

Abstract
Cyclosporin A (CsA) blocks T cell activation by interfering with the Ca2+-dependent phosphatase, calcineurin. Proinflammatory responses to bacteria that are activated by Ca2+-fluxes in airway cells are a potential target for CsA. Although local immunosuppression may be advantageous to control airway inflammation, it could also increase susceptibility to bacterial pneumonia and invasive infection. As aerosolized CsA is currently under study in lung transplantation, we examined its direct effects on airway cells as well as in a murine model of pneumonia. Epithelial interleukin-6 production was very effectively inhibited by CsA, whereas CXCL8 production, the major PMN chemokine, was only modestly diminished. Responses to a TLR2 agonist Pam3Cys were more sensitive to CsA inhibition than those activated by Pseudomonas aeruginosa. CsA substantially blocked activation of nuclear factor of activated T cells and cAMP-responsive element–binding protein (P < 0.001), inhibited CCAAT/enhancer-binding protein by 50% (P < 0.05), and minimally blocked activator protein-1 and nuclear factor-κB responses to bacteria in epithelial cells. The in vitro effects were confirmed in a mouse model of P. aeruginosa infection with similar rates of PMN recruitment, pneumonia and mortality in CsA treated and control mice. These studies indicate that airway epithelial signaling is a potential target for CsA, and such local immunosuppression may not increase susceptibility to invasive infection.
Keywords: cyclosporin A, Pseudomonas aeruginosa, airway cells, pneumonia, aerosol
Cyclosporin A (CsA) is a potent immunosuppressant originally developed to inhibit T cell responses that limit the success of human organ transplantation. CsA functions by targeting Ca2+-dependent signaling pathways. A primary target is nuclear factor of activated T cells (NFAT)-dependent gene transcription in T cells, which, along with activated protein-1 (AP-1), is particularly important for the activation of interleukin (IL)-2 expression and T cell proliferation (1, 2). CsA binds to the cyclophilins and forms a complex with the Ca2+-dependent phosphatase, calcineurin, thereby blocking its activity (3, 4). By blocking calcineurin, CsA inhibits the dephosphorylation of NFAT and thus prevents its mobilization and activation within the cell nucleus (5). Proinflammatory responses that are activated by Ca2+-fluxes, or that involve transcription factors that are Ca2+-dependent, are a potential target for CsA.
Airway epithelial cells express calcineurin as well as several Ca2+-dependent transcription factors and actively participate in immune signaling (6, 7). Many pulmonary pathogens including Staphylococcus aureus and Pseudomonas aeruginosa activate Ca2+ fluxes in epithelial cells upon contact with specific receptors. For example, recognition of asialoGM1/toll-like receptor 2 (TLR2) on airway cells initiates 100 nM Ca2+ fluxes sufficient to activate nuclear factor-κB (NF-κB) and generate CXCL8 and GM-CSF expression (8, 9). Several other Ca2+-dependent transcription factors are also activated by bacterial ligands leading to local cytokine expression and mucin production (10). Peptidoglycan activates the leucine zipper proteins, cAMP-responsive element binding protein (CREB)/ATF, and AP-1 (11). CREB is expected to respond to changes in cyclic nucleotides released at the surface of the airway in response to Ca2+ fluxes. In addition, CREB functions as a co-activator of CCAAT/enhancer binding protein (C/EBP), which regulates the expression of IL-6 (12). Although it has been recognized that diverse bacteria can activate signaling by airway epithelial cells through a common Ca2+-dependent pathway, it is not clear exactly which transcription factors are involved or if any of these are potential targets for immunosuppression by CsA.
As CsA blocks the activation and proliferation of T cells, it has been tested in clinical trials to control the excessive inflammation associated with rheumatoid arthritis, psoriasis, and inflammatory bowel disease (13–15). Excessive proinflammatory signaling is a major component of airway pathology in cystic fibrosis (CF) as well as many other respiratory diseases (16, 17). The delivery of CsA by aerosol in lung transplantation is currently being studied, not only to avoid systemic toxicities of the drug, but to decrease local immunoproliferative responses and bronchiolitis obliterans, a major complication of lung transplantation (18). Because cyclosporin acts through inhibition of the Ca2+-dependent phosphatase calcineurin, which is expressed in the airway epithelium, it seemed likely that cyclosporin could affect epithelial signaling in response to bacterial ligands. However, interfering with the induction of chemokine expression and the influx of polymorphonuclear leukocytes (PMNs) in response to bacteria could render the host susceptible to invasive pulmonary infection.
In the studies detailed in this article, we examined the effects of CsA on proinflammatory signaling mediated by airway epithelial cells in vitro and in a mouse model of acute infection. The consequences of CsA inhibition of Ca2+-dependent transcription factors were tested by monitoring the ability of mice treated with aerosolized CsA to clear airway infection.
MATERIALS AND METHODS
Cell Culture and Reagents
1HAEo− cells, SV40 immortalized human airway epithelial cells whose properties have been well characterized (19) were grown in Minimum Essential Medium with Earle's salts supplemented with 10% fetal calf serum (Invitrogen, Carlsbad, CA). Human bronchial airway epithelial cells were obtained from Cambrex Bio Science (Walkersville, MD). Unless specified, reagents were purchased from Sigma (St. Louis, MO). All media used were supplemented with 100 units/ml penicillin, 100 μg/ml streptomycin, 50 μg/ml gentamicin, and 4 μg/ml amphotericin B.
CXCL8 and IL-6 Assays
CXCL8 and IL-6 were measured by enzyme-linked immunosorbant assay (R&D Systems, Minneapolis, MN, and Pierce Endogen, Rockford, IL) as previously described (8) following exposure of confluent monolayers of 1HAEo− cells, weaned from serum for 24 h in 96-well plates, to heat-killed P. aeruginosa PAO1 (108 cfu), 0.1 μM thapsigargin (Molecular Probes, Eugene, OR), or 5 μg/ml Pam3Cys-Ser-Lys4 (Pam3Cys) (EMC, Hopkinton, MA). The effects of various inhibitors were tested by pretreating the cells for 1 h with 5 μM BAPTA/AM (Molecular Probes) or for 24 h with 100 nM cyclosporin (Sigma). Supernatants were harvested and duplicate wells were treated with trypan blue to assess epithelial cell viability during the assay, which was > 75%, and normalized to protein content using the Micro BCA Protein Assay Kit (Pierce Endogen). Each CXCL8 and IL-6 data point was determined in quintuplicate, and a mean and standard deviation were calculated.
Ca2+ Imaging
Subconfluent monolayers of 1HAEo− airway epithelial cells grown on glass coverslips were loaded with 2 μM Fluo-3/AM (Molecular Probes) and 0.02% pluronic acid at room temperature for 45 min, then washed twice with phosphate-buffered saline (PBS). Fluorescent images were collected by confocal microscopy using a Zeiss LSM 510 META system (Carl Zeiss Microimaging Inc., Thornwood, NY) and light intensity analysis performed using ImageJ software (U.S. National Institutes of Health, Bethesda, MD). Images were collected at 6- to 12-s intervals following the application of P. aeruginosa PAO1 (108 cfu), 15 μM thapsigargin (Molecular Probes), and 15 μg/ml Pam3Cys (EMC). Ca2+ fluxes are represented by changes in fluorescence intensity. Recordings were monitored from seven cells/field, and at least three fields were examined for each condition.
Luciferase Assays
1HAEo− cells grown in 24-well plates to 70–80% confluence were washed with PBS and transiently transfected in OptiMEM media (Invitrogen) using FuGENE 6 (Roche, Indianapolis, IN) and the luciferase reporter plasmids: 0.5 μg/ml pNFAT-luc, 0.5 μg/ml pNF-κB-luc, 0.5 μg/ml pcEBP-luc, 0.5 μg/ml pAP1-luc, or 0.4 μg/ml pRL-luc and 0.1 μg/ml pCREB (Stratagene, La Jolla, CA). After stimulation, cells were washed, lysed, and harvested using Passive Lysis Buffer (Promega, Madison, WI). Luciferase assays were performed using the reagents and protocol for the Luciferase Assay System (Promega). After standardization by protein content, data were plotted as the mean of quadruplicate samples.
Nuclear Lysates
1HAEo− cells, grown in 6-well plates to 80% confluence and weaned from serum overnight, were stimulated with P. aeruginosa PAO1 (108 cfu) for various time intervals, washed, and lysed by the addition of 20 mM HEPES, 0.5% NP-40, 10% glycerol, 40 mM NaCl, 3 mM MgCl2, 1 mM DTT, and a cocktail of protease inhibitors, for 10 min on ice followed by centrifugation at 500 × g for 5 min. The remaining pellets were first solubilized in 400 μl of 20 mM HEPES, 40 mM NaCl, 3 mM MgCl2, 1 mM DTT, and 0.32 M sucrose followed by centrifugation at 500 × g for 7 min, then solubilized in 100 μl of 20 mM HEPES, 1.5 mM NaCl, 3 mM MgCl2, 1 mM DTT, and 25% glycerol followed by the addition of 100 μl of 800 mM KCl, 1.5 mM MgCl2, 1 mM DTT, 25% glycerol, 0.5 mM EGTA, and 1% NP-40. The solutions were then incubated with rotation at 4°C for 30 min followed by centrifugation at 12 000 × g for 20 min. Supernatants, containing nuclear proteins, were collected for Western hybridization.
Western Hybridization
Proteins were separated on 4–12% NUPAGE gels (Invitrogen), transferred to Immobilon-P (Millipore, Bedford, MA), and blocked in 5% skim milk overnight. Immunodetection was done with rabbit polyclonal antibody to anti-NFATc4 (Santa Cruz Biotech, Santa Cruz, CA), anti–phosphorylated CREB (Cell Signaling Technology, Beverly, MA), anti–phosphorylated B C/EBP (Cell Signaling Technology), anti–phosphorylated NF-κB (Santa Cruz Biotech), and anti–phosphorylated ATF-2 (Cell Signaling Technology). Anti-rabbit IgG conjugated to horseradish peroxidase (Santa Cruz Biotech) was used as the secondary antibody and detected with Chemiluminescence Reagent Plus (Perkin Elmer Life Sciences, Boston, MA).
Mouse Model of Pneumonia and Aerosolized Cyclosporin A Treatment
Five-day-old BALBc mice received either aerosolized cyclosporin A (CsA) (treatment group), an equivalent volume of aerosolized ethanol (drug delivery vehicle control group: DDV control), or neither (control). A dose of 25 mg/kg of CsA was dissolved in 5 ml of ethanol and aerosolized to 1- to 3-μm particles using a MiniHEART Hi-Flo continuous nebulizer (Westmed, Tucson, AZ) into a chamber containing four mice. On Day 6 of life, both groups were intranasally inoculated with either 1–3 × 108 cfu of P. aeruginosa PA14, a highly virulent clinical isolate (20), in 10 μl of PBS or PBS alone and received another treatment dose of either aerosolized CsA or ethanol. On Day 7, the mice, with weights ranging from 3–4 g, were killed with pentobarbital. For bacteriologic analysis, one lung was plated on LB agar plates. Pneumonia was defined as the recovery of > 103 cfu per lung (21). For analysis of cell populations in the lungs, cell suspensions were obtained from homogenates of the other lung. The red blood cells were lysed, and the remaining cells were suspended in PBS containing 10% normal mouse serum and incubated for 30 min at 37°C. Cells were then double-stained with PE-labeled anti-CD45 and FITC-labeled anti-Ly6G (PMNs), anti-F4/80 (macrophages), anti-CD11 (dendritic cells), anti-CD4, or anti-CD8 (T cells) (BD Pharmingen, San Diego, California). Negative control samples were incubated with irrelevant, isotype-matched antibodies. Cells were gated based on their FSC/SSC profile and analyzed for the double expression of CD45 and Ly6G, F4/80, CD11, CD4, or CD8. Cyclosporin levels in lung homogenates were measured using a Cyclosporin Monoclonal Whole Blood assay (Abbott Laboratories, Abbott Park, IL) from representative mice treated with aerosolized cyclosporin. Lung sections from representative uninfected mice in the control and treatment group were fixed, stained with hematoxylin and eosin, and examined by light microscopy.
Statistical Analysis
Continuous variables were compared using a nonparametric t test. Categorical variable proportions were compared using Fisher's exact test or Pearson's χ2 test. The two-tailed significance level, α, was < 0.05.
RESULTS
CXCL8 and IL-6 Induction Are Ca2+-Dependent
CsA is expected to have an effect on the activation of Ca2+-dependent signaling pathways. To demonstrate the role of Ca2+ release in bacterial induction of CXCL8 and IL-6 expression, we tested the effect of BAPTA on airway epithelial cells stimulated by intact bacteria (P. aeruginosa) and a TLR2 agonist, Pam3Cys. In primary human bronchial cells, both CXCL8 and IL-6 induction by P. aeruginosa were inhibited by BAPTA by > 80% (P < 0.001) and induction by Pam3Cys was inhibited by 90% (P < 0.001) (Figures 1A and 1B). Although CsA effectively blocked IL-6 induction by P. aeruginosa and Pam3Cys and CXCL8 induction by Pam3Cys (P < 0.001), it had a more modest effect in inhibiting CXCL8 production in response to P. aeruginosa (25% decrease, P < 0.05) (Figures 1A and 1B). In 1 HAEo− cells, BAPTA inhibition produced similar results (Figures 1C and 1D). To verify that these stimuli, in fact, activate Ca2+ fluxes, 1HAEo− cells were loaded with Fluo-3 and fluorescence intensity was recorded. The expected Ca2+ flux was noted following exposure to Pam3Cys and P. aeruginosa PAO1, and was similar to that induced by the positive control, thapsigargin, an inhibitor of the Ca2+ ATPase that transiently releases Ca2+ from intracellular stores and prevents its reaccumulation (Figure 1E).
Figure 1.

CXCL8 and IL-6 induction are Ca2+-dependent. (A, B) Human bronchial epithelial cells in primary culture were pretreated for 1 h with 5 μM BAPTA, 24 h with 100 nM cyclosporin (CsA), or were left untreated (Control). CXCL8 and IL-6 were assayed by enzyme-linked immunosorbent assay in the unstimulated condition (Unstim) as well as 4 h after exposure to heat-killed P. aeruginosa PAO1 (108 cfu) or 1 h after exposure to 5 ug/ml Pam3Cys. Results are normalized to PAO1 (0.03 ng/μg protein ± 0.0006 for CXCL8; 0.2 pg/μg protein ± 0.05 for IL-6) indicated as 100% for the purpose of comparison. *P < 0.05, **P < 0.001 compared with control for each stimuli. (C, D) 1HAEo− cells treated as above. CXCL8 and IL-6 were assayed by enzyme-linked immunosorbent assay in the unstimulated condition (Unstim) as well as 4 h after exposure to heat-killed P. aeruginosa PAO1 (108 cfu) or 0.1 μM thapsigargin (Thaps) or 1 h after exposure to 5 ug/ml Pam3Cys. Results are normalized to PAO1 (0.05 ng/μg protein ± 0.003 for CXCL8; 0.9 pg/μg protein ± 0.17 for IL-6) indicated as 100% for the purpose of comparison. (E) Subconfluent 1HAEo− cells were loaded with Fluo3 and stimulated with P. aeruginosa PAO1 (108 cfu), 15 μM thapsigargin (Thaps), and 15 μg/ml Pam3Cys as indicated. Fluorescence intensity, representing changes in Ca2+ fluxes, were recorded by a Zeiss Axiovert microscope.
Epithelial Activation of NFAT
The prime target of CsA in immune cells is the activation of NFAT. NFATc4 is an NFAT family member present in lung tissue (1). NFATc4 is stimulated in response to P. aeruginosa, as shown by screening nuclear lysates of the airway cells for NFATc4 at timed intervals and confirming nuclear translocation by 30 min of bacterial exposure (Figure 2A). Luciferase reporter assays were performed to monitor the response of NFAT to bacterial stimulation. In response to the release of intracellular Ca2+ by thapsigargin, there was a > 10-fold activation in the NFAT luciferase reporter construct, which was substantially inhibited in the presence of CsA (P < 0.001) (Figure 2A). Exposure to heat-killed P. aeruginosa stimulated a 2-fold increase in NFAT activation, which was also inhibited by CsA. The specificity of these responses was confirmed in parallel experiments in which the airway cells were transfected with an expression plasmid encoding a green fluorescent protein–VIVIT peptide (GFP-VIVIT), which encodes a high-affinity calcineurin-binding protein that blocks the docking site for NFAT (Figure 2B) (22). NFAT activation induced by thapsigargin was completely blocked in cells that expressed pVIVIT. As has been shown in T cells (23), NFAT activation was dependent upon p38 mitogen-activated protein kinases (MAPK) activity, as NFAT activation was significantly inhibited by the p38 inhibitor SB202190 (P < 0.05) (Figure 2B).
Figure 2.

Epithelial activation of NFAT. (A) 1HAEo− cells transfected with an NFAT luciferase reporter construct and either pretreated for 24 h with 100 nM cyclosporin (CsA) or untreated (Control). Following stimulation for 4 h with 0.1 μM thapsigargin (Thaps) or heat-killed P. aeruginosa PAO1 (108 cfu), luciferase activity was measured and reported as fold activity compared with the unstimulated (Unstim) condition. 1HAEo− cells stimulated with P. aeruginosa PAO1 (108 cfu) were lysed at timed intervals (min) and nuclear lysates screened for the presence of NFATc4. (B, C) 1HAEo− cells transfected with an NFAT luciferase reporter construct and also with the GFP control vector (Control), the expression vector GFP-VIVIT (VIVIT plasmid), or pretreated for 2 h with 6 μM of p38 inhibitor SB202190 (p38 inhibitor). Following stimulation for 4 h with 0.1 μM thapsigargin (Thaps), luciferase activity was measured and reported as fold activity compared with the unstimulated (Unstim) condition. Supernatants were collected and CXCL8 and IL-6 assayed by enzyme-linked immunosorbent assay. Results are normalized to PAO1 (0.49 ng/μg protein ± 0.013 for CXCL8; 2.56 pg/μg protein ± 0.39 for IL-6) indicated as 100% for the purpose of comparison. *P < 0.05, **P < 0.001 compared with control for each stimulus.
The contribution of NFAT activation to airway epithelial CXCL8 and IL-6 expression was examined. Whereas both thapsigargin and heat-killed bacteria elicited CXCL8 and IL-6 production, there was little to no inhibition of chemokine production in the cells expressing pVIVIT, indicating that neither CXCL8 nor IL-6 expression are NFAT-dependent (Figure 2C). Thus, we concluded that the release of Ca2+ from intracellular stores in airway cells is sufficient to activate NFAT and that this response can be inhibited by CsA. However, NFAT is not necessary for either CXCL8 or IL-6 production in response to bacterial stimulation.
Activation of Other Transcription Factors in Response to P. aeruginosa
The release of Ca2+ from store-operated channels at the cell surface in response to bacteria is likely to initiate the activity of a number of transcription factors. We used luciferase reporter constructs to screen for the involvement of AP-1 and NF-κB, transcription factors previously shown to respond to bacterial ligands (8, 11), as well as CREB and C/EBP or NF-IL-6, transcriptional factors closely linked to Ca2+-associated signaling pathways (24, 25). The p65 component of NF-κB was clearly mobilized to the epithelial cell nucleus in response to P. aeruginosa stimulation, as was P-ATF-2, the transcription factor associated with AP-1 (Figures 3A and 3B). P. aeruginosa–induced NF-κB activation was decreased by 40% in CsA-treated cells, and AP-1 activity was similarly inhibited (P < 0.05).
Figure 3.

Activation of other transcription factors in response to P. aeruginosa. (A–D) 1HAEo− cells stimulated with P. aeruginosa PAO1 (108 cfu) were lysed at timed intervals (min) and nuclear lysates screened for the presence of P-NF-κB, P-ATF-2, P-CREB, and P-C/EBP β. 1HAEo− cells were transfected with a NF-κB, AP-1, CREB, or C/EBP luciferase reporter construct and either pretreated for 24 h with 100 nM cyclosporin (CsA) or untreated (Control). Following stimulation for 4 h with heat-killed P. aeruginosa PAO1 (108 cfu), luciferase activity was measured and reported as fold activity compared with the unstimulated (Unstim) condition. *P < 0.05, **P < 0.001 compared with control for the PAO1 stimuli.
In response to P. aeruginosa, there was immediate phosphorylation of CREB, as confirmed by Western blot, and 4-fold induction in activity, a response that was blocked by 80% by cyclosporin (P < 0.001) (Figure 3C). The kinetics of the C/EBP β translocation, a C/EBP isoform known to be present in the lung (26), were similar, and activation was inhibited by ∼ 50% by CsA (P < 0.05) (Figure 3D). These experiments suggest that CsA is capable of inhibiting the activation of major transcription factors that are involved in the immediate epithelial response to bacterial infection.
Effects of CsA in the Host Response to P. aeruginosa Infection In Vivo
CsA is currently being used as an aerosol for immunosuppressive treatment in lung transplant recipients (18). Delivery of the drug by this route could affect epithelial cell signaling as detailed above, as well as parenchymal T cells, while avoiding systemic immunosuppression and toxicities of the drug. However, local delivery of CsA may impede epithelial signaling and leave the host at greater risk to invasive pulmonary infection. To test the local effects of inhaled CsA, we evaluated the responses of mice to an intranasal inoculum of P. aeruginosa PA14, monitoring the influx of inflammatory cells and the ultimate outcome of infection. There was no obvious toxicity observed in uninfected mice treated with CsA (25 mg/kg) when comparing hematoxylin and eosin–stained lung sections from CsA-treated and control mice (Figure 4A), and adequate levels of CsA (average 21 μg/gram of lung tissue) were measured in whole lung homogenates (27). Mice in the DDV control group were comparable to mice in the control group with regards to pneumonia and mortality rates as well as lung cell populations (Figures 4B–4G). CsA treatment did not increase susceptibility to acute infection, as comparable numbers of mice in both the control group (1/5 or 20%) and DDV control group (3/16 or 19%) developed pneumonia, as compared with 2/19 or 11% of the CsA-treated group (P = 0.5, P = 0.6). Mortality rates between the groups were also not statistically different; 3/16 or 19% of the DDV control group (1/5 or 20% of the control group) died as compared with 1/19 or 5% of the CsA-treated mice (P = 0.3, P = 0.4) (Figure 4B). In addition, the control mice, DDV control mice, and the CsA-treated mice were able to recruit PMNs in response to infection. In the control mice, there was an increase from a median of 19% (uninfected) to 42% (PA14-infected) PMNs (% of CD45+ cells, P < 0.05). In the DDV control mice, there was an increase from a median of 14% (uninfected) to 34% (PA14-infected) PMNs (% of CD45+ cells, P < 0.05). In the CsA-treated group, the increase was from 12% (uninfected) to 25% (PA14-infected) PMNs (P < 0.05) (Figure 4C). The CsA-treated, infected mice had a statistically significant decrease in macrophage (P < 0.05) and dendritic (P < 0.001) cell populations as compared with uninfected mice. A similar decrease was seen in the untreated mice but was not statistically significant (Figures 4D and 4E). Therefore, we concluded that CsA treatment did not increase the risk of acute pneumonia or death in the treated animals, nor did it prevent an appropriate neutrophil response to infection.
Figure 4.
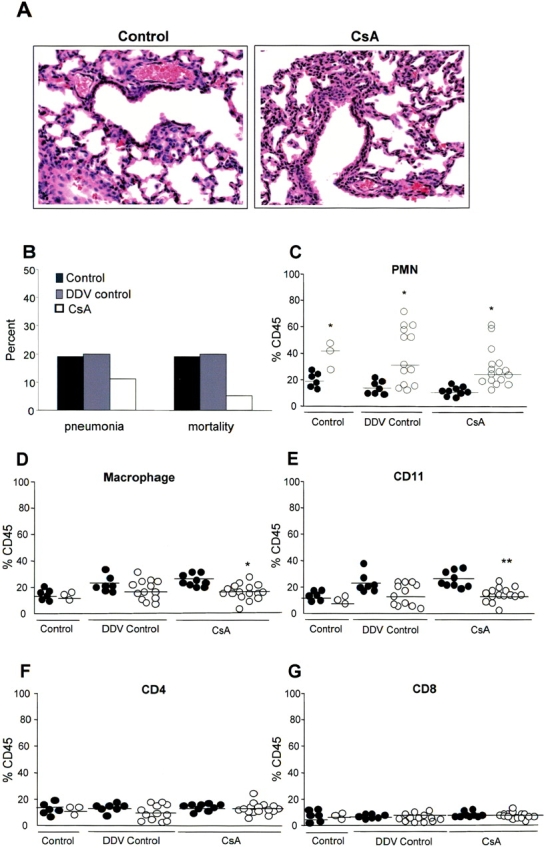
Effects of CsA in the host response to P. aeruginosa infection in vivo. (A) Hematoxylin and eosin staining of lung sections from representative uninfected mice in the control and cyclosporin-treated (CsA) group. (B) Percentage of P. aeruginosa PAO1-infected mice developing pneumonia (defined as the recovery of > 103 cfu per lung) and dying (mortality) in the control, ETOH-treated (DDV control), and cyclosporin-treated (CsA) groups. (C–G) Flow cytometry analysis of PMN, macrophage, CD11, CD4, and CD8 cell populations of lung homogenates expressed as a percentage of CD45+ cells. For each cell population, P. aeruginosa PA14-infected mice (PA14, filled circles) were compared with uninfected mice inoculated with PBS alone (PBS, open circles) in the control, the ETOH-treated (DDV control), and cyclosporin-treated (CsA) groups. *P < 0.05, **P < 0.001.
DISCUSSION
Bacteria activate a number of signaling pathways in airway cells, including several that are potential targets for inhibition by CsA. Both gram-negative and gram-positive bacteria as well as several isolated bacterial components are capable of activating Ca2+ fluxes in airway cells (8). Ca2+-dependent signals appear to be involved in the induction of CXCL8 and IL-6 expression, as suggested by the inhibition of chemokine production in the presence of BAPTA. We expected that CsA, which targets the Ca2+-dependent phosphatase, calcineurin, would inhibit proinflammatory signaling. TLR2-induced signaling, as stimulated by Pam3Cys, is more susceptible to CsA inhibition in primary airway epithelial cells, as compared with CXCL8 signaling stimulated by P. aeruginosa, which is likely to be mediated by several other pathways as well. NFAT, the expected target of CsA in immune cells, is abundant in the form of NFATc4 in airway cells, is mobilized to the nucleus by bacterial stimulation, and is significantly inhibited by CsA. NFAT has been implicated in the expression of surfactant in airway cells (28), but does not appear to be necessary for CXCL8 or IL-6 transcription.
The minimal effect of cyclosporin in inhibiting bacterial-induced CXCL8 production may be attributed to its similarly modest effects on NF-κB and AP-1 activation, reportedly the most important transcription factors in the activation of CXCL8 expression (29). In contrast, CREB activity was substantially reduced by CsA treatment of the airway cells. This is consistent with reports of CsA inhibition of other ATF/CREB promoter elements in the cyclinD1 promoter (30) and effects on CRE–reporter constructs in pancreatic islet cells and Jurkat cell lines (31, 32). CREB is phosphorylated at various sites by a number of kinases, including MAPKs and Ca2+/calmodulin-dependent protein kinase (CaMKs), which are activated by increases in Ca2+ and may be indirectly affected by CsA/cyclophilin/calcineurin interactions (33). Bacterial induction of the mucin MUC 2 gene (MUC 2) expression has been shown to involve ATP release and the induction of Ca2+ fluxes at the epithelial surface (10). This pathway is likely to involve CREB as well as NF-κB, and is a potential candidate for suppression by CsA.
The in vitro studies also suggested that bacterial-induced C/EBP activity can be blocked by CsA. Specific C/EBP isoforms are known to stimulate both the IL-6 and CXCL8 promoters (34–36). IL-6 expression is activated by exposure of the epithelial cells to bacterial ligands and blocked by CsA in primary airway epithelial cells, suggesting that C/EBP is involved in IL-6 production.
The in vivo experiments indicated that, for at least the immediate period of time following infection with a large inoculum of organisms, CsA-treated mice are not at increased risk for invasive infection. In uninfected mice, the distribution of CD45+ cells in the lungs of the CsA-treated animals did not differ significantly from the DDV control group. Consistent with the limited inhibition of chemokine expression in vitro, the CsA-treated mice, despite high tissue levels of CsA, were nonetheless able to recruit PMNs into the lungs after infection with P. aeruginosa. In response to infection, both CsA-treated and untreated mice had a decrease in the macrophage and dendritic cell populations, which is likely explained by the increased percentage of CD45+ cells representing PMNs. The CsA-treated mice thus handled P. aeruginosa infection at least as well as the DDV control group.
Delivery of CsA by aerosol is a novel use of an established immunosuppressive drug. Systemic cyclosporin is known to increase the risk of mortality in an animal model of septic shock and increase susceptibility to diverse infections, especially those controlled by T cells, in transplant patients (37, 38). In our murine model of P. aeruginosa pneumonia, however, aerosolized cyclosporin did not increase mortality. The difference in mortality may be due to the method of administration of cyclosporin; systemic cyclosporin directly affects T cells, whereas aerosolized cyclosporin may predominantly affect lung epithelial cell signaling. Recent data from a randomized trial of inhaled cyclosporin in lung transplant recipients by Iacono and coworkers (39) showed that the addition of aerosolized cyclosporin to standard systemic immunosuppression prolonged survival and survival free of bronchiolitis obliterans, a predominantly epithelial process and a major cause of death in lung transplant recipients. Surprisingly, there was no difference between the treatment and placebo group in the rate of acute rejection, a complication characterized by lymphocytic infiltration, specifically T cells. In our model of pneumonia, aerosolized cyclosporin did not affect the percentage of CD4 and CD8 cells in the lungs either in the presence or absence of infection. Although the use of aerosolized cyclosporin appears to be safe in the mouse in the setting of acute infection, a common complication after lung transplant (40, 41), our model only examined the short-term administration of cyclosporin.
Administration of cyclosporin by aerosol represents a novel delivery of a potent immunosuppressive drug that has several targets in airway epithelial cells, particularly NFAT, CREB, and C/EBP. Further studies are needed to elucidate the effects of cyclosporin inhibition of these targets in epithelial cells.
Acknowledgments
The authors thank Dr. Anjana Rao for her generous gift of the eukaryotic expression vector GFP-VIVIT and the GFP control vector, as well as Novartis for their gift of cyclosporin A used in the aerosolization treatment of the mice.
This work was supported by an NIH grant (HL60293) to A.P. and by a Cystic Fibrosis Foundation Fellowship to V.W.
Conflict of Interest Statement: None of the authors have a financial relationship with a commercial entity that has an interest in the subject of this manuscript.
References
- 1.Rao A, Luo C, Hogan PG. Transcription factors of the NFAT family: regulation and function. Annu Rev Immunol 1997;15:707–747. [DOI] [PubMed] [Google Scholar]
- 2.Chow CW, Rincon M, Davis RJ. Requirement for transcription factor NFAT in interleukin-2 expression. Mol Cell Biol 1999;19:2300–2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Friedman J, Weissman I. Two cytoplasmic candidates for immunophilin action are revealed by affinity for a new cyclophilin: one in the presence and one in the absence of CsA. Cell 1991;66:799–806. [DOI] [PubMed] [Google Scholar]
- 4.Liu J, Farmer JD Jr, Lane WS, Friedman J, Weissman I, Schreiber SL. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell 1991;66:807–815. [DOI] [PubMed] [Google Scholar]
- 5.Rao A. NFATp, a cyclosporin-sensitive transcription factor implicated in cytokine gene induction. J Leukoc Biol 1995;57:536–542. [DOI] [PubMed] [Google Scholar]
- 6.Chen Y, Zhao M, Fu M, Yao W, Tang C. The role of calcineurin in the lung fibroblasts proliferation and collagen synthesis induced by basic fibroblast growth factor. Chin Med J (Engl) 2003;116:857–862. [PubMed] [Google Scholar]
- 7.Kumar A, Knox AJ, Boriek AM. CCAAT/enhancer-binding protein and activator protein-1 transcription factors regulate the expression of interleukin-8 through the mitogen-activated protein kinase pathways in response to mechanical stretch of human airway smooth muscle cells. J Biol Chem 2003;278:18868–18876. [DOI] [PubMed] [Google Scholar]
- 8.Ratner AJ, Bryan R, Weber A, Nguyen S, Barnes D, Pitt A, Gelber S, Cheung A, Prince A. Cystic fibrosis pathogens activate Ca2+-dependent mitogen-activated protein kinase signaling pathways in airway epithelial cells. J Biol Chem 2001;276:19267–19275. [DOI] [PubMed] [Google Scholar]
- 9.Saba S, Soong G, Greenberg S, Prince A. Bacterial stimulation of epithelial G-CSF and GM-CSF expression promotes PMN survival in CF airways. Am J Respir Cell Mol Biol 2002;27:561–567. [DOI] [PubMed] [Google Scholar]
- 10.McNamara N, Khong A, McKemy D, Caterina M, Boyer J, Julius D, Basbaum C. ATP transduces signals from ASGM1, a glycolipid that functions as a bacterial receptor. Proc Natl Acad Sci USA 2001;98:9086–9091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gupta D, Wang Q, Vinson C, Dziarski R. Bacterial peptidoglycan induces CD14-dependent activation of transcription factors CREB/ATF and AP-1. J Biol Chem 1999;274:14012–14020. [DOI] [PubMed] [Google Scholar]
- 12.Kovacs KA, Steinmann M, Magistretti PJ, Halfon O, Cardinaux JR. CCAAT/enhancer-binding protein family members recruit the coactivator CREB-binding protein and trigger its phosphorylation. J Biol Chem 2003;278:36959–36965. [DOI] [PubMed] [Google Scholar]
- 13.Tugwell P, Pincus T, Yocum D, Stein M, Gluck O, Kraag G, McKendry R, Tesser J, Baker P, Wells G. Combination therapy with cyclosporine and methotrexate in severe rheumatoid arthritis. The Methotrexate-Cyclosporine Combination Study Group. N Engl J Med 1995;333:137–141. [DOI] [PubMed] [Google Scholar]
- 14.Heydendael VM, Spuls PI, Opmeer BC, de Borgie CA, Reitsma JB, Goldschmidt WF, Bossuyt PM, Bos JD, de Rie MA. Methotrexate versus cyclosporine in moderate-to-severe chronic plaque psoriasis. N Engl J Med 2003;349:658–665. [DOI] [PubMed] [Google Scholar]
- 15.Lichtiger S, Present DH, Kornbluth A, Gelernt I, Bauer J, Galler G, Michelassi F, Hanauer S. Cyclosporine in severe ulcerative colitis refractory to steroid therapy. N Engl J Med 1994;330:1841–1845. [DOI] [PubMed] [Google Scholar]
- 16.Weber AJ, Soong G, Bryan R, Saba S, Prince A. Activation of NF-kappaB in airway epithelial cells is dependent on CFTR trafficking and Cl- channel function. Am J Physiol Lung Cell Mol Physiol 2001;281:L71–L78. [DOI] [PubMed] [Google Scholar]
- 17.ten Brinke A, Zwinderman AH, Sterk PJ, Rabe KF, Bel EH. “Refractory” eosinophilic airway inflammation in severe asthma: effect of parenteral corticosteroids. Am J Respir Crit Care Med 2004;170:601–605. [DOI] [PubMed] [Google Scholar]
- 18.Iacono AT, Corcoran TE, Griffith BP, Grgurich WF, Smith DA, Zeevi A, Smaldone GC, McCurry KR, Johnson BA, Dauber JH. Aerosol cyclosporin therapy in lung transplant recipients with bronchiolitis obliterans. Eur Respir J 2004;23:384–390. [DOI] [PubMed] [Google Scholar]
- 19.DiMango E, Ratner AJ, Bryan R, Tabibi S, Prince A. Activation of NF-kappaB by adherent Pseudomonas aeruginosa in normal and cystic fibrosis respiratory epithelial cells. J Clin Invest 1998;101:2598–2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.He J, Baldini RL, Deziel E, Saucier M, Zhang Q, Liberati NT, Lee D, Urbach J, Goodman HM, Rahme LG. The broad host range pathogen Pseudomonas aeruginosa strain PA14 carries two pathogenicity islands harboring plant and animal virulence genes. Proc Natl Acad Sci USA 2004;101:2530–2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tang HB, DiMango E, Bryan R, Gambello M, Iglewski BH, Goldberg JB, Prince A. Contribution of specific Pseudomonas aeruginosa virulence factors to pathogenesis of pneumonia in a neonatal mouse model of infection. Infect Immun 1996;64:37–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aramburu J, Yaffe MB, Lopez-Rodriguez C, Cantley LC, Hogan PG, Rao A. Affinity-driven peptide selection of an NFAT inhibitor more selective than cyclosporin A. Science 1999;285:2129–2133. [DOI] [PubMed] [Google Scholar]
- 23.Wu CC, Hsu SC, Shih HM, Lai MZ. Nuclear factor of activated T cells c is a target of p38 mitogen-activated protein kinase in T cells. Mol Cell Biol 2003;23:6442–6454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Enslen H, Sun P, Brickey D, Soderling SH, Klamo E, Soderling TR. Characterization of Ca2+/calmodulin-dependent protein kinase IV: role in transcriptional regulation. J Biol Chem 1994;269:15520–15527. [PubMed] [Google Scholar]
- 25.Yukawa K, Tanaka T, Tsuji S, Akira S. Expressions of CCAAT/Enhancer-binding proteins beta and delta and their activities are intensified by cAMP signaling as well as Ca2+/calmodulin kinases activation in hippocampal neurons. J Biol Chem 1998;273:31345–31351. [DOI] [PubMed] [Google Scholar]
- 26.Lekstrom-Himes J, Xanthopoulos KG. Biological role of the CCAAT/enhancer-binding protein family of transcription factors. J Biol Chem 1998;273:28545–28548. [DOI] [PubMed] [Google Scholar]
- 27.Blot F, Tavakoli R, Sellam S, Epardeau B, Faurisson F, Bernard N, Becquemin MH, Frachon I, Stern M, Pocidalo JJ, et al. Nebulized cyclosporine for prevention of acute pulmonary allograft rejection in the rat: pharmacokinetic and histologic study. J Heart Lung Transplant 1995;14:1162–1172. [PubMed] [Google Scholar]
- 28.Dave V, Childs T, Whitsett JA. Nuclear factor of activated T cells regulates transcription of the surfactant protein D gene (Sftpd) via direct interaction with thyroid transcription factor-1 in lung epithelial cells. J Biol Chem 2004;279:34578–34588. [DOI] [PubMed] [Google Scholar]
- 29.Mastronarde JG, Monick MM, Mukaida N, Matsushima K, Hunninghake GW. Activator protein-1 is the preferred transcription factor for cooperative interaction with nuclear factor-kappaB in respiratory syncytial virus-induced interleukin-8 gene expression in airway epithelium. J Infect Dis 1998;177:1275–1281. [DOI] [PubMed] [Google Scholar]
- 30.Schneider G, Oswald F, Wahl C, Greten FR, Adler G, Schmid RM. Cyclosporine inhibits growth through the activating transcription factor/cAMP-responsive element-binding protein binding site in the cyclin D1 promoter. J Biol Chem 2002;277:43599–43607. [DOI] [PubMed] [Google Scholar]
- 31.Eckert B, Schwaninger M, Knepel W. Calcium-mobilizing insulin secretagogues stimulate transcription that is directed by the cyclic adenosine 3′,5′-monophosphate/calcium response element in a pancreatic islet beta-cell line. Endocrinology 1996;137:225–233. [DOI] [PubMed] [Google Scholar]
- 32.Kruger M, Schwaninger M, Blume R, Oetjen E, Knepel W. Inhibition of CREB- and cAMP response element-mediated gene transcription by the immunosuppressive drugs cyclosporin A and FK506 in T cells. Naunyn Schmiedebergs Arch Pharmacol 1997;356:433–440. [DOI] [PubMed] [Google Scholar]
- 33.Shaywitz AJ, Greenberg ME. CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu Rev Biochem 1999;68:821–861. [DOI] [PubMed] [Google Scholar]
- 34.Vales LD, Friedl EM. Binding of C/EBP and RBP (CBF1) to overlapping sites regulates interleukin-6 gene expression. J Biol Chem 2002;277:42438–42446. [DOI] [PubMed] [Google Scholar]
- 35.Hu HM, Tian Q, Baer M, Spooner CJ, Williams SC, Johnson PF, Schwartz RC. The C/EBP bZIP domain can mediate lipopolysaccharide induction of the proinflammatory cytokines interleukin-6 and monocyte chemoattractant protein-1. J Biol Chem 2000;275:16373–16381. [DOI] [PubMed] [Google Scholar]
- 36.Zhu YM, Bradbury DA, Pang L, Knox AJ. Transcriptional regulation of interleukin (IL)-8 by bradykinin in human airway smooth muscle cells involves prostanoid-dependent activation of AP-1 and nuclear factor (NF)-IL-6 and prostanoid-independent activation of NF-kappaB. J Biol Chem 2003;278:29366–29375. [DOI] [PubMed] [Google Scholar]
- 37.Iwamura H, Sato M, Wakitani K. Comparative study of glucocorticoids, cyclosporine A, and JTE-607 [(-)-Ethyl-N[3,5-dichloro-2-hydroxy-4-[2-(4-methylpiperazin-1-yl)ethoxy]be nzoyl]-L-phenylalaninate dihydrochloride] in a mouse septic shock model. J Pharmacol Exp Ther 2004;311:1256–1263. [DOI] [PubMed] [Google Scholar]
- 38.Nucci M, Andrade F, Vigorito A, Trabasso P, Aranha JF, Maiolino A, De Souza CA. Infectious complications in patients randomized to receive allogeneic bone marrow or peripheral blood transplantation. Transpl Infect Dis 2003;5:167–173. [DOI] [PubMed] [Google Scholar]
- 39.Iacono AT, Johnson BA, Grgurich WF, Corcoran TE, Youssef JG, Smith-Seiler DA, Smaldone GC, Zeevi A, Yousem SA, Fung JJ, et al. A randomized trial of cyclosporine inhalation solution (CyIS) in lung transplant recipients. Pediatr Pulmonol Suppl 2004;27:A375. [Google Scholar]
- 40.Zander DS, Baz MA, Visner GA, Staples ED, Donnelly WH, Faro A, Scornik JC. Analysis of early deaths after isolated lung transplantation. Chest 2001;120:225–232. [DOI] [PubMed] [Google Scholar]
- 41.Dowling RD, Zenati M, Yousem SA, Pasculle AW, Kormos RL, Armitage JA, Griffith BP, Hardesty RL. Donor-transmitted pneumonia in experimental lung allografts: successful prevention with donor antibiotic therapy. J Thorac Cardiovasc Surg 1992;103:767–772. [PubMed] [Google Scholar]