

Abstract
Lung surfactant lowers surface tension and adjusts interfacial rheology to facilitate breathing. A novel instrument, the interfacial stress rheometer (ISR), uses an oscillating magnetic needle to measure the shear viscosity and elasticity of a surfactant monolayer at the air–water interface. The ISR reveals that calf lung surfactant, Infasurf, exhibits remarkable fluidity, even when exposed to air pollution residual oil fly ash (ROFA), hydrogen peroxide (H2O2), or conditioned media from resting A549 alveolar epithelial cells (AEC). However, when Infasurf is exposed to a subphase of the soluble fraction of ROFA- or H2O2-treated AEC conditioned media, there is a prominent increase in surfactant elasticity and viscosity, representing two-dimensional gelation. Surfactant gelation is decreased when ROFA-AEC are pretreated with inhibitors of cellular reactive oxygen species (ROS), or with a mitochondrial anion channel inhibitor, as well as when A549-ρ0 cells that lack mitochondrial DNA and functional electron transport are investigated. These results implicate both mitochondrial and nonmitochondrial ROS generation in ROFA-AEC–induced surfactant gelation. A549 cells treated with H2O2 demonstrate a dose-dependent increase in lung surfactant gelation. The ISR is a unique and sensitive instrument to characterize surfactant gelation induced by oxidatively stressed AEC.
Keywords: alveolar epithelium, interfacial rheology, residual oil fly ash, hydrogen peroxide
Elevated levels of air pollution particles are associated with increased morbidity and mortality resulting from acute and chronic cardiopulmonary injury (1–4). Inhalation of particulate air pollutants can trigger generation of reactive oxygen and nitrogen species in the airway epithelium, leading to expression of multiple proinflammatory cytokines and airway pathobiology (5). Air pollutants have also been recognized to influence the structure and function of the lung surfactant system (6, 7). Oxidative inactivation of surfactant by cellular reactive oxygen species (ROS), leading to surfactant lipid peroxidation, protein carbonylation, and functional impairment of surfactant protein A, are thought to be possible mechanisms underlying air pollutant–induced surfactant damage (7–11). Oxidative damage to lung epithelial cells is a component of acute lung injury and associated respiratory failure (12).
Residual oil fly ash (ROFA), an emission source air pollution particle produced by burning low-grade oil, contains metal salts including iron, vanadium and nickel, sulfates, silicates, and carbon- and nitrogen-containing compounds (13). Exposure of lung epithelial cells to ROFA initiates phosphorylation-dependent inflammatory signaling, activation of specific transcription factors such as nuclear factor-κB, ATF-2, c-jun, and CREB, leading to increased expression and release of proinflammatory cytokines such as interleukin (IL)-6 and IL-8, macrophage inflammatory protein-2, and tumor necrosis factor-α (13–15). In addition, the metals in ROFA catalyze the production of cellular ROS, such as hydrogen peroxide (H2O2), superoxide anion (O2−·), hydroxyl radical (·OH), and nitric oxide (NO) through mechanisms that involve mitochondrial signaling and the ionic state of iron (16–18). A ROFA-induced oxidative burst in alveolar epithelial cells triggers DNA damage and apoptosis (18). The expression of epithelial cell inflammatory mediators and the production of cellular ROS following ROFA exposure contribute to the functional inactivation of lung surfactant (11, 16, 19). Furthermore, chronic exposure of rats to air pollutants (inhaled cigarette smoke) was associated with decreased expression of surfactant proteins (20). Oxidative inactivation of lung surfactants has been associated with lipid peroxidation and protein carbonyl formation (11). In addition, A549 alveolar epithelial cells (AEC) exposed to hydrogen peroxide generate ROS that induce inflammatory signaling, DNA damage, and apoptosis (21–24). Thus, inhaled air pollutants and hydrogen peroxide can each damage lung surfactant, yet the specific biological mechanisms through which lung epithelial cell oxidative stresses affect lung surfactant rheology are not well characterized.
Interfacial rheology refers to the constitutive relationships between interfacial mechanical stresses and interfacial deformations. In a complex interface, such as the one produced by spreading lung surfactant at the air–water interface, these relationships can be highly nonlinear and characterized by interfacial viscosities and frequency-dependent, interfacial moduli. Such interfaces are termed “viscoelastic,” referring to flow responses that are intermediate between a purely viscous response and one that is purely elastic. The interfacial stress rheometer (ISR) is a novel instrument that uses an oscillating magnetic needle to measure the shear viscosity and elasticity of a surfactant monolayer at the air–water interface (25–27).
Using the ISR, we demonstrate that calf lung surfactant, Infasurf, exhibits remarkable fluidity under control conditions, or when exposed to the soluble fraction of ROFA, hydrogen peroxide (H2O2), or conditioned media from resting A549 AEC. However, when Infasurf is exposed to a liquid subphase of the soluble fraction of ROFA- or H2O2-treated AEC conditioned media, there is a prominent increase in surfactant elasticity and viscosity, representing two-dimensional gelation of lung surfactant. Surfactant gelation is prominent without significant changes in surface pressure, establishing the ISR as a sensitive, novel instrument for studying surfactant damage by cellular oxidative stress.
MATERIALS AND METHODS
ROFA
The ROFA used has a mass median aerodynamic diameter of 1.95 ± 0.18 μm and was acquired from Southern Research Institute (Birmingham, AL)/NHEERL, EPA (Research Triangle Park, NC). ROFA contains iron, vanadium and nickel, sulfates, silicates, and carbon- and nitrogen-containing compounds (13). For this study, the soluble fraction of ROFA was used, as the metal in the insoluble fraction of ROFA would aggregate along the magnetic field lines of the ISR, which disrupted the measurement. To obtain the soluble fraction, ROFA suspensions were centrifuged for 5 min at 13,000 rpm, and then filtered through a 0.1-μm filter (Pall Gelman Laboratory, Ann Arbor, MI), as previously described (18). Concentrations of metals in this soluble fraction have been previously determined (16).
Infasurf
The calf lung surfactant extract, Infasurf, was a generous gift from Ony Inc. (Amherst, NY). Spreading solutions were made by dissolving 20 μl of Infasurf in 1.5 ml of 90/10 (vol/vol%) chloroform/methanol solutions. Chloroform, methanol, and other chemicals were obtained from Sigma Chemical Co. (St. Louis, MO).
Cell Culture
A549 cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA) and maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with L-glutamine (0.3 μg/ml), nonessential amino acids, penicillin (100 U/ml), streptomycin (200 μg/ml), and 10% fetal bovine serum (FBS; GIBCO, Grand Island, NY). For each experiment, we used a seeding density of 3.0 × 105 cells/ml/well plated onto six-well plates (Costar, Cambridge, MA). The cells were grown to confluence over 24 h in a humidified 95% air–5% CO2 incubator at 37°C. Before the generation of conditioned media for analysis, cells were washed once in serum-free media. Conditioned media to be analyzed in the ISR was prepared in the absence of any FBS, to eliminate rheological effects associated with adsorption of serum proteins to the surfactant monolayer. For the H2O2 experiments A549 AEC were grown in a 50-mm cell culture dish.
For experiments involving ROFA, we selected a single dose of 100 μg/ml ROFA and exposed A549 cells to the soluble component of ROFA for 16 h. In prior dose–response studies we determined that treatment of A549 AEC for 24 h with 100 μg/ml soluble air pollution particulate matter triggered substantial A549 cell inflammatory signaling, DNA damage, and apoptosis (18).
ROS Inhibitor Studies
A549 cells were pretreated for 15 min and then cotreated during 16 h ROFA exposure with iron chelators, phytic acid (500 μM) or deferoxamine (1 mM), or a free radical scavenger, sodium benzoate (50 mM).
Production and Characterization of A549-ρ0 Cells Lacking Mitochondrial DNA
A549-ρ0 cells were generated by slow chemical elimination of mitochondrial DNA by culturing in medium supplemented with ethidium bromide (25 ng/ml), sodium pyruvate (1 mM), and uridine (50 μg/ml), as described elsewhere (28, 29). The lack of normal oxidative phosphorylation in these A549-ρ0 cells was demonstrated by comparing antimycin-A–induced ROS formation in A549 and A549-ρ0 cells by using dichlorofluorescein assay.
Dichlorofluorescein Measurements of ROS
The production of ROS in A549 and A549-ρ0 cells was determined using 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA; Molecular Probes, Eugene, OR). After the cells were treated with either control media, soluble ROFA, inhibitors + soluble ROFA or antimycin (1 μg/ml), the cells were washed with phosphate-buffered saline (PBS) and then loaded for 30 min with DCFH-DA (10 μM) in DMEM without phenol red. The acetoxymethyl group on DCFH-DA is cleaved by nonspecific esterases within the cell, resulting in a nonfluorescent charged molecule that does not cross the cell membrane. Intracellular ROS irreversibly oxidize the DCFH-DA to generate fluorescent dichlorofluorescein (DCF). After treatment, the media were removed, the cells were lysed and centrifuged to eliminate debris, and the fluorescence in the supernatant was assessed using a fluorometer (excitation 500 nm/emission 530 nm). Data were normalized to the cells exposed to control media corrected per microgram of protein (Bio-Rad protein assay).
ISR
The ISR is a unique oscillating magnetic needle viscometer that measures the dynamic rheology of surfactants at an air–liquid interface (Figure 1A) (25–27). The design of the ISR is based on applying a controlled shear stress to an interface while simultaneously measuring the resultant deformation (strain) as a function of time. Conventionally, a large Langmuir trough filled with a liquid (subphase) is used, on top of which a monolayer of interest is spread (25–27). However, for our study, a Langmuir trough was modified to smaller size (a rectangular glass trough 7.6 cm long, 5 cm wide, and 0.5 cm tall; volume = 19 ml), to accommodate the relatively smaller amount of cell supernatant (20 ml) for each of our study conditions. At the bottom of the trough resides a rectangular, u-shaped glass channel with a sidewall height that is the same as the depth of the liquid subphase. Floating at the air–liquid interface inside the glass channel and parallel to its walls is a slender magnetized rod. The Langmuir trough is located inside and along the central axis of a pair of Helmholtz coils, which create a magnetic field gradient at the air–liquid interface. The magnetic field gradient applies a stress to the magnetized rod, causing it to move along the interface. As the rod moves, the portion of the surfactant film between the rod and channel wall undergoes a shear deformation. In addition, by sinusoidally varying the current sent to the Helmholtz coils, the magnetic field gradient (applied stress) also varies sinusoidally, causing the magnetized rod to oscillate back and forth. By simultaneously monitoring the supplied current (oscillating applied shear stress) and resulting rod position (oscillating shear strain) as a function of time, rheological properties of the surfactant film can be calculated. The interfacial storage modulus (Gs') is a measure of interfacial film elasticity and interfacial loss modulus (Gs”) is a measure of interfacial film viscosity. For the purposes of this paper, only the interfacial storage modulus (Gs') will be discussed. For reference, a clean air–water interface will have a Gs' value of 0 mN/m as it lacks any structure with the ability to resist an applied stress and will continually deform. On the other hand, interfacial films consisting of highly entangled, interconnected networks, with structures that have the ability to store the energy of an applied stress and resist deformation, can have large Gs' values on the order of 100 mN/m.
Figure 1.
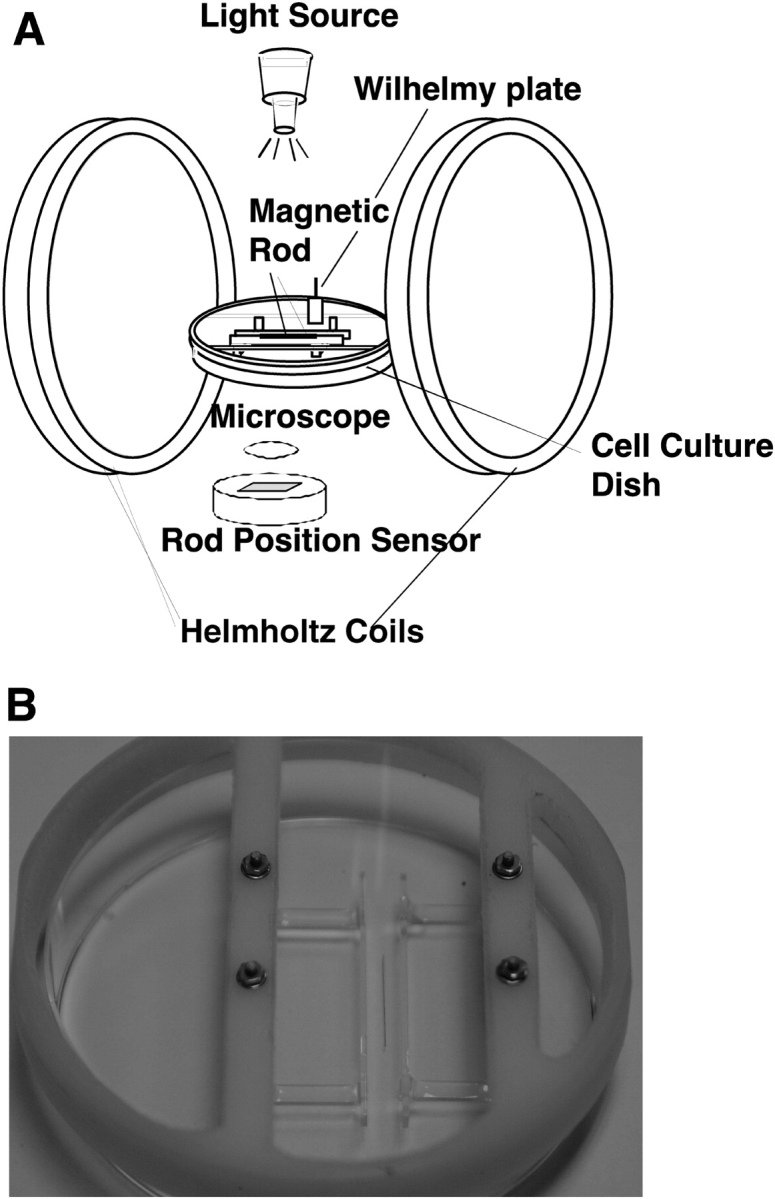
ISR used to measure lung surfactant rheology. (A) A surfactant monolayer is spread on a liquid subphase and surface pressure is continuously monitored by a Wilhelmy plate connected to a force transducer. A magnetized steel needle floating within the monolayer is displaced by varying the current delivered to the Helmholtz coils. By simultaneously monitoring the supplied current (oscillating applied shear stress) and resulting rod position (oscillating shear strain) as a function of time, rheological properties of the surfactant film are calculated. The interfacial storage modulus (Gs') is a measure of interfacial film elasticity and interfacial loss modulus (Gs”) is a measure of interfacial film viscosity. (B) An in situ cell for the ISR allows measurements of time-dependent changes in surfactant rheology to be conducted directly above a monolayer of cultured A549 epithelial cells.
These experiments were conducted by spreading a monolayer of Infasurf (20 μl of the calf lung surfactant extract (Infasurf; Ony Inc.) dissolved in 1.5 ml of 90/10 vol/vol% chloroform/methanol solution (Sigma) on a 20-ml liquid subphase of either PBS, PBS + ROFA (100 μg/ml ROFA, soluble portion), or conditioned media from A549 AEC. The Infasurf monolayers were spread to a surface pressure of 25 mN/m and the frequency of the oscillatory shear stress applied to the floating magnetic needle was 0.25 Hz, representing a typical respiratory rate. Data were collected at room temperature every 30 min for 3–4 h.
Experiments on the dose–response of A549 AEC to hydrogen peroxide were conducted using an in situ cell in which studies of surfactant rheology were conducted directly above the conditioned media released from the cultured AEC (Figure 1B). Data were collected every 30 min for 6 h.
Fluorescence Microscopy
The fluorescently-labeled phospholipid, rhodamine B 1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine, triethylammonium salt (rhodamine DHPE), was purchased from Molecular Probes. Rhodamine DHPE was added to the Infasurf spreading solution to a concentration of 1.25 weight% of the total phospholipids. Fluorescence microscopy images of the Infasurf monolayer were obtained after 2 h of exposure to the subphase using a Nikon Microphot SA microscope using a ×40 extra-long working objective and were recorded using a Pentamax intensified CCD camera (Princeton Instruments, Trenton, NJ). Image analysis was performed using Metamorph software (Universal Imaging Corporation, Downington, PA). This imaging technique can detect contrast differences in fluorescence between condensed and fluid monolayer domains because the bulky head group of the rhodamine-labeled phospholipids is excluded from the condensed phases of the film. As a result, details of the monolayer's structural morphology can be visualized.
Statistics
All data are reported as mean ± SEM. Statistical analysis was done by one-way ANOVA, and Tukey tests. Results were considered significant when P < 0.05.
RESULTS
Interfacial Rheology of Lung Surfactant
Infasurf monolayers exhibit remarkable fluidity.
We first performed a series of kinetic ISR experiments on Infasurf monolayers spread either on PBS solution, or on DMEM cell culture medium without fetal bovine serum (Figure 1). For both liquid subphases, the measured rheological responses were constant for the 4-h duration of the experiment (Figure 2). Furthermore, the elastic storage modulus, Gs', and the viscous loss modulus, Gs”, both remained extremely low, at 0.001 mN/m (Figures 2A and 2B, open squares), indicative of a very fluid-like film, not significantly different from a clean air–water interface.
Figure 2.

Lung surfactant gelation is induced by ROFA-treated A549 AEC. Elastic storage modulus, Gs' (A) and viscous loss modulus, Gs” (B) are shown. A monolayer of lung surfactant spread on a subphase of DMEM media alone (open squares), or soluble ROFA (100 μg/ml) in DMEM (filled squares) shows very low elasticity (Gs') and viscosity (Gs”) representative of remarkable microfluidity. Conditioned supernatant from resting A549 AEC (open triangles) preserves the fluidity of lung surfactant. Conditioned media from ROFA-treated AEC (100 μg/ml for 16 h incubation) induces significant increases in elasticity and viscosity by 3.5 h, representing lung surfactant gelation (*P < 0.005, n = 3).
ROFA Alone and A549 Conditioned Media Alone Do Not Significantly Alter Infasurf Rheology
ROFA can generate iron-derived ROS through Fenton reactions (11), and these ROS can cause lipid peroxidation and disulfide bond breakage, each of which can alter the surface tension–lowering properties of lung surfactant. Here, we examined the effects of addition of the soluble fraction of ROFA to serum-free DMEM on the interfacial rheology of an Infasurf monolayer. Soluble ROFA produces negligible effects on the elasticity of surfactant: the elastic and viscous moduli are unchanged from control (Figures 2A and 2B, filled boxes). Having established that soluble ROFA alone did not significantly alter the rheology of Infasurf, we next examined the effects of cultured A549 AEC supernatant. AEC supernatant also produced insignificant changes in Infasurf rheology (Figures 2A and 2B, open triangles).
ROFA–AEC Interaction Induces Two-Dimensional Gelation of Lung Surfactant
In contrast to the above results, we observed that conditioned media from A549 AEC exposed to soluble ROFA for 16 h induced significant increases in the elastic storage modulus, Gs' (Figure 2A, filled triangles), and the viscous loss modulus, Gs” (Figure 2B, filled triangles). These increases in the elastic and viscous storage moduli represent two-dimensional gelation of lung surfactant. Lung surfactant gelation after exposure to soluble ROFA depends absolutely on the presence and reaction of living epithelial cells. We consistently observed increasing surfactant gelation with increasing duration of exposure to ROFA-AEC conditioned media (Figure 2). Because of the requirements for large amounts of A549 conditioned media for these studies of lung surfactant rheology, we elected to stimulate cells with a single dose of ROFA most likely to alter surfactant rheology. We chose 100 μg/ml of soluble ROFA because that dose has been previously established to induce inflammatory mediator expression in cultured human airway epithelial cells (14). Furthermore, doses between 75 and 150 μg of ROFA were shown to induce acute lung injury in rats (16). The in vitro concentration of 100 μg/ml of soluble ROFA is possibly higher than that in the environment (there is little firm data to address this question) but would be achieved in vivo in an occupational setting (13). In exploratory studies, we investigated different durations of exposure of A549 cells to ROFA (data not shown), and selected 16 h as the optimal time for induction of lung surfactant gelation.
ROFA-AEC–Induced Surfactant Gelation Involves Iron-Catalyzed ROS
We previously demonstrated dose-dependent induction of DNA damage and apoptosis in A549 cells exposed to ambient air pollution particulate matter (18). Here, we determined the protective effects of iron chelators, phytic acid (500 μM) or deferoxamine (1 mM), or a free radical scavenger, sodium benzoate (50 mM), on ROFA-AEC surfactant gelation (Figure 3). We measured the interfacial rheology of lung surfactant using conditioned media from A549 cells pre- and cotreated with each ROS inhibitor during the 16-h exposure to the soluble fraction of ROFA. ROS inhibitors alone produced no surfactant gelation, and A549 cells treated with each inhibitor in the absence of ROFA exposure produced no surfactant gelation (data not shown). Each of the ROS inhibitors was very effective in preventing ROFA-AEC surfactant gelation (Figure 3, open circles). For clarity, we present only the effects measured on the elastic modulus, Gs'. We observed similar protective effects of ROS inhibitors on changes in the viscous modulus, Gs” (data not shown). Collectively, these data suggest that ROFA-AEC surfactant gelation is mediated through ROS.
Figure 3.

Inhibitors of ROS generation and transport decrease ROFA-AEC lung surfactant gelation. (A) A549 cells were pretreated for 15 min and then cotreated during exposure to soluble ROFA (100 μg/ml for 16 h) with ROS inhibitors, phytic acid (PA, 500 μM), deferoxamine (DF, 1 mM), or sodium benzoate (NaB, 50 mM) and the conditioned supernatants were evaluated by interfacial storage modulus measurements, Gs' (elasticity). Each inhibitor significantly decreased ROFA-AEC gelation of lung surfactant (*†‡P < 0.005, n = 3). (B) Transport of mitochondrially-generated ROS into the cytoplasm was inhibited by the mitochondrial anion channel blocker, DIDS (before and during exposure to soluble ROFA), and the conditioned supernatant evaluated. DIDS significantly inhibited ROFA-AEC surfactant gelation (*P < 0.005, n = 3). y-Axis is logarithmic scale.
Mitochondrial-Derived ROS Contribute to ROFA-AEC Surfactant Gelation
We next characterized the contribution of mitochondrial-derived ROS to ROFA-AEC surfactant gelation by coincubating A549 cells with the mitochondrial anion channel inhibitor, 4,4′ diisothiocyanatostilbene-2, 2′disulfonic acid (DIDS) (10 μM) during ROFA exposure (30–32). A549 cells treated with DIDS alone showed no increase in surfactant gelation (Figure 3B, filled squares). ROFA-AEC surfactant gelation (Figure 3B, filled triangles) was significantly prevented by the mitochondrial ROS inhibitor, DIDS (Figure 3B, open triangles). These data suggest that mitochondrial-derived ROS contribute to ROFA-AEC surfactant gelation.
A549-ρ0 Cells Exhibit Decreased ROS Production and ROFA-Induced Surfactant Gelation
A549-ρ0 cells that lack mitochondrial DNA and functional electron transport were generated by long-term culture in the presence of ethidium bromide, as described elsewhere (28, 29). We used DCF fluorescence to compare ROS production in wild-type A549 and A549-ρ0 cells (Figure 4A). Antimycin-A blocks complex III of the respiratory chain in the mitochondria, leading to generation of O2−. Wild-type A549 cells with an intact electron transport chain showed substantial increases in DCF fluorescence with antimycin-A, or ROFA. Compared with wild-type, the A549-ρ0 cells showed almost no increase in DCF fluorescence in response to antimycin-A. Compared with wild-type, the A549-ρ0 cells showed less than half the increase in DCF fluorescence after ROFA treatment. Inhibitors of ROS, phytic acid, deferoxamine, and sodium benzoate significantly decreased ROS production and DCF fluorescence in both wild-type and A549-ρ0 cells. These data suggest that both mitochondrial- and non–mitochondrial-derived sources contribute to ROFA-induced ROS production. Despite complete loss of mitochondrial DNA and deficient respiratory chain function, we observed a small but significant increase in surfactant gelation after exposure of A549-ρ0 cells to ROFA (Figure 4B, filled squares versus open squares). We also observed that surfactant gelation activity was substantially decreased from ROFA-treated A549-ρ0 cells compared with wild-type A549 cells (Figure 4B, filled squares versus filled triangles). Taken together, these data suggest that non–mitochondrial- and mitochondrial-derived ROS contribute to ROFA-AEC surfactant gelation.
Figure 4.

Comparison of ROS generation and surfactant gelation between wild-type A549 and A549-ρ0 cells. (A) A549-ρ0 cells lack mitochondrial DNA and functional electron transport. Measurement of basal and stimulated ROS production in A549 and A549-ρ0 cells by DCF fluorescence. A549 cells treated with ROFA or antimycin-A (1 μg/ml) generate significant ROS (*P < 0.005). Antimycin-A does not significant trigger ROS generation, but ROFA treatment does trigger moderate ROS production in A549-ρ0 cells (†P < 0.005). ROS inhibitors partially block ROFA-induced ROS generation in both cells lines (A549 + ROFA versus A549 + PA + DF + NaB + ROFA, ‡P < 0.005; A549-ρ0 + ROFA versus A549-ρ0 + PA + DF + NaB + ROFA, §P < 0.005) (n = 3). (B) Minimal surfactant gelation is induced by ROFA-treatment of A549-ρ0 cells. Following ROFA treatment, A549-ρ0 cells show substantially less increase in elastic storage modulus, Gs', than A549 cells (*P < 0.005). ROFA-treated A549-ρ0 cells showed minimal yet significant increase in Gs' compared with nonstimulated A549-ρ0 cells (†P < 0.0001), (n = 3). y-Axis is logarithmic scale.
Fluorescence Microscopy of Infasurf Monolayers Reveals Changes in Structural Domains that Correlate with Rheology
We used epifluorescence microscopy of fluorescently labeled phospholipid to characterize changes in the monolayer's structural morphology over time. Infasurf monolayers spread above a subphase of FBS-free DMEM and examined after 2 h exposure showed uniformly bright fields with small, solid-like domains sparsely spread throughout (Figure 5A). Surfactant monolayers spread above conditioned media from A549 AEC showed the appearance of a few dark filaments (Figure 5B). In contrast, surfactant monolayers spread above ROFA-AEC conditioned media showed the development of a microscopic structural network of highly interconnected dark filaments (Figure 5C). The time course of appearance of the network of interconnected filaments correlated well with the increases in elasticity and viscosity measured with the ISR (Figures 2, 3, and 4, and data not shown). These results suggest that the development of an interconnected network within the monolayer is associated with ROFA-AEC–induced surfactant gelation.
Figure 5.

Epifluorescence microscopy of lung surfactant monolayers reveals a network of interconnected filaments that correlates with two-dimensional gelation measured by the ISR. Lung surfactant monolayers were spread with inclusion of fluorescently-labeled phospholipid, rhodamine DHPE, and examined microscopically after 2 h. (A) Monolayers spread on DMEM media show uniformly bright fields with small, solid-like domains sparsely spread throughout. (B) Monolayers spread on conditioned supernatant from resting A549 AEC show appearances of few dark filaments. (C) Monolayers spread on ROFA-treated A549 cell supernatant show development of a microscopic structural network of highly interconnected dark filaments.
Hydrogen Peroxide Induces Dose-Dependent Increase in A549-Amplified Surfactant Gelation
We next compared surfactant gelation induced by ROFA-AEC treatment with H2O2-AEC treatment. These studies were conducted using a new in situ ISR cell (Figure 1B), in which the surfactant monolayer is spread directly on top of the conditioned media released from A549 cells cultured in a 50-mm dish. The advantage of this ISR cell is the ability to monitor, in real-time, surfactant gelation resulting from oxidative damage to living A549 cells. In control studies, we determined that H2O2, at doses up to 1 mM, when present in the subphase in the absence of A549 cells, produced small increases in surfactant elasticity and viscosity (Figure 6, filled triangles, and data not shown). We then determined that exposure of A549 cells in situ to increasing concentrations of H2O2 resulted in dose- and time-dependent increases in surfactant gelation (Figure 6, open diamonds, crosses). The magnitude of surfactant gelation achieved by exposure of A549 cells in situ to 0.5 mM and 1 mM H2O2 exposure for 6 h was comparable to that achieved with the 16 h ROFA-AEC conditioned media. The surface pressure did not change significantly during exposure of A549 cells to 1 mM H2O2 (Figure 6, open circles). These results indicate that distinct oxidative stresses on living A549 AEC, including exposure to ROFA and H2O2, trigger surfactant gelation. Most importantly, the presence of living AEC greatly amplifies surfactant gelation by oxidative stresses.
Figure 6.

Dose- and time-dependent increase in surfactant gelation by hydrogen peroxide treatment of A549 AEC. A549 AEC were cultured in 50-mm sterile dishes, and used for rheology studies when 80% confluent. Cells were washed in FBS-free DMEM, placed into the ISR then exposed at time zero to H2O2 (0, 0.5, or 1 mM). A surfactant monolayer was spread on the AEC supernatant, and kinetic measurements of elasticity, Gs', were recorded every 30 min up to 6 h. Surface pressure was continuously monitored with a Wilhemy balance throughout the duration of the experiment (AEC exposed to 1 mM H2O2, open circles). Conditioned supernatant from resting AEC preserves the fluidity of lung surfactant (open squares). Conditioned media from H2O2-treated AEC induces increases in elasticity representing lung surfactant gelation at 5.5 h (open diamonds and stars) (1 mM H2O2-AEC versus resting AEC, *P < 0.005, n = 3; 1 mM H2O2-AEC versus media + 1 mM H2O2, †P < 0.005, n = 3).
DISCUSSION
This study represents the first application of a unique and sensitive instrument, the ISR, to study the rheology of natural lung surfactant. Normal calf lung surfactant, Infasurf, reveals remarkable fluidity, with elastic storage and viscous loss moduli close to the lower limits of detection. A liquid subphase of conditioned supernatant from resting A549 AEC preserves this microfluidity of lung surfactant. Soluble ROFA alone in the subphase produces no changes in surfactant rheology. In contrast, a subphase of conditioned media from ROFA-treated A549 AEC confers dramatic changes in surfactant rheology, leading within 2 h to profound increases in elasticity and viscosity, representing two-dimensional gelation of the surfactant monolayer. The time course of lung surfactant gelation is paralleled by the development of an interconnected network of filaments within the monolayer.
A major finding of this study is that surfactant gelation induced by ROFA-treated AEC is substantially decreased by inhibition of ROS production. ROFA contains soluble salts of metals, including iron and vanadium, and it has been established that iron-catalyzed ROS generation contributes to cellular inflammation, DNA damage, and apoptosis in epithelial cells (13). We demonstrate that ROS inhibitors, phytic acid, deferoxamine, and sodium benzoate, each substantially decrease ROFA-AEC surfactant gelation. We extended these studies to distinguish the relative contributions of mitochondrial and nonmitochondrial sources of ROS to ROFA-AEC surfactant gelation. Mitochondria are an important source of intracellular ROS (32, 33). The mitochondrial electron transport chain generates ROS, which are then transported into the cytoplasm through voltage-dependent anion channels. The mitochondrial anion channel inhibitor, DIDS, protects cells from oxidative injury by blocking the egress of mitochondrial ROS into the cytoplasm (30). We show that DIDS substantially decreases ROFA-AEC surfactant gelation, implicating mitochrondrial ROS in surfactant gelation.
As an alternative demonstration of the contribution of mitochondrial ROS to the ROFA-AEC surfactant gelation, we generated and studied A549-ρ0 cells that lack mitochondrial DNA and a functional electron transport chain (29). These cells showed substantial decreases in ROS production, measured by DCF fluorescence, after stimulation with antimycin-A or ROFA. The remaining ROS production triggered by ROFA in the A549-ρ0 cells must arise through non–mitochondrial-dependent mechanisms. This residual, non–mitochondrial-dependent ROS production conferred moderate surfactant gelation.
Using a different form of cellular oxidative stress, H2O2, we demonstrated dose- and time-dependent increase in surfactant gelation. Our use of a new in situ cell that enables studies above living, oxidatively stressed A549 cells, allows direct kinetic studies of surfactant gelation. We observed reproducible surfactant gelation in response to two distinct cellular oxidative stressors, H2O2, and soluble ROFA. In each case, we demonstrate that living A549 AEC initiate or substantially amplify surfactant gelation by oxidative stressors.
The exact physical mechanism of lung surfactant gelation caused by oxidatively stressed A549 AEC remains unknown. ROS species such as superoxide anion, O2−, H2O2, and nitric oxide, have been shown to cause direct damage to lung surfactant through mechanisms that include lipid peroxidation (11). However, these ROS species are relatively short-lived, and would be expected to be quenched through interactions with cellular thiol–containing reducing agents, such as glutathione (34). In our studies on ROFA-AEC, the mediators released by oxidatively stressed A549 cells into the conditioned media remained active in surfactant gelation after transfer of the media between laboratories for the ISR studies. Our development of the in situ ISR cell used for the H2O2 studies decreases the time delay associated with transfer of conditioned media, and facilitates kinetic studies of surfactant gelation due to mediators released directly from A549 cells. It is possible that surfactant gelation develops as a consequence of protein adsorption from the subphase onto the surfactant monolayer. However, we have not detected any significant increases in total protein released into the conditioned media following oxidative stress. It is possible that specific proteins are secreted after cellular oxidative stresses that are capable of damaging surfactant. An example of such a specific surfactant-damaging protein is soluble phospholipase A2, which can increase the production of lysophosphatidylcholine (lysoPC). LysoPC can intercalate into surfactant monolayers, and, due to the altered angle of its side chains, interfere with the packing of the surfactant phospholipids, leading to increases in surface tension (35).
An alternative mechanism for our observations that cellular oxidative stresses lead to surfactant gelation involves conformational changes in surfactant proteins. Johansson described that surfactant protein C (SP-C) possess a central domain of poly-valines that normally adopts an α-helical conformation within the surfactant monolayer (36). Palmitoylation of SP-C on two adjacent cysteine residues in the N-terminal domain enhances the lipid solubility of SP-C (36). Under certain conditions, that may include oxidative stress, the α-helix of SP-C unwinds and forms a β sheet, and this misfolded SP-C molecule is prone to form aggregates with other misfolded SP-C molecules. Johansson suggests that misfolding and aggregation of SP-C is analogous to amyloid β-protein, which can precipitate as fibrils and lead to Alzheimers's dementia, and to the prion protein PrP, which can misfold and cause neurodegenerative spongiform encephalopathies (37). An important question, therefore, is to determine whether lung surfactant inactivation under cellular oxidative stress involves misfolding and aggregation of SP-C and how such a process might present as two-dimensional gelation in rheology studies.
Our studies on surfactant rheology and surfactant gelation induced by cellular oxidative stresses hold important implications for diverse acute and chronic lung diseases. Our studies establish that AEC ROS generation after oxidative stress is closely linked to the release of mediators that induce surfactant gelation. Situations in which AEC oxidative injury is known to occur include inhalational exposure to hyperoxia (e.g., during mechanical ventilation for respiratory failure), to ozone, and to paraquat (38). Furthermore, septic shock represents a pathophysiologic condition in which activated leukocytes release oxidants, including nitric oxide, in the lung. Oxidative damage to AEC in these diverse situations may all result in surfactant gelation. The consequences of surfactant gelation will be to greatly increase the work of breathing. Gelled surfactant will not respread as effectively as normal surfactant during lung inflation, leading to diminished lowering of surface tension within alveoli. Decreases in surface tension–lowering by gelled surfactant would promote alveolar collapse and mismatches of ventilation and perfusion. Repeated cycles of alveolar collapse and reinflation associated with gelled and functionally inactive lung surfactant will exacerbate alveolar epithelial injury.
The contribution of airborne pollutants to chronic lung diseases of the airways has been well established. Less clear is the contribution of airborne pollution to chronic alveolar diseases. Our study suggests that chronic exposure to airborne pollution may induce cellular oxidative stress capable of triggering gelation of lung surfactant. Genetic abnormalities in lung surfactant protein C have been associated with familial idiopathic pulmonary fibrosis (39). How chronic air pollutants might affect surfactant rheology in vivo and long-term susceptibility to surfactant-related interstitial lung diseases are important questions for future studies.
Our discovery that lung surfactant rheology is exquisitely sensitive to AEC oxidative stresses will serve to focus new efforts on further understanding the delicate interrelationships between AEC and lung surfactant. Future measurements of lung surfactant rheology will guide evaluations of novel therapeutic interventions to prevent, attenuate or reverse surfactant gelation and lung injury.
Acknowledgments
None of the authors have a financial relationship with a commercial entity that has an interest in the subject of this manuscript.
This work was supported by the Stanford University Bio-X program (P.N.K./G.G.F.), and NIH grants R01-HL62588 (P.N.K.) and HL010487 (D.U.).
References
- 1.United Nations Environment Programme, and WHO. Air pollution in the world's megacities. A Report from the U.N. Environment Programme and WHO. Environment 1994;36:5–37. [Google Scholar]
- 2.Samet JM, Dominici F, Curriero FC, Coursac I, Zeger SL. Fine particulate air pollution and mortality in 20 US cities, 1987–1994. N Engl J Med 2000;343:1742–1749. [DOI] [PubMed] [Google Scholar]
- 3.Pope CA III, Burnett RT, Thun MJ, Calle EE, Krewski D, Ito K, Thurston GD. Lung cancer, cardiopulmonary mortality, and long-term exposure to fine particulate air pollution. JAMA 2002;287:1132–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Buckeridge DL, Glazier R, Harvey BJ, Escobar M, Amrhein C, Frank J. Effect of motor vehicle emissions on respiratory health in an urban area. Environ Health Perspect 2002;110:293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Martin LD, Krunkosky TM, Dye JA, Fischer BM, Jiang NF, Rochelle LG, Akley NJ, Dreher KL, Adler KB. The role of reactive oxygen and nitrogen species in the response of airway epithelium to particulates. Environ Health Perspect 1997;105:1301–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eskelson CD, Chvapil M, Strom KA, Vostal JJ. Pulmonary phospholipidosis in rats respiring air containing diesel particulates. Environ Res 1987;44:260–271. [DOI] [PubMed] [Google Scholar]
- 7.Juvin P, Fournier T, Boland S, Soler P, Marano F, Desmonts JM, Aubier M. Diesel particles are taken up by alveolar type II tumor cells and alter cytokines secretion. Arch Environ Health 2002;57:53–60. [DOI] [PubMed] [Google Scholar]
- 8.Possmayer F, Yu SH, Weber JM, Harding PG. Pulmonary surfactant. Can J Biochem Cell Biol 1984;62:1121–1133. [DOI] [PubMed] [Google Scholar]
- 9.Gilliard N, Heldt GP, Loredo J, Gasser H, Redl H, Merritt TA, Spragg RG. Exposure of the hydrophobic components of porcine lung surfactant to oxidant stress alters surface tension properties. J Clin Invest 1994;93:2608–2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Muller B, Seifart C, Barth PJ. Effect of air pollutants on the pulmonary surfactant system. Eur J Clin Invest 1998;28:762–777. [DOI] [PubMed] [Google Scholar]
- 11.Andersson S, Kheiter A, Merritt TA. Oxidative inactivation of surfactants. Lung 1999;177:179–189. [DOI] [PubMed] [Google Scholar]
- 12.Lang JD, McArdle PJ, O'Reilly PJ, Matalon S. Oxidant-antioxidant balance in acute lung injury. Chest 2002;122:314S–320S. [DOI] [PubMed] [Google Scholar]
- 13.Ghio AJ, Silbajoris R, Carson JL, Samet JM. Biologic effects of oil fly ash. Environ Health Perspect 2002;110:89–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Silbajoris R, Ghio AJ, Samet JM, Jaskot R, Dreher KL, Brighton LE. In vivo and in vitro correlation of pulmonary MAP kinase activation following metallic exposure. Inhal Toxicol 2000;12:453–468. [DOI] [PubMed] [Google Scholar]
- 15.Samet JM, Silbajoris R, Huang T, Jaspers I. Transcription factor activation following exposure of an intact lung preparation to metallic particulate matter. Environ Health Perspect 2002;110:985–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dreher KL, Jaskot RH, Lehmann JR, Richards JH, McGee JK, Ghio AJ, Costa DL. Soluble transition metals mediate residual oil fly ash induced acute lung injury. J Toxicol Environ Health 1997;50:285–305. [PubMed] [Google Scholar]
- 17.Kadiiska MB, Mason RP, Dreher KL, Costa DL, Ghio AJ. In vivo evidence of free radical formation in the rat lung after exposure to an emission source air pollution particle. Chem Res Toxicol 1997;10:1104–1108. [DOI] [PubMed] [Google Scholar]
- 18.Upadhyay D, Panduri V, Ghio A, Kamp DW. Particulate matter induces alveolar epithelial cell DNA damage and apoptosis: role of free radicals and the mitochondria. Am J Respir Cell Mol Biol 2003;29:180–187. [DOI] [PubMed] [Google Scholar]
- 19.Roberts ES, Richards JH, Jaskot R, Dreher KL. Oxidative stress mediates air pollution particle-induced acute lung injury and molecular pathology. Inhal Toxicol 2003;15:1327–1346. [DOI] [PubMed] [Google Scholar]
- 20.Subramaniam S, Whitsett JA, Hull W, Gairola CG. Alteration of pulmonary surfactant proteins in rats chronically exposed to cigarette smoke. Toxicol Appl Pharmacol 1996;140:274–280. [DOI] [PubMed] [Google Scholar]
- 21.Holm BA, Hudak BB, Keicher L, Cavanaugh C, Baker RR, Hu P, Matalon S. Mechanisms of H2O2-mediated injury to type II cell surfactant metabolism and protection with PEG-catalase. Am J Physiol 1991;261:C751–C757. [DOI] [PubMed] [Google Scholar]
- 22.Nanavaty UB, Pawliczak R, Doniger J, Gladwin MT, Cowan MJ, Logun C, Shelhamer JH. Oxidant-induced cell death in respiratory epithelial cells is due to DNA damage and loss of ATP. Exp Lung Res 2002;28:591–607. [DOI] [PubMed] [Google Scholar]
- 23.Dandrea T, Hellmold H, Jonsson C, Zhivotovsky B, Hofer T, Warngard L, Cotgreave I. The transcriptosomal response of human A549 lung cells to a hydrogen peroxide-generating system: relationship to DNA damage, cell cycle arrest, and caspase activation. Free Radic Biol Med 2004;36:881–896. [DOI] [PubMed] [Google Scholar]
- 24.Upadhyay D, Bundesmann M, Panduri V, Correa-Meyer E, Kamp DW. Fibroblast growth factor-10 attenuates H2O2-induced alveolar epithelial cell DNA damage: role of MAPK activation and DNA repair. Am J Respir Cell Mol Biol 2004;31:107–113. [DOI] [PubMed] [Google Scholar]
- 25.Brooks CF, Fuller GG, Frank CW, Robertson CR. An interfacial stress rheometer to study rheological transitions in monolayers at the air-water interface. Langmuir 1999;15:2450–2459. [Google Scholar]
- 26.Naumann CA, Brooks CF, Fulller GG, Knoll W, Frank CW. Viscoelastic properties of lipopolymers at the air-water interface: a combined interfacial stress rheometer and film balance study. Langmuir 1999;15:7752–7761. [Google Scholar]
- 27.Brooks CF, Thiele J, Frank CW, O'Brien DF, Knoll W, Fuller GG, Robertson CR. Surface shear rheology of a polymerizable lipopolymer monolayer. Langmuir 2002;18:2166–2173. [Google Scholar]
- 28.King MP, Attardi G. Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation. Science 1989;246:500–503. [DOI] [PubMed] [Google Scholar]
- 29.King MP, Attardi G. Isolation of human cell lines lacking mitochondrial DNA. Methods Enzymol 1996;264:304–313. [DOI] [PubMed] [Google Scholar]
- 30.Beavis AD, Davatol-Hag H. The mitochondrial inner membrane anion channel is inhibited by DIDS. J Bioenerg Biomembr 1996;28:207–214. [DOI] [PubMed] [Google Scholar]
- 31.Han D, Williams E, Cadenas E. Mitochondrial respiratory chain-dependent generation of superoxide anion and its release into the intermembrane space. Biochem J 2001;353:411–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Han D, Antunes F, Canali R, Rettori D, Cadenas E. Voltage-dependent anion channels control the release of the superoxide anion from mitochondria to cytosol. J Biol Chem 2003;278:5557–5563. [DOI] [PubMed] [Google Scholar]
- 33.Han Z, Johnston C, Reeves WH, Carter T, Wyche JH, Hendrickson EA. Characterization of a Ku86 variant protein that results in altered DNA binding and diminished DNA-dependent protein kinase activity. J Biol Chem 1996;271:14098–14104. [DOI] [PubMed] [Google Scholar]
- 34.Comhair SA, Erzurum SC. Antioxidant responses to oxidant-mediated lung diseases. Am J Physiol Lung Cell Mol Physiol 2002;283:L246–L255. [DOI] [PubMed] [Google Scholar]
- 35.Holm BA, Keicher L, Liu MY, Sokolowski J, Enhorning G. Inhibition of pulmonary surfactant function by phospholipases. J Appl Physiol 1991;71:317–321. [DOI] [PubMed] [Google Scholar]
- 36.Johansson J. Structure and properties of surfactant protein C. Biochim Biophys Acta 1998;1408:161–172. [DOI] [PubMed] [Google Scholar]
- 37.Johansson J, Weaver TE, Tjernberg LO. Proteolytic generation and aggregation of peptides from transmembrane regions: lung surfactant protein C and amyloid beta-peptide. Cell Mol Life Sci 2004;61:326–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Berisha HI, Pakbaz H, Absood A, Said SI. Nitric oxide as a mediator of oxidant lung injury due to paraquat. Proc Natl Acad Sci USA 1994;91:7445–7449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nogee LM. Alterations in SP-B and SP-C expression in neonatal lung disease. Annu Rev Physiol 2004;66:601–623. [DOI] [PubMed] [Google Scholar]