAHs (nitro-PAHs) account for > 50% of the total mutagenicity of urban ambient air samples (2, 3), where nitronaphthalenes in combination with methyl-nitronaphthalenes are responsible for the majority of the vapor-phase mutagenicity (4). Nitro-PAHs are mainly found in diesel engine exhaust gas. However, they are also formed from their parent PAHs through atmospheric photochemical reactions (2) and can therefore be derived from the combustion of any fossil fuels. Increased concerns about nitro-PAH toxicity have arisen with the recent reports of a higher ratio of nitro-PAH:PAH generated by the purportedly “cleaner” modern diesel engines (3).
Like their parent PAHs, nitro-PAHs are inert compounds that require metabolic activation to electrophiles, mainly by cytochrome P450 monooxygenases (P450 s), to cause cellular toxicity. In the airways, activation of volatile PAHs and nitro-PAHs occurs mainly in the nonciliated epithelial cells (“Clara cells”), which contain the highest levels of P450 activity in the lung (5). 1-NN initially targets the Clara cell, and the formation of protein adducts has been suggested as a mechanism of toxicity (6). In addition to being a more potent and less species-selective pneumotoxicant than its parent PAH, naphthalene (7, 8), 1-NN toxicity is synergized by prior long-term exposure to ozone in rat (9, 10). The airways of rats exposed to ozone for more than 90 d undergo remodeling, which render rats resistant to further ozone exposure (11). We have shown in previous studies that these ozone-resistant rats are significantly more susceptible to 1-NN than filtered air–exposed controls (9, 10). Tropospheric ozone is mainly produced during the formation of photochemical smog and is driven by the presence of PAHs and ultraviolet radiation (2). The levels of NO and NO2 that drive the nitration reaction of PAHs are also high during photochemical smog, and thus ozone and nitro-PAHs commonly coexist in urban areas.
In this study, we have used in vivo proteomics approaches to test the hypothesis that the development of tolerance to oxidant stress produced by ozone elevates the potential for protein adduction by electrophilic metabolites of cytotoxic PAHs. The pattern of protein adduct formation by 1-NN metabolites with and without prior long-term ozone exposure was compared, and eight adducted proteins were identified. The results of this study will be used as the basis for the development of protein biomarkers, which subsequently can be used in human exposure assessments. Finally, the findings of this work provide new insight regarding the synergistic interaction of ozone and 1-NN, two air pollutants prevalent in photochemical smog.
MATERIALS AND METHODS
Animals
Male Sprague-Dawley rats were purchased from Harlan (San Diego, CA). Animals were fed ad libitum and housed in an AAALAC-accredited facility in HEPA filtered cages at the California National Primate Research Center, University of California, Davis, for at least 5 d before use. Twenty-two rats (68–71 d old) were randomly assigned to six groups. Groups 1 (n = 5), 3 (n = 4), and 5 (n = 4) were exposed to filtered air for 90 d, and Groups 2 (n = 5), 4 (n = 4), and 6 (n = 4) were exposed to 0.8 ppm ozone for 8 h/d for 90 d as previously described (10). On Day 91, the rats in Groups 1 and 2 received a single interperitoneal injection of 50 mg/kg 14C-labeled 1-NN in corn oil (specific activity 2,214 dpm/nmol). Groups 3 and 4 received an intraperitoneal injection of 50 mg/kg 1-NN in corn oil, and Groups 5 and 6 received an intraperitoneal injection of corn oil vehicle only. The rats were removed from ozone exposure for at least 24 h before injection. Two hours after injection, the animals received a lethal intraperitoneal dose of pentobarbital. Bronchiolar lavage fluid and airway epithelial proteins were collected through lysis-lavage as previously described (12). Liver tissue was collected, and all samples were flash frozen on dry ice and stored at −80°C until use. An additional 10 rats (65–72 d old) were given a single intraperitoneal injection of 50 mg/kg 14C-labeled 1-NN in corn oil (specific activity 214 dpm/nmol). The rats were given a lethal dose of pentobarbital intraperitoneal at 1, 2, 4, 6, or 8 h after injection (n = 2), and the various tissues were collected as described above.
Chemicals
Dextrose, acetic acid, methanol, and acetone were obtained from Fisher Chemicals (Pittsburgh, PA). SeaPlaque low melting point agarose was purchased from FMC Bioproducts (Rockland, ME), and ultrapure urea was obtained from US Biological Corp (Cleveland, OH). Omnipure TRIS, glycine, and SDS were purchased from EM Sciences (Gibbstown, NJ). CHAPS, protease inhibitor cocktail III, and Triton X-100 were acquired from Calbiochem (LaJolla, CA). Bradford protein assay reagent and PVDF Sequi-blot membranes were obtained from Bio-Rad (Hercules, CA). Bovine serum albumin (BSA), thiourea, tributylphosphine (TBP), and dithiothreitol (DTT) were ordered from Sigma-Aldrich (St. Louis, MO). IsoGel low EEO agarose was ordered from BMA (Rockland, ME), 14C-1-NN and 1-NN were synthesized as previously described (13). Duracryl was obtained from Genomic Solutions (Ann Arbor, MI), and Rhinohide and SYPRO Ruby protein stain were purchased from Molecular Probes (Eugene, OR). All other electrophoresis materials and equipment were obtained from Amersham Biosciences (Piscataway, NJ). All solutions were prepared with deionized water (resistivity 18.1 MΩ/cm).
Two-Dimensional Electrophoresis
Liver samples were homogenized with a Polytron homogenizer in lysis buffer (2 M thiourea, 7 M urea, 4% wt/vol CHAPS, 0.5% wt/vol Triton X-100, 1% wt/vol (65 mM) DTT, and 2% vol/vol protease inhibitor cocktail III). The homogenate was centrifuged for 75 min at 100,000 × g at 15°C, and the protein concentration of the supernatant was determined with the method of Bradford (14). Proteins in the dextrose bronchiolar lavage samples were concentrated using BioMax centrifugal filters and tubes according to the manufacturer's instructions (Millipore, Bedford, MA). Lysis-lavage samples (containing airway epithelial cell proteins) were used directly without modifications. Lysis-lavage samples and liver samples were separated on IPG strips of pH ranges 4.5–5.5, 5.5–6.7, and 6–9 with a total protein load of 1,000 μg, 800 μg, and 700 μg, respectively. Only four of the samples obtained by lavaging the airways with dextrose solution (12) (bronchiolar lavage) contained sufficient protein to allow two-dimensional electrophoresis (2DE) analysis (two filtered air, two ozone), and these were separated on IPG strips of pH 4–7 using all isolated proteins (229–400 μg protein/strip). Proteins were prepared for isoelectric focusing on IPG strips of all pH ranges except 6–9 as follows: IPG buffer for the respective pH range was added to a final concentration of 1% vol/vol, and the protein sample was diluted to 350 μl with lysis buffer. The protein sample was loaded onto the IPG strip through rehydration at room temperature overnight, and IEF was performed using a Multiphor II Electrophoresis unit and an EPS 3501 XL power supply at 20°C with the following gradient protocol: 0–50 V, 1 min; 50 V, 1 h; 50–1,000 V, 3 h; 1,000–3,500 V, 3 h; 3,500 V, 19 h (total 74.9 kVh). IPG strips of pH 6–9 were rehydrated overnight with DeStreak Rehydration Buffer supplemented with 1% vol/vol IPG buffer pH 6–11 and 2% vol/vol protease inhibitor cocktail III. Protein samples were diluted to 346 μl with lysis buffer without DTT, and 4 μl DeStreak Reagent was added. The sample was loaded onto the IPG strip during IEF through the wick loading method (15) using 5.5-cm-long electrode strips, and the following gradient program was used: 0–1,000 V, 2.5 h; 1,000–3,500 V, 2.5 h; 3,500 V, 21 h (total 80.4 kVh). After IEF, the IPG strips were stored at −80°C until use in the second dimension.
Before loading the IPG strips onto 20 cm × 25 cm 10% T Duracryl or Rhinohide acrylamide gels, the strips were incubated 2 × 15 min in equilibration buffer (50 mM tris-HCl pH 8.8, 6 M urea, 30% glycerol, 2% SDS). Reduction and alkylation of sulfhydryls was performed through incorporation of 65 mM DTT in the first incubation, and 10 mM iodoacetamide in the second. The strips were sealed using 0.5% IsoGel agarose. The second dimension separation was performed in a Hoefer ISO-DALT 2D Electrophoresis system connected to an EPS 3501 XL power supply at 10°C, 14 mA/gel until the dye front had migrated 17 cm (∼ 18 h) in 25 mM Tris, 192 mM glycine, and 0.1% SDS. After electrophoresis, the protein gels were either fixed in 500 ml of 20% ethanol/7% acetic acid for > 1 h or blotted to PVDF membranes (14C-labeled samples only). Electroblotting to Sequi-blot PVDF membranes (0.2 μm pore size) was performed using the ISO-DALT system at 10°C, 250 mA for 19 h in 25 mM Tris, 192 mM glycine, 10% (vol/vol) methanol. The membranes were then washed with 3 × 30 min with water followed by 30 min in 60% methanol and dried in a vacuum desiccator before visualization of proteins.
Protein Visualization and Quantification in 2DE Gels
All staining was performed in polymethylpentane staining dishes wrapped in aluminum foil to prevent photo bleaching of the stains. SYPRO Ruby staining was performed at a 10-fold dilution in 20% ethanol overnight in a volume of 500 ml. The gels were destained for 15 min in 20% ethanol/7% acetic acid, followed by 2 × 15 min equilibration in water prior to visualization with a Typhoon 8600 fluorescent scanner (Molecular Dynamics, Sunnyvale, CA) using the 532-nm laser and the 610-bp30 filter, PMT 600 for acidic range gels and PMT 650 for basic range gels. Total protein abundance in protein spots was determined using ImageQuant software (v 5.1; Molecular Dynamics). A standard curve was created by merging three replicate 1-dimensional SYPRO Ruby stained gels containing a dilution series of three different proteins with varying pI and molecular weights (BSA, aldolase, and lysozyme; Sigma-Aldrich), with a resulting limit of detection of 2 ng protein.
Phosphorimaging
Low energy Storage Phosphorimaging Screens (Fuji Medical Systems USA, Inc., Stamford, CT) were exposed to PVDF membranes for 67 d. The plates were simultaneously exposed to protein standards created through dot-blotting a dilution series of 14C-BSA (Sigma-Aldrich) to a strip of PVDF membrane. The 14C-BSA was diluted with cold BSA, and the protein content in each dot-blot was kept constant. The radioactive protein spots were visualized using a Typhoon laser scanner and quantified using Image Quant software and a standard curve from the 14C-BSA standard strip. The limit of detection was 0.0064 DPM, which is equivalent to 3 fmol adducted 1-NN.
Protein Identification by MALDI-TOF/TOF
Protein spots in the 2DE gels corresponding to the protein spots detectable on the phosphorimaging screens were excised from the gel. The gel plugs were incubated at room temperature in 25 mM NH4HCO3 for 5 min, followed by acetonitrile for 5 min. These two steps were repeated. Proteins were then reduced with 10 mM DTT in 25 mM NH4HCO3 (60°C) for 10 min, allowed to cool for 15 min, and alkylated with 100 mM iodoacetamide in 25 mM NH4HCO3 for 35 min at room temperature. The reduction and alkylation steps were repeated once. Finally, proteins were digested for 4 h in 25 mM NH4HCO3 containing sequencing-grade, modified trypsin (Cat # V5111; Promega, Madison, WI) at a final trypsin concentration of 5 ng/μl (37°C). The reaction was stopped through the addition of 0.3% formic acid. Samples were aspirated and dispensed three times using a C18 ZipTipu-C18 (Millipore) and eluted with a solution of 70% acetonitrile, 0.2% formic acid, and 5 mg/ml MALDI matrix (α-cyano-4-hydroxycinnamic acid). A 1.3-μl sample was spotted onto the MALDI target and analyzed using an Applied Biosystems 4700 Proteomics Analyzer (Applied Biosystems Inc., Foster City, CA) with TOF/TOF Optics. MALDI-MS data were collected in the m/z range of 700–4,000 using trypsin autolysis peaks of m/z 842.51 and 2,211.10 D as internal standards. Data were analyzed using Applied Biosystems GPS Explorer Software. MASCOT searches were performed on all MS and MS/MS data sets, and a probability-based Mowse score > 100 (equivalent to P = 0.01) was considered a statistically significant hit.
Protein Identification by LC-MS/MS
An HPLC system (Paradigm MG4; Michrom Bio Resources, Auburn, CA) directly coupled with an ion trap mass spectrometer (LCQ Deca XP plus; Finnigan, San Jose, CA) was used for microcapillary 1-D LC-MS/MS data acquisition. A homemade fritless reverse phase (RP) microcapillary column (0.1 mm × 180 mm) was packed with C18 Aqua: 5 μm 300 Å (Phenomenex, Torrance, CA) (16). Tryptic peptide mixtures were desalted and concentrated using the RP trap column (0.15 mm × 30 mm) before chromatographic separation by the microcapillary RP column with a flow rate of 300 nl/min. Eluted peptides were then directly sprayed into the mass spectrometer. MS/MS spectra were acquired for the most intense peptide ion from the previous MS spectra with dynamic exclusion for 3 min. A 2.5-h gradient (0–10% B for 10 min, 10–45% B for 110 min, 45–100% B for 20 min, 100% B for 10 min) was used (buffer A = 5% acetonitrile/0.1% formic acid, buffer B = 80% acetonitrile/0.1% formic acid). SEQUEST searches were performed for each MS/MS data set against the sub-database “Rattus” from the NCBI protein nr database, and proteins were filtered out using DTASelect software (17). ΔCn > 0.08 and Xcorr > 2.0 were considered a statistically significant hit. Proteins with at least two peptides matching the above criteria were filtered out, and at least one MS/MS spectra was manually reconfirmed for the proteins with < 4 peptides identified.
Determination of Total 14C-1-NN Protein Binding
Total protein-bound radioactivity was measured after removal of residual, unbound 14C-1-NN and metabolites from the protein samples with acetone precipitation or dialysis. Dialysis was performed in 0.1% SDS and 1 mM EDTA using 2,000 molecular weight cut-off DispoDialyzer tubes (Spectrum Laboratories, Rancho Dominguez, CA). The first dialysis was performed at room temperature to avoid precipitation of urea. Subsequently, dialysis was performed at 4°C until the radioactivity of the dialysis bath did not exceed background levels. NaOH was added to achieve a final concentration of 2 N to dissolve all precipitated proteins. The protein concentration was determined using the method of Bradford (14), and the radioactivity was determined by scintillation counting.
Acetone precipitation was performed through the addition of 2 vols of cold acetone followed by centrifugation at 4,000 × g for 10 min at room temperature. The supernatant was discarded, and the pellet resuspended in acetone. The procedure was repeated until the DPM of the supernatant did not exceed background levels. The pellet was then resuspended in 2 N NaOH, the protein concentration was determined with the method of Bradford (14), and the radioactivity was counted. The first four supernatants were collected, evaporated to dryness, and reconstituted for protein concentration determination. No leakage of protein into the supernatant occurred.
Statistical Analyses
Mean and standard deviation values were calculated on all data sets, and t tests were performed using Microsoft Excel (Redmond, WA).
RESULTS
Time-Course of 1-NN Protein Adduct Formation
The time-course of the total protein adduct formation by 1-NN metabolites was quantified in rat airway epithelium and liver samples (Figure 1). Maximum binding occurred as early as 2 h after injection in the airway epithelium. In the liver, maximum binding occurred much later, at ∼ 6 h after injection. Furthermore, the maximum level of bound metabolite in the liver (nmol 1-NN/mg protein) was much lower (4-fold) than that observed in the airway epithelium (Figure 1).
Figure 1.

The time-course of total protein binding in vivo of 1-NN metabolites was quantified in rat airway epithelium (left) and liver (right) after exposure to 50 mg/kg 14C-1-NN. Each diamond represents an individual animal, and each horizontal line represents the average value of that time-point (n = 2). The maximum protein binding occurred as early as 2 h after injection in the airway epithelium compared with ∼ 6 h after injection in the liver. The extent of binding was, on average, 4-fold higher in the airway than in the liver.
Protein Adduct Formation and Protein Abundance
Protein adduct formation by 1-NN metabolites in vivo in filtered air–exposed (Exposure Group 1) and ozone-tolerant (Exposure Group 2) animals was compared qualitatively in liver, airway epithelium, and bronchiolar lavage fluid. A total of 14 protein spots were found to be adducted in the airway epithelium (Figures 2–3); four in the pI range 4.5–5.5, ten in the pI range 5.5–6.71, and none in the pI range 6–9. Protein # 14 was only adducted in rats exposed to ozone prior to the 1-NN exposure (Figure 2). No protein adducts were detected in liver samples. However, proteins 5–10 were adducted in the four bronchiolar lavage samples (obtained by dextrose lavage) where sufficient protein was present to allow 2DE analysis (results not shown).
Figure 2.
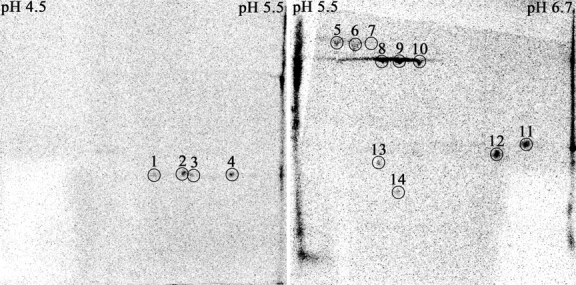
Images from storage phosphor screens exposed for 67 d to PVDF membranes containing airway epithelial cell proteins from rats exposed to ozone for 90 d followed by a single intraperitoneal injection of 50 mg/kg 14C-1-NN (Exposure Group 2). The 14 spots that were selected for identification are circled. Three different pH ranges were used to achieve maximum selectivity and sensitivity, but since no 14C activity was detected in the basic range (pH 6–9), this image is not shown.
Figure 3.

2DE gels containing airway epithelial cell proteins from rats exposed to ozone for 90 d followed by a single intraperitoneal injection of 50 mg/kg 1-NN are displayed above (Exposure Group 4). Three different pH ranges were used to achieve maximum sensitivity. The proteins were visualized with SYPRO Ruby protein stain and a Typhoon fluorescent scanner. The 14 protein spots determined to be adducted by 1-NN metabolites through phosphorimaging are circled and labeled with the respective protein ID number. Note that the abundance of spots number 12 and 13 are below the detection limit for SYPRO Ruby and, therefore, no spots are visible. The circles indicate the gel area that was excised for protein identification. No spots were found to be adducted in the basic region.
Although long-term ozone exposure seemed to induce a slight increase in protein adduction, the difference was not statistically significant (Figure 4). A quantitative evaluation of each individual adducted protein spot isolated from airway epithelium is displayed in Figure 5. Both total protein abundance in 2DE gel images and total 1-NN metabolite binding in PVDF membranes were quantified. The approximate specific activity (nmol adduct/nmol protein) of each individual adducted protein in ozone treated (O3) versus filtered air (FA)-treated animals is displayed in Table 1. Long-term ozone exposure appeared to alter the ratio of protein adduction of several protein spots, most notably spots # 13 (↑94% over the filtered air controls), and # 14 (only adducted in ozone-treated animals). No significant alteration in protein abundance in response to ozone treatment and/or 1-NN treatment was observed, although a general trend of slightly elevated levels in spots # 1–11 was observed in ozone-treated control animals, but not ozone + 1-NN–treated animals (data not shown).
Figure 4.

No significant difference in total protein adduct formation by 1-NN metabolites was observed between airway epithelial proteins obtained from filtered air–exposed animals (FA, Exposure Group 1) versus ozone-exposed animals (Exposure Group 2) 2 h after injection of 50 mg/kg 14C-1-NN. The values are expressed as mean + SD (n = 5).
Figure 5.

The total protein abundance with the corresponding quantity of adducted protein is displayed for each of the 14 adducted proteins in filtered air–exposed (blue and red) and ozone-exposed (green and yellow) animals, respectively. The values are expressed as the mean pmol of individual protein on gels loaded with identical amounts of protein (n = 5) + SD. The abundances of spots number 12 and 13 were below the detection limit of the 2DE analysis. The values displayed here are estimated from the detection limit of the 2DE analysis, and therefore lack error bars. The total abundance of protein # 6 is 40 ± 15 pmol for filtered air–exposed animals and 51 ± 40 pmol for ozone-exposed animals.
TABLE 1.
Identification and quantification of proteins adducted by 1-NN metabolites
| Frequency*
|
Adduction†
|
MALDI
|
LC-MS
|
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Spot | Protein | FA | O3 | FA | O3 | Score‡ | pept§ | % cov¶ | pept§ | Accession No. | Main Function |
| 1 | HSPB1 | 4 | 2 | 0.01 | 0.01 | 323 | 12 | 28.2 | 4 | sptP42930 | Stress |
| 2 | Ser-Arg-rich SR protein | 4 | 2 | 0.01 | 0.03 | 184 | 30 | – | – | trmQ9JKL7 | Splicing regulation |
| HSPB1 | 96 | 13 | 35.9 | 4 | sptP42930 | Stress | |||||
| 3 | HSPB1 | 4 | 2 | 0.01 | 0.01 | 322 | 12 | 44.7 | 9 | sptP42930 | Stress |
| 4 | HSPB1 | 4 | 2 | 0.01 | 0.02 | 346 | 12 | 43.2 | 10 | sptP42930 | Stress |
| 5 | Serum albumin precursor | 5 | 4 | 0.01 | 0.01 | 271 | 27 | 11.5 | 8 | sptP02770 | Transport |
| 6 | Serum albumin precursor | 5 | 5 | < 0.01 | < 0.01 | 239 | 27 | 19.2 | 13 | sptP02770 | Transport |
| 7 | Serum albumin precursor | 5 | 4 | 0.01 | 0.01 | 292 | 25 | – | – | sptP02770 | Transport |
| 8 | Aldehyde dehydrogenase | 5 | 5 | 0.05 | 0.06 | 446 | 27 | – | – | sptP11884 | Ethanol oxidation |
| 9 | Selenium-binding protein 2 | 5 | 5 | 0.07 | 0.09 | 120 | 17 | – | – | gpNP_543168 | Unknown |
| 10 | Selenium-binding protein 2 | 4 | 5 | 0.02 | 0.03 | ND | ND | 28.0 | 17 | gpNP_543168 | Unknown |
| 11 | Triosephosphate isomerase | 5 | 5 | 0.04 | 0.04 | 388 | 18 | 20.1 | 5 | sptP48500 | Glycolysis |
| 12 | Biliverdin reductase B | 5 | 5 | 1.00 | 1.00 | ND | ND | 9.2 | 2 | gbXM_214823 | Oxidative stress |
| 13 | Peroxiredoxin 6 | 5 | 5 | 0.36 | 0.70 | ND | ND | 44.2 | 9 | sptO35244 | Oxidative stress |
| 14 | Calreticulin (vasostatin) | 0 | 4 | 0.00 | 0.01 | 182 | 13 | 4.6 | 2 | sptP18418 | Multiple |
Definition of abbreviations: gb, GenBank; gp, GenPept; HSPB1, heat shock protein B1; ND, not detected; spt, SWISS-prot database; trm, TrEMBL database.
Number of animals where the respective protein was adducted (n = 5).
Average nmol adduction of each protein (n = frequency)/average abundance in nmol (n = 5).
The probability based Mowse score.
The number of peptides used for database matching.
% sequence coverage.
Spots # 12 and 13 were not detectable by SYPRO Ruby staining. Instead, the protein abundance was estimated to be just below the average limit of detection for most proteins (2 ng/spot), which is the highest abundance of protein expected to be present in these protein spots. It is important to note that the abundance of protein could be considerably lower than the detection limit, which would result in a much higher degree of metabolite binding than that reported in Table 1 and Figure 5. For protein spot # 12, this estimation resulted in more than 1.0 nmol adduct/nmol protein, indicating that protein # 12 either is not detected with high sensitivity by SYPRO Ruby, or that multiple sites on each protein are adducted. Protein spots # 8, 10, and 11 also had a relatively large portion of the total protein abundance adducted, but no difference could be seen between ozone-treated and filtered air–treated animals.
Identification of Adducted Proteins
Identification of the 14 adducted airway epithelial cell protein spots was attempted primarily by MALDI TOF/TOF, but in some cases also by ESI LC-MS/MS. The 14 adducted protein spots represented 8 different protein species. The identities of these proteins together with their primary function, frequency of adduction in individual test subjects, and the degree of protein adduction (nmol 1-NN/nmol protein) are presented in Table 1. The number of matched peptides and probability based Mowse score for the MALDI-TOF/TOF data, and the % sequence coverage for the LC-MS/MS data, are also displayed in this table. The sequence information for the peptides used for identification is given in the supplemental information (Tables E1 and E2 in the online supplement). The identification of protein spot # 2 resulted in possible protein species with the MALDI-TOF/TOF method (Table 1).
DISCUSSION
Earlier work has shown that the mechanism of toxicity for 1-NN is dependent on activation by P450 s in the airway epithelium to form electrophilic metabolites, and that these metabolites bind covalently to proteins (6). The toxicity of 1-NN to the airway epithelium is also exacerbated in rat by prior long-term exposure to ozone (9, 10). Although some increase in P450-dependent metabolism of this substrate has been observed in ozone-tolerant animals (10), the precise mechanism for this synergistic interaction has not been elucidated. In this study, we used proteomic approaches to identify airway epithelial proteins which are adducted by 1-NN metabolites in vivo and quantitatively examined how the adduction pattern is affected by prior ozone exposure of sufficient length to produce tolerance. A total of 14 different protein spots representing 8 different gene products were found to be adducted by 1-NN metabolites, and one protein, calreticulin, was only adducted in ozone-tolerant rats. The number of proteins found to be adducted in this study is much lower than in previous in vitro and in situ studies (18), which suggests that the list may not be exhaustive. Nevertheless, the in vivo approach used in this study makes their relevance to toxicity more probable. However, protein adduct formation is not necessarily linked to toxicity. It was shown as early as 1988 that various macromolecules in the cell can act as a sink for electrophilic metabolites without causing toxicity (19). The adduction pattern in nontarget cells/tissues can provide valuable information about the mechanism of toxicity. The liver is considered a nontarget tissue for 1-NN in uninduced animals (20). No specific, adducted proteins were detected in the liver assessed by 2DE/phosphorimaging, even though activation of 1-NN with subsequent protein adduct formation has previously been shown in this organ (6, 20). After intraperitoneal administration, a portion of the 1-NN is metabolized in the liver; the remaining compound (∼ 67% of the dose) eventually reaches the lung through the systemic circulation (21). It is possible that the toxicity and covalent binding of reactive metabolites observed in the lung arises from stable electrophilic metabolites produced in the liver that are delivered to the lung through the blood stream. However, the time-course of protein adduct formation (Figure 1) showed that the adduction occurs much earlier and to a much greater extent in the airway (∼ 2 h after injection) than in the liver (∼ 6 h after injection), which indicates that metabolism of 1-NN in the liver and subsequent transfer of reactive metabolites to the lung via the blood stream is not likely to be important for the toxicity seen in the airway. The time point for maximum protein binding in the lung also coincides well with the peak blood concentration following intraperitoneal administration of 1-NN, which occurs 91 min after injection (21).
Although any soft nucleophile (a nucleophile with low electronegativity and high polarization) can be a putative target for electrophilic metabolites such as 1-NN, sulfhydryls of cysteine residues have been shown to be the most likely target for similar bioactivated compounds (22). Moreover, reactive 1-NN metabolites form adducts with glutathione as shown both in vitro and in vivo (20, 21), and glutathione is likely to act as a major sink for the activated 1-NN metabolites. However, proteins with a high abundance of cysteines are also likely to act as sinks for electrophilic 1-NN metabolites, possibly without resulting toxicity. Seven of the adducted protein spots representing four protein species contained multiple cysteine residues: serum albumin precursor (spots # 5–6) has 35 cysteines, aldehyde dehydrogenase 2 (spot # 8) and triosephosphate isomerase (spot # 11) have 9 cysteines, and selenium-binding protein (spots # 9–10) has 10 cysteines. Based solely on the known functions of these four proteins, adduction does not appear to be important in 1-NN toxicity or to the synergistic response of 1-NN in ozone tolerant rats. Serum albumin is a blood protein involved in the transport of nutrients and electrolytes, and it is involved in the regulation of the osmotic pressure of blood. Serum albumin would not be present in an intact cell, and this adducted protein is probably detected as a contaminant from leakage through the capillary bed. Therefore, adduction of this protein is not likely to be the cause of toxicity in a selectively targeted cell type such as the Clara cell. However, albumin is covalently modified by both naphthalene oxide and naphthoquinone, and adducted albumin has successfully been used as a biomarker of naphthalene exposure (23).
Aldehyde dehydrogenase 2 (ADH2) is a mitochondrial enzyme with acetaldehyde as its main substrate (for review, see Ref. 24). Although ADH2 is also adducted by 1-NN metabolites in airways of Rhesus macaque in vitro (unpublished data) and one of its cysteines (Cys321) is located in the active site, it is not likely that 0.05 nmol adduct/nmol protein of this enzyme would cause any significant toxicity because a large portion of the human population is deficient in this particular ADH isozyme (24).
Triosephosphate isomerase (TPI) is an enzyme involved in glycolysis, where it interconverts D-glyceraldehyde 3-phosphate to dihydroxyacetone phosphate. At least one of the cysteine residues, Cys126, appears to be essential for correct folding and stability of the protein (25). However, the effects of inefficient glycolysis may not be as severe in the lung as in other cell types such as in the brain and red blood cells that solely depend on glucose as their energy source.
The function of selenium-binding proteins (SBP) is largely unknown, although it has been proposed that human SBP1 (92% homologous with rat SBP2) is involved in the late stages of intra-Golgi protein transport (26). The relatively high adduction rate of SBP2 in combination with the adduction by 1-NN metabolites in the airways of Rhesus macaque in vitro (unpublished data), and by naphthalene metabolites in mouse airways in situ (18) suggests that adduction of this protein may be of some importance to 1-NN toxicity. The adduction ratio was also higher in ozone-exposed animals than in filtered air–exposed animals, but the lack of information about the function of SBP2 and its 10 cysteine residues makes it impossible to draw any conclusions about its significance in the mechanism of toxicity.
The microenvironment of each cysteine also influences its reactivity, because the immediate surroundings of a particular cysteine residue dictates its pKa. All of the adducted proteins in this study were found in the acidic range of the 2DE gels, indicating that proteins with an acidic pI provide better targets for the electrophilic 1-NN metabolites. Carboxylic acid residues of acidic proteins will predominantly exist in their carboxylate state at physiological pH, which causes adjacent thiols to exist mainly as the much more reactive thiolate. However, the fact that no basic proteins were found to be adducted may also be explained by the inability to focus basic proteins on single pH-unit IPG strips, and the resulting lower sensitivity for proteins with basic pIs compared with proteins with acidic pIs.
Information about structure and function alone will not provide sufficient evidence to determine whether adduction of a protein plays a role in causing toxicity, as the degree of adduction is equally important. To our knowledge, this study is the first attempt to quantify the extent of adduction of specific proteins in vivo. Absolute quantification of protein abundance is difficult to calculate in proteomics studies due to the binding mechanism of most postelectrophoretic protein stains, including SYPRO Ruby. The binding is dependent on the number of positively charged amino acid residues of the protein, and results in large variations in the sensitivity of detection between different proteins. The method used in this study calculated a standard curve by merging standard curves from three different proteins with representative pIs and molecular weights (MWs). However, this approach results in a large degree of uncertainty in the accuracy of the calculated absolute abundance. The same is true for converting specific activity (nmol adduct/nmol protein) into absolute abundance of adducted protein, since this conversion is dependent on the number of 1-NN metabolites covalently bound to each protein molecule. Thus, the calculated specific activities of adducts presented in Table 1 should be viewed as providing a range rather than absolute values. However, the relative change in adduction for each individual protein between filtered air– and ozone-exposed animals is reliable because these are calculated on the same protein species, and the sensitivity of detection with SYPRO Ruby and the number of covalently bound 1-NN metabolites per protein can be assumed to remain constant for each individual protein.
The two most extensively adducted proteins, peroxiredoxin 6 and biliverdin reductase, are intimately associated with the first line of antioxidant defense system of the cell. Biliverdin reductase reduces water-soluble biliverdin to the insoluble antioxidant bilirubin. In many respects, bilirubin acts in a similar fashion to glutathione. However, the lipophilic bilirubin is more prone to protect membrane proteins and lipids against oxidation than the hydrophilic glutathione (27). Bilirubin is generally present at a much lower concentration in the cell (20–50 nM) compared with glutathione (1–10 mM). However, the rapid reduction of biliverdin to bilirubin affords 10,000-fold amplification of its antioxidant capacity. Depletion of biliverdin reductase has been shown to cause increased formation of reactive oxygen species and subsequent cell death in vitro (27), and the observed adduction of essentially all of the protein is therefore a potential mechanism of toxicity for 1-NN.
Peroxiredoxin 6 (prx 6), the second most highly adducted protein in the study, is also involved in antioxidant defense through detoxification of peroxides (for review, see Ref. 28). Prx 6 is also adducted by naphthalene metabolites in mouse airways in situ (18) and rhesus macaque airways in vitro (unpublished data). In addition to its peroxidase activity, prx 6 has also been shown to possess phospholipase A2 activity (29), which is important in regulation of the inflammatory response. In contrast to biliverdin reductase, the adduction of prx 6 was heavily affected by prior long-term ozone exposure. The adduction pattern of two other proteins, heat shock protein (HSP) B1 and calreticulin, also were strongly affected by long-term ozone exposure. Although neither of the proteins were highly adducted by 1-NN metabolites (0.01–0.03 nmol adduct/nmol protein), the alteration in adduction pattern after ozone exposure in combination with their functions make them interesting targets. Both are multifunctional proteins with chaperone activity, thus indirectly associated with the antioxidant defense system of the cell. HSPB1, also known as HSP27, belongs to the family of small HSPs, which normally are present at low levels located in large aggregates in the cytosol (for review, see Ref. 30). During stress it is released from the aggregate through phosphorylation and protects the cell in numerous ways. Besides preventing protein aggregation in the stressed cell through its function as a molecular chaperone, it can also act as a direct inhibitor of protein synthesis to prevent any misfolding of newly synthesized proteins which may add to the aggregation. Indirectly, this inhibits apoptosis, as aggregation of misfolded proteins can induce apoptosis. HSP27 has been shown to inhibit apoptosis directly through migration to the mitochondria and interaction with caspases, and to stabilize the cytoskeleton through binding to actin filaments and inhibition of the otherwise dynamic polymerization process of actin. Undoubtedly, HSP27 plays an important protective role during the assault from toxicants such as 1-NN. Only a small percentage of the total HSPB1 present was found to be adducted by 1-NN metabolites, and merely a loss of function due to adduction is not likely to cause toxicity. However, it is possible that adduction of a protein can cause alterations of the function as opposed to just a loss of function, and even adduction of a small percentage of the protein could then have a substantial biological impact. HSPB1 has been identified as a target for reactive 1-NN metabolites in airways of Rhesus macaque in vitro (unpublished data). In addition, HSP25 (the murine homolog of HSPB1) is increased substantially in mice treated with other Clara cell toxicants like naphthalene (31). In filtered air–exposed animals, the four2 HSPB1 protein spots were adducted in four out of five animals, while the corresponding protein spots were adducted only in two out of five ozone-tolerant animals.
In contrast, calreticulin was adducted in four out of five ozone-exposed animals, but in none of the five filtered air–exposed animals. Calreticulin is a multifunctional protein primarily found in the lumen of the endoplasmic reticulum (ER), where it acts as a molecular chaperone and is involved in the assembly of major histocompatibility complex 1, folding of glycoproteins, and modulation of Ca2+ release (for review, see Ref. 32). Calreticulin is also exported to the cell surface, where it is involved in regulation of the immune response, cell migration and adhesiveness, and inhibition of angiogenesis. The latter three functions have been attributed to the N-terminal domain (aa 1–180) of calreticulin, also known as vasostatin (33). Vasostatin is a 20-kD protein, which matches the location of the protein spot on the 2DE gel very well, in contrast to the full-length calreticulin which is much too large (56 kD). Thirteen of the 14 peptide fragments used for identification of calreticulin are located within the vasostatin domain of calreticulin. Thus, it is most likely vasostatin and not calreticulin that is adducted by 1-NN metabolites after ozone exposure. The vasostatin domain also contains calreticulin's three cysteine residues. In many cases, it has not been elucidated which domain is responsible for various functions attributed to calreticulin, and thus a discussion of the functions of calreticulin may be applicable to vasostatin. Notably, a lung-specific function has been reported, where calreticulin binds to SP-A and SP-D in the airway lumen (34). These two collectins have an anti-inflammatory role in the unchallenged lung. During stress, the collectins activate the production of proinflammatory agents and apoptotic cell ingestion by macrophages through binding to the calreticulin/CD91 complex on the surface of epithelial cells (35). Calreticulin has also been shown to function as a hapten and elicit an immunogenic response after adduction by halothane metabolites (36).
In summary, these studies have identified eight proteins adducted by 1-NN metabolites. The two most extensively adducted proteins, peroxiredoxin 6 and biliverdin reductase, are intimately associated with the first-line antioxidant defense system of the cell. In addition, prior ozone exposure altered the protein adduction pattern of three proteins, peroxiredoxin 6, HSPB1, and calreticulin. Although these are all multifunctional proteins, the alteration of shared functions including antioxidant defense, molecular chaperoning, and regulation of the immune response are most likely to exacerbate the toxicity of 1-NN as previously observed in ozone-exposed animals (9, 10). Although the immediate effects of adduction of each of these proteins is not well understood, this study has established a framework for elucidating the mechanisms of toxicity for these two toxicants. We have shown that oxidant exposure changes the pattern of adduct formation, and that these changes correlate with increased toxicity observed in previous pathologic studies. To further understand the mechanism of toxicity and synergism of ozone and 1-NN, we are currently conducting another in vivo study to determine the quantitative alterations of the airway proteome over time after 1-NN exposure, and how this is affected by prior long-term ozone exposure.
Supplementary Material
Acknowledgments
The authors extend special thanks to David Quilici at the Nevada Proteomics Center at the University of Nevada, Reno and Young Jin Lee at the Molecular Structure Facility at UC Davis for assistance in protein identification and interpretation of mass spectra. They also thank Dr. Brian Tarkington at the California Primate Center Exposure facility for assistance with ozone exposures, Dr. Lei Putney for kindly lending us laboratory space during the sample collection, and Dr. Suzette Smiley-Jewell and Dr. Craig Wheelock for extensive help with editing of the manuscript.
This study was supported by NIH grants ES04311, ES04699, ES00628 and ES09681. UC Davis is an NIEHS Center of Environmental Health (ES 05,707), and support of core facilities used in this work is gratefully acknowledged. Å.W. was supported by the Superfund, the American-Scandinavian Foundation, the University of California Toxic Substances Research and Teaching Program, and the Japan Society for Promotion of Science. The Nevada Proteomics Center is supported by Award P20 RR16464 from the National Center for Research Infrastructure through the Biomedical Research Infrastructure (BRIN) Program.
This article has an online supplement, which is accessible from this issue's table of contents at www.atsjournals.org
Conflict of Interest Statement: None of the authors have a financial relationship with a commercial entity that has an interest in the subject of this manuscript.
Footnotes
Protein spots # 8–10 are very abundant proteins, which can cause streaking in some 2DE gels. Although some streaking occurred in the image selected for Figure 2, it is clear from Figure 3 and from the other replicates (data not shown) that they in fact are individual protein spots.
One of the four HSPB1 spots (spot # 2) was identified as Ser-Arg rich protein with a higher probability based MOWSE score than HSPB1. However, the MW and pI of a Ser-Arg rich protein does not match the location of the protein spot in the 2D gel. In addition, the other 3 protein spots located in the same spot train were unambiguously identified as HSPB1, and since the MW and pI of HSPB1 matches the spot location very well, we assume that this is the correct identification.
References
- 1.Wong GW, Lai CK. Outdoor air pollution and asthma. Curr Opin Pulm Med 2004;10:62–66. [DOI] [PubMed] [Google Scholar]
- 2.Atkinson R, Arey J. Atmospheric chemistry of gas-phase polycyclic aromatic hydrocarbons: formation of atmospheric mutagens. Environ Health Perspect 1994;102:117–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schauer C, Niessner R, Poschl U. Analysis of nitrated polycyclic aromatic hydrocarbons by liquid chromatography with fluorescence and mass spectrometry detection: air particulate matter, soot, and reaction product studies. Anal Bioanal Chem 2004;378:725–736. [DOI] [PubMed] [Google Scholar]
- 4.Gupta P, Harger WP, Arey J. 1996. The contribution of nitro- and methylnitronaphthalenes in the vapor phase mutagenicity of ambient air samples, 30 ed. Elsevier Science Ltd, Oxford.
- 5.Serabjit-Singh CJ, Wolf CR, Philpot RM, Plopper CG. Cytochrome p-450: localization in rabbit lung. Science 1980;207:1469–1470. [DOI] [PubMed] [Google Scholar]
- 6.Verschoyle RD, Carthew P, Wolf CR, Dinsdale D. 1-Nitronaphthalene toxicity in rat lung and liver: effects of inhibiting and inducing cytochrome P450 activity. Toxicol Appl Pharmacol 1993;122:208–213. [DOI] [PubMed] [Google Scholar]
- 7.West JA, Pakehham G, Morin D, Fleschner CA, Buckpitt AR, Plopper CG. Inhaled naphthalene causes dose dependent Clara cell cytotoxicity in mice but not in rats. Toxicol Appl Pharmacol 2001;173:114–119. [DOI] [PubMed] [Google Scholar]
- 8.Paige R, Wong V, Plopper C. Dose-related airway-selective epithelial toxicity of 1-nitronaphthalene in rats. Toxicol Appl Pharmacol 1997;147:224–233. [DOI] [PubMed] [Google Scholar]
- 9.Paige RC, Wong V, Plopper CG. Long-term exposure to ozone increases acute pulmonary centriacinar injury by 1-nitronaphthalene: II. Quantitative histopathology. J Pharmacol Exp Ther 2000;295:942–950. [PubMed] [Google Scholar]
- 10.Paige RC, Royce FH, Plopper CG, Buckpitt AR. Long-term exposure to ozone increases acute pulmonary centriacinar injury by 1-nitronaphthalene: I. Region-specific enzyme activity. J Pharmacol Exp Ther 2000;295:934–941. [PubMed] [Google Scholar]
- 11.Paige R, Plopper CG. Acute and chronic effects of ozone in animal models: air pollution and health. London: Academic Press; 1999.
- 12.Wheelock AM, Zhang L, Tran MU, Morin D, Penn S, Buckpitt AR, Plopper CG. Isolation of rodent airway epithelial cell proteins facilitates in vivo proteomics studies of lung toxicity. Am J Physiol Lung Cell Mol Physiol 2004;286:L399–L410. [DOI] [PubMed] [Google Scholar]
- 13.Crivello JV. Nitrations and oxidations with inorganic nitrate salts in trifluoroacetic anhydride. J Org Chem 1981;46:3056–3062. [Google Scholar]
- 14.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 1976;72:248–254. [DOI] [PubMed] [Google Scholar]
- 15.Sabounchi-Schutt F, Astrom J, Olsson I, Eklund A, Grunewald J, Bjellqvist B. An immobiline DryStrip application method enabling high-capacity two-dimensional gel electrophoresis. Electrophoresis 2000;21:3649–3656. [DOI] [PubMed] [Google Scholar]
- 16.Gatlin CL, Kleemann GR, Hays LG, Link AJ, Yates JR III. Protein identification at the low femtomole level from silver-stained gels using a new fritless electrospray interface for liquid chromatography-microspray and nanospray mass spectrometry. Anal Biochem 1998;263:93–101. [DOI] [PubMed] [Google Scholar]
- 17.Tabb DL, McDonald WH, Yates JR III. DTASelect and Contrast: tools for assembling and comparing protein identifications from shotgun proteomics. J Proteome Res 2002;1:21–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lin CY, Isbell MA, Morin D, Boland BC, Salemi MR, Jewell WT, Weir AJ, Fanucchi MV, Baker GL, Plopper CG, et al. Characterization of a structurally intact in situ lung model and comparison of naphthalene protein adducts generated in this model vs lung microsomes. Chem Res Toxicol 2005;18:802–813. [DOI] [PubMed] [Google Scholar]
- 19.Lau SS, Monks TJ. The contribution of bromobenzene to our current understanding of chemically-induced toxicities. Life Sci 1988;42:1259–1269. [DOI] [PubMed] [Google Scholar]
- 20.Watt KC, Morin DM, Kurth MJ, Mercer RS, Plopper CG, Buckpitt AR. Glutathione conjugation of electrophilic metabolites of 1-nitronaphthalene in rat tracheobronchial airways and liver: identification by mass spectrometry and proton nuclear magnetic resonance spectroscopy. Chem Res Toxicol 1999;12:831–839. [DOI] [PubMed] [Google Scholar]
- 21.Halladay JS, Sauer JM, Sipes IG. Metabolism and disposition of [(14)C]1-nitronaphthalene in male Sprague-Dawley rats. Drug Metab Dispos 1999;27:1456–1465. [PubMed] [Google Scholar]
- 22.Mason DE, Liebler DC. Characterization of benzoquinone-peptide adducts by electrospray mass spectrometry. Chem Res Toxicol 2000;13:976–982. [DOI] [PubMed] [Google Scholar]
- 23.Waidyanatha S, Zheng Y, Serdar B, Rappaport SM. Albumin adducts of naphthalene metabolites as biomarkers of exposure to polycyclic aromatic hydrocarbons. Cancer Epidemiol Biomarkers Prev 2004;13:117–124. [DOI] [PubMed] [Google Scholar]
- 24.Sladek NE. Human aldehyde dehydrogenases: potential pathological, pharmacological, and toxicological impact. J Biochem Mol Toxicol 2003;17:7–23. [DOI] [PubMed] [Google Scholar]
- 25.Gonzalez-Mondragon E, Zubillaga RA, Saavedra E, Chanez-Cardenas ME, Perez-Montfort R, Hernandez-Arana A. Conserved cysteine 126 in triosephosphate isomerase is required not for enzymatic activity but for proper folding and stability. Biochemistry 2004;43:3255–3263. [DOI] [PubMed] [Google Scholar]
- 26.Porat A, Sagiv Y, Elazar Z. A 56-kDa selenium-binding protein participates in intra-Golgi protein transport. J Biol Chem 2000;275:14457–14465. [DOI] [PubMed] [Google Scholar]
- 27.Baranano DE, Rao M, Ferris CD, Snyder SH. Biliverdin reductase: a major physiologic cytoprotectant. Proc Natl Acad Sci USA 2002;99:16093–16098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fujii J, Ikeda Y. Advances in our understanding of peroxiredoxin, a multifunctional, mammalian redox protein. Redox Rep 2002;7:123–130. [DOI] [PubMed] [Google Scholar]
- 29.Chen JW, Dodia C, Feinstein SI, Jain MK, Fisher AB. 1-Cys peroxiredoxin, a bifunctional enzyme with glutathione peroxidase and phospholipase A2 activities. J Biol Chem 2000;275:28421–28427. [DOI] [PubMed] [Google Scholar]
- 30.Concannon CG, Gorman AM, Samali A. On the role of Hsp27 in regulating apoptosis. Apoptosis 2003;8:61–70. [DOI] [PubMed] [Google Scholar]
- 31.Williams KJ, Cruikshank MK, Plopper CG. Pulmonary heat shock protein expression after exposure to a metabolically activated Clara cell toxicant: relationship to protein adduct formation. Toxicol Appl Pharmacol 2003;192:107–118. [DOI] [PubMed] [Google Scholar]
- 32.Johnson S, Michalak M, Opas M, Eggleton P. The ins and outs of calreticulin: from the ER lumen to the extracellular space. Trends Cell Biol 2001;11:122–129. [DOI] [PubMed] [Google Scholar]
- 33.Pike SE, Yao L, Jones KD, Cherney B, Appella E, Sakaguchi K, Nakhasi H, Teruya-Feldstein J, Wirth P, Gupta G, et al. Vasostatin, a calreticulin fragment, inhibits angiogenesis and suppresses tumor growth. J Exp Med 1998;188:2349–2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gardai SJ, Xiao YQ, Dickinson M, Nick JA, Voelker DR, Greene KE, Henson PM. By binding SIRPalpha or calreticulin/CD91, lung collectins act as dual function surveillance molecules to suppress or enhance inflammation. Cell 2003;115:13–23. [DOI] [PubMed] [Google Scholar]
- 35.Vandivier RW, Ogden CA, Fadok VA, Hoffmann PR, Brown KK, Botto M, Walport MJ, Fisher JH, Henson PM, Greene KE. Role of surfactant proteins A, D, and C1q in the clearance of apoptotic cells in vivo and in vitro: calreticulin and CD91 as a common collectin receptor complex. J Immunol 2002;169:3978–3986. [DOI] [PubMed] [Google Scholar]
- 36.Butler LE, Thomassen D, Martin JL, Martin BM, Kenna JG, Pohl LR. The calcium-binding protein calreticulin is covalently modified in rat liver by a reactive metabolite of the inhalation anesthetic halothane. Chem Res Toxicol 1992;5:406–410. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.