

Abstract
Apoptosis of distal lung epithelial cells plays a pivotal role in the pathogenesis of acute lung injury. In this context, proteinases, either circulating or leukocyte-derived, may contribute to epithelial apoptosis and lung injury. We hypothesized that apoptosis of lung epithelial cells induced by leukocyte elastase is mediated via the proteinase activated receptor (PAR)-1. Leukocyte elastase, thrombin, and PAR-1–activating peptide, but not the control peptide, induced apoptosis in human airway and alveolar epithelial cells as assessed by increases in cytoplasmic histone-associated DNA fragments and TUNEL staining. These effects were largely prevented by a specific PAR-1 antagonist and by short interfering RNA directed against PAR-1. To ascertain the mechanism of epithelial apoptosis, we determined that PAR-1AP, thrombin, and leukocyte elastase dissipated mitochondrial membrane potential, induced translocation of cytochrome c to the cytosol, enhanced cleavage of caspase-9 and caspase-3, and led to JNK activation and Akt inhibition. In concert, these observations provide strong evidence that leukocyte elastase mediates apoptosis of human lung epithelial cells through PAR-1–dependent modulation of the intrinsic apoptotic pathway via alterations in mitochondrial permeability and by modulation of JNK and Akt.
Keywords: acute lung injury, proteinases, inflammation, signal transduction
Activated neutrophils can release proteinases such as elastase and other proinflammatory molecules that contribute to the pathogenesis of the inflammatory tissue injury seen in acute lung injury (ALI) and acute respiratory distress syndrome (ARDS) (1, 2). In support of this notion, leukocyte elastase levels are increased in serum and bronchoalveolar lavage (BAL) fluid in clinical studies and animal models of ALI/ARDS (3–5). We have recently reported that leukocyte elastase released by activated neutrophils induces apoptosis of gastrointestinal epithelial cells in an in vitro model (6, 7). These observations are relevant to the pathogenesis of both acute (ALI/ARDS) and chronic (emphysema) lung injury, where inappropriate apoptosis of lung epithelial cells may contribute to the pathogenesis of pulmonary dysfunction (8–13).
Apoptosis, or programmed cell death, is fundamental to physiologic processes such as embryogenesis and in the remodeling of tissues that occurs during normal repair processes (9). Cells dying from apoptosis tend to be disposed of in a noninflammatory manner, whereas death by necrosis is often associated with the activation of proinflammatory pathways in the surrounding environment. The signaling pathways leading to apoptosis can generally be divided into the extrinsic (surface receptor–mediated) and intrinsic (mitochondrial-dependent) pathways (14). The former pathway is triggered following ligand binding to “death” receptors of the TNF superfamily such as TNFR1, Fas, and TRAIL receptors (15). In contrast, the intrinsic pathway can be executed independently of death receptors and involves alteration of mitochondrial permeability (16). Apoptotic pathways can be modulated by various inflammatory molecules and elucidation of these mechanisms may lead to novel therapeutic interventions that inhibit key steps in the apoptosis pathways.
In addition to leukocyte-derived proteases, serum proteases, and in particular thrombin, may play an important role in ALI/ARDS. Indeed, thrombin has been implicated in the pathogenesis of a diverse array of inflammatory diseases, such as inflammatory bowel disease, ventilator-associated pneumonia, and myocardial injury (17–19). The importance of the coagulation cascade in the setting of ALI/ARDS is reinforced by studies revealing fibrin and microthrombi in the lungs of these patients, increased procoagulant activity in the early exudative phase of lung injury, and increased BAL fibrinopeptide A levels (20, 21).
Thrombin can influence cell function through a group of cell surface receptors known as proteinase-activated receptors (PARs). These are G protein–coupled receptors that are activated by cleavage of their extracellular N-terminus by proteinases (22, 23). This cleavage creates a neo-amino terminus or “tethered ligand” that binds to and activates the receptor. There are four known PARs, and all are expressed by human lung epithelial cells (24). Although thrombin activates PAR-1, -3, and -4, PAR-1 is considered the archetypal receptor for thrombin. Synthetic peptides such as S(T)FLLR-NH2, which mimic the tethered ligand sequence of PAR-1, can function independently of receptor cleavage (23, 25). PAR activation has been linked to a number of responses including platelet aggregation, vasodilation, vasoconstriction, increased vascular permeability, increased intestinal permeability, granulocyte chemotaxis, and intestinal epithelial apoptosis (26–28). Although generally proinflammatory, there is evidence that PAR-2 activation may attenuate pulmonary inflammation (29, 30) Our knowledge of the effects of PAR activation on lung epithelial function is incomplete.
In the current study, we focused on the role of PAR-1 in the regulation of lung epithelial apoptosis. We report that leukocyte elastase, through PAR-1 activation, promotes epithelial apoptosis via a mitochondrial-dependent pathway. We also examined the signal transduction pathways downstream of PAR-1 modulated by elastase and other receptor agonists. In the present study, we report that elastase modulates the activity of Akt and the amino-terminal c-Jun kinase (JNK) and thus regulates apoptosis.
MATERIALS AND METHODS
Cell Culture
Primary human small airway epithelial (HSAE) cells (Cambrex, Walkersville, MD) were grown in Small Airway Cell Basal Medium (SABM) supplemented with growth factors and antibiotics according to the manufacturer's instructions. BEAS-2B, a human bronchial epithelial cell line, was provided by Dr. Reen Wu (University of California, Davis). Cells were grown in 1:1 mix of Dulbecco's modified Eagle's medium (DMEM) and Ham's F-12 Nutrient Mix (DMEM/F12) (GIBCO/BRL, Grand Island, NY) supplemented with the following: 10 μg/ml of human recombinant insulin, 25 ng/ml of recombinant human epidermal growth factor, 5 μg/ml of transferrin, 2% (vol/vol) penicillin-streptomycin (all from GIBCO/BRL, Grand Island, NY) and 0.1 μM of hydrocortisone (Sigma, St. Louis, MO). Primary human alveolar type II cells were isolated as described (31) and grown in DMEM with 10% fetal calf serum. A549 cells (American Type Tissue Collection, Gaithersburg, MD), neoplastic lung cells with features of alveolar type II cells, were grown in DMEM with 10% fetal bovine serum. Epithelial cells were seeded on tissue culture plastic plates coated with human type VI collagen (Sigma), and grown to confluence at 37°C in 5% CO2.
Reagents
Human leukocyte elastase from human sputum was from EPC (Owensville, MO). PAR-1–activating peptide (PAR-1AP: Thr-Phe-Leu-Leu-Arg, TFLLR-NH2) and PAR-1 control peptide (Arg-Leu-Leu-Phe-Thr, RLLFT-NH2) were obtained from the Alberta Peptide Institute (Edmonton, AB, Canada). Thrombin from human plasma was from Sigma-Aldrich. SP600125, a selective JNK inhibitor, was obtained from A.G. Scientific, Inc. (San Diego, CA). The PAR-1 antagonist, SCH79797, was obtained from Tocris-Cookson Inc. (Ellisville, MO).
Antibodies
Primary antibodies included: anti–thrombin R (mouse monoclonal) (Santa Cruz Biotechnology, Santa Cruz, CA); anti-Akt (rabbit polyclonal); anti-phospho Ser-473 Akt (rabbit polyclonal); anti-SAPK/JNK (rabbit polyclonal); anti–phospho SAPK/JNK (rabbit polyclonal); anti–caspase 3 (rabbit polyclonal); anti–caspase 9 (rabbit polyclonal); anti–cytochrome c (rabbit polyclonal) (all from Cell Signaling Technology, Beverly, MA); anti-actin (murine monoclonal) (ICN, Aurora, OH); and anti-mitochondria COX IV (mouse monoclonal) (Abcam, Cambridge, MA).
Apoptosis Analysis
Human lung epithelial apoptosis was quantified 4 and 12 h after treatment with leukocyte elastase, PAR-1 AP, control peptide, and thrombin using the Cell Death Detection ELISA kit (Roche, Mannheim, Germany) that specifically detects the histone region (H1, H2A, H2B, H3, and H4) of mono- and oligonucleosomes that are released during apoptosis. Absorbance at 405 nm in a 96-well plate was measured using a microplate reader (THERMO max; Molecular Devices, Sunnyvale, CA). Apoptosis was measured in duplicate from 105 lung epithelial cells from each treatment group, and expressed as the absorbance ratio relative to control (28). Terminal deoxynucleotidyl transferase-mediated nick end labeling (TUNEL) was performed on cells grown on coated glass slides (Lab-Tek, Naperville, IL) using an in situ Cell Death Detection Kit, Fluorescein (Roche) according to the manufacturer's instructions. Monolayers were mounted with fluorescent mounting media (DAKO, Carpinteria, CA) onto slides and visualized using a fluorescence microscope (LEICA DM-IRB2) controlled by Open Lab software (Improvision Inc., Lexington, MA).
RNA Extraction and Reverse Transcription–Polymerase Chain Reaction
Total RNA was isolated from cultured lung epithelial cells using Trizol reagent (TRI Reagent; Sigma) following the manufacturer's protocol. Purity was checked by the A260/A280 ratio. Total RNA was reverse transcribed using random hexamer priming and SuperScript II Rnase H- Reverse Transcriptase (RT) (SuperScript First-Strand Synthesis System for RT-PCR; Invitrogen, Carlsbad, CA). cDNA was prepared from 5 μg RNA in a 20-μl volume, adding 50 units SuperScript II RT, 2 μl 10× RT buffer, 40 units RNaseOut Recombinant RNase Inhibitor, 5 mM MgCl2, 10 mM dithiothreitol, 0.5 mM dNTP mix, and 7.5 ng/μl random hexamers. A negative control reaction lacking RT was also performed for each RNA sample. The random hexamer primer was annealed for 10 min at 25°C. cDNA synthesis was performed for 50 min at 42°C, followed by 15 min at 70°C to terminate the reaction. One microliter of RNAse H was added to each tube and incubated for 20 min at 37°C, and cDNA was stored at −20°C until used. PCR was performed in 100 μl of reaction solution, containing 0.2 mM dNTPs, 1.5 mM MgCl2, 2.5 U/100 μl of Taq polymerase (Taq DNA polymerase; Fermentas, Burlington, ON, Canada), 0.5 μM oligonucleotide primers, human PAR-1, Accession No. M62424, sense 5′-TGTGAACTGATCATGTTTATG-3′, antisense 5′-TTCGTAAGATAAGAGATATGT-3′, PCR product 708 bp; human GAPDH, Accession No. M33197, sense 5′-TCAACGACCACTTTGTCAAGCTCA-3′, antisense 5′-GCTGGTGGTCCAGGGGTCTTACT-3′, PCR product 120 bp and first-strand cDNA products. After an initial denaturation step (3 min at 94°C), PCR mixtures were amplified by 30 cycles (30 s at 94°C, 30 s at 55°C, 30 s at 72°C). The final extension period was of 7 min at 72°C. The PCR products were analyzed by electrophoresis using a 2% agarose gel with ethidium bromide.
Measurement of Intracellular Ca2+
Human lung epithelial cells were cultured on 25 mm2 microscope cover slips coated with human type VI collagen. Coverslips were placed in a sterile microscope chamber (Attofluor; Molecular Probes, Eugene, OR) and epithelial cells loaded with 3 μM Fura-2/AM (Molecular Probes) in DMEM/F12 medium at 37°C, 5% CO2 for 25 min, and then washed three times with the same medium. Dynamic changes in intracellular calcium concentration ([Ca2+]i) in individual cells was measured using a Nikon inverted fluorescent microscope equipped with a CCD camera (Orca; Hamamatsu, Bridgewater, NJ) as previously described in detail (32). Images were acquired and analyzed using SimplePCI software (Compix, Cranberry Township, PA). Cellular fluorescence was monitored for at least 2 min to ensure stability before cells were challenged with agonists including leukocyte elastase, PAR-1AP, or PBS buffer control. Data were recorded for 15 min for each sample.
siRNA Design and Transfection
The different siRNAs were designed corresponding to the human PAR-1 gene sequence (Accession No. M62424). Sequences were chosen on the basis of guidelines from QIAGEN (Mississauga, ON, Canada). siRNA sequences were found using BLAST software from the National Center for Biotechnology Information (NCBI). A strategy using the “search for short nearly exact matches” mode against all human sequences deposited in the GenBank and RefSeq databases that also did not have significant homology (> 17 contiguous nucleotides of identity) to genes other than the targets (33) yielded appropriate sequences. PAR-1 siRNAs (listed in Table 1) were synthesized and annealed by QIAGEN. A quantity of 50 nM of control and PAR-1 siRNAs was transfected into BEAS-2B cells (30–50% confluence) with Lipofectamine 2000 (Invitrogen, Carlsbad, CA) on the basis of manufacturer's instructions. After 72 h incubation, cells were used for experiments as described below and harvested for flow cytometry and Western blot analysis.
TABLE 1.
Sequences of siRNA nucleotides used to silence PAR-1
| siRNA Name | DNA Target Sequence | siRNA Sequence | Length |
|---|---|---|---|
| PAR-1A | AACATCATGGCCATCGTTGTG | CAUCAUGGCCAUCGUUGUGTT TTGUAGUACCGGUAGCAACAC |
21 |
| PAR-1C | AACGTCCTCCTGATTGCGCAT | CGUCCUCCUGAUUGCGCAUTT TTGCAGGAGGACUAACGCGUA |
21 |
Flow Cytometry
Adherent lung epithelial cells which were transfected with PAR-1 and control siRNA were detached from culture dishes with a cell scraper. Cells were sedimented by centrifugation, washed with PBS once, and resuspended in HBSS-BSA 2% (wt/vol) at a final concentration of 2 × 106 cells/ml. Cells were immunolabeled with either an FITC-conjugated anti–thrombin receptor-mouse monoclonal antibody or a control FITC-conjugated-mouse IgG1 (Santa Cruz Biotechnology, Santa Cruz, CA), both at 1:500 dilution, for a 1 h at 4°C. Cells were then washed by FCM buffer (Santa Cruz Biotechnology, Santa Cruz, CA), and resuspended in 1% paraformaldehyde. Flow cytometry was performed on a FACScan using CELL-quest software (Becton Dickinson, Palo Alto, CA). Values are expressed as relative fluorescence index.
Cell Fractionation
Lung epithelial cells cultured in 60-mm plastic culture dishes were incubated with leukocyte elastase 0.1 U/ml and PAR-1AP 100 μM for 0, 2, 4, 12 h. They were washed once with PBS, scraped into PBS, pelleted at 1,000 × g for 5 min, and resuspended in hypotonic buffer (10 mM NaCl, 5 mM MgCl2, 10 mM Tris-HCl [pH 7.5], 100 μM PMSF). Cells were allowed to swell on ice for 10 min and homogenized with a tight pestle using a 21-G needle (10 strokes) before lysis by additional of isotonic homogenizing buffer (2.5× MS buffer, 525 mM mannitol, 175 mM sucrose, 12.5 mM Tris-HCl [pH 7.5], and 2.5 mM EDTA [pH 7.5]). After mixing, cell fragments were sedimented at 1,300 × g. Supernatants were collected and sedimented at 17,000 × g for 15 min. After centrifugation, pellets were resuspended in 1× MS buffer and used as the heavy membrane fraction containing mitochondria. The supernatants were further centrifuged at 100,000 × g for 1 h, and resulting supernatants were used as the cytosol fraction.
SDS-PAGE and Immunoblotting
For SDS-PAGE of whole cell extracts, cells were collected with boiling lysis buffer (2% SDS, 10% glycerol, 65 mM Tris/HCl, pH 6.8, 50 mM dithiothreitol) supplemented with a protease inhibitor cocktail (Roche). Subcellular fractions were also collected with the same lysis buffer as described above. SDS-PAGE and immunoblotting were performed as described previously in detail (7). The data were analyzed by densitometry using NIH Image.
Mitochondrial Membrane Potential Assay
Lung epithelial cells were cultured on glass chamber slides (Lab-Tek, Naperville, IL) and incubated with PBS (as a negative control), 1 μM of valinomycin (as a positive control) (SIGMA, St. Louis, MO), leukocyte elastase, thrombin, and PAR-1AP for 30 min, 1 h, or 3 h. They were labeled with JC-1 using the DePsipher kit (Trevigen for R&D Systems Inc, Minneapolis, MN) according to the manufacturer's instructions. After labeling, cells were observed using fluorescence microscopy (LEICA DM-IRB), and Open lab (Improvision Inc., Lexington, MA) was used for data acquisition and analysis.
Statistical Analysis
Parametric data were compared by using t tests for mean values or ANOVA with correction for multiple comparisons (Fisher's PLSD test) when more than two variables were analyzed using STATview software.
RESULTS
Leukocyte Elastase Induces Apoptosis of Lung Epithelial Cells
We have recently reported that leukocyte elastase induces apoptosis of intestinal epithelial cells (7). To determine if there are analogous effects of elastase on lung epithelial cells, we quantified apoptosis in epithelial cells of both airway and alveolar origin in response to leukocyte elastase by monitoring the presence of histone associated mono- and oligonucleosomes. These studies revealed that exposure of the airway epithelial cell line BEAS-2B as well as primary cultures of human small airway epithelial (HSAE) and human alveolar type II cells to elastase-induced apoptosis in a dose-dependent manner that reached a maximum at 0.3 U/ml (Figures 1A–1C). Similar responses were noted in human A549 cells (data not shown). To confirm these observations by an independent method, we quantified lung epithelial cell apoptosis using TUNEL staining (Figures 1D–1F). These studies confirmed that treatment of both airway (BEAS-2B and HSAE) epithelial cells and alveolar type II epithelial cells with leukocyte elastase induced apoptosis. We conclude that leukocyte elastase induces apoptosis of lung epithelial cell lines and primary cultures of lung epithelial cells of both airway and alveolar origin in physiologic concentrations and in a dose-dependent manner.
Figure 1.
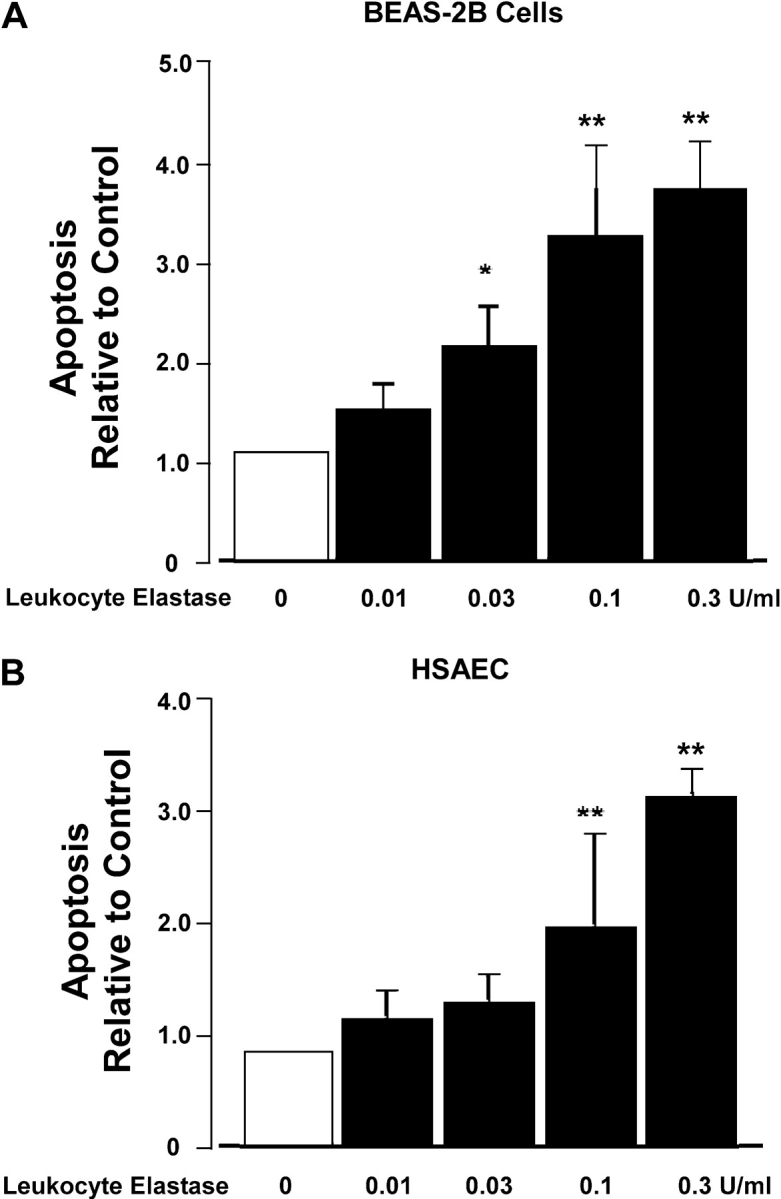


Leukocyte elastase (LE) induces lung epithelial apoptosis in a dose-dependent manner. Cell death detection assay (A–C) and TUNEL staining (D–F) were used for the detection of apoptosis. Three different lung epithelial cell types BEAS-2B (A and D), human small airway epithelial (HSAE) cells (B and E), and primary human alveolar type II (HAEC) cells (C and F) were used. In experiments with HSAE cells, many cells lifted off the coverslip during washing. Therefore, we collected cells in the supernatant and applied them to slides using a cytocentrifuge. The graphs (A–C) represent the absorbance values relative to control (buffer only). Values are mean ± SD; *P < 0.05 and **P < 0.01 compared with control. (A) n = 10; (B and C) n = 4. For TUNEL labeling (D–F), apoptotic cells demonstrate a greater intensity of fluorescence. Representative data each from one of three experiments are shown.
Expression of Functional PAR-1 by Human Lung Epithelial Cells
To determine the mechanisms of leukocyte elastase–induced apoptosis, we focused on PAR-1, a membrane receptor known to be modulated by proteolytic cleavage. Using a combination of RT-PCR and Western analysis, we determined that BEAS-2B, HSAE cells, and primary alveolar type II cells (not shown) express PAR-1 mRNA and protein (Figures 2A and 2B). To test the functionality of PAR-1, we measured intracellular calcium concentration using the calcium-sensitive fluorescent dye, FURA-2AM. The epithelial cells were challenged with either 0.1 U/ml leukocyte elastase or 100 μM PAR-1AP. As illustrated in Figures 2C and 2D, a reproducible response was observed in each case characterized by a rapid and transient rise of cytosolic Ca2+, typical of receptor-mediated signaling events (24). These data indicate that exposure of cells to either leukocyte elastase or PAR-1AP induces rapid and transient intracellular calcium flux typical of receptor-mediated events; the similarity of these responses suggests that both may be mediated by PAR-1.
Figure 2.

Expression of PAR-1 in lung epithelia as determined by RT-PCR and flow cytometry. (A) PAR-1 mRNA is detected in BEAS-2B and HSAE cells using RT-PCR. Upper panel represents PAR-1 and lower panel represents GAPDH expression, the latter used as a “housekeeping” gene. (B) Detection of PAR-1 cell surface receptor expression by FACS analysis. Cells were incubated with PAR-1 mouse monoclonal antibody FITC-conjugated (b) or with normal mouse IgG1 FITC conjugated (a). Representative data each from one of four experiments are shown. (C and D) Functionality of PAR-1 as determined by [Ca2+]i fluxes. Cells were challenged leukocyte elastase (LE) 0.1 U/ml or PAR-1AP 100 μM (arrow). Representative data each from one of three independent experiments are shown.
PAR-1 Agonists Induce Apoptosis of Lung Epithelial Cells
To provide more definitive evidence that apoptosis of lung epithelial cells induced by leukocyte elastase is mediated via PAR-1, BEAS-2B, HSAE and alveolar type II cells were treated with the prototypical PAR-1 activating proteinase, thrombin. Responses were compared with treatment with PAR-1 activating peptide (AP), a control inactive peptide of similar amino acid composition, and with leukocyte elastase. Figure 3 illustrates that thrombin, leukocyte elastase, and PAR-1AP but not the inactive peptide induced apoptosis of all three epithelial cell types. Apoptotic changes were detected after 4 h and reached a maximum at 12 h. The magnitude of the response to PAR-1AP (3-fold) and to thrombin (3.6-fold) at 12 h was similar to the response observed with leukocyte elastase. Apoptosis of lung epithelial cells under these conditions was confirmed by TUNEL staining (Figures 3C, 3F, and 3I). Taken together, these observations indicate that direct activation of PAR-1 induces apoptosis of lung epithelial cells. Moreover, the responses are similar to those induced by leukocyte elastase suggesting that the effects of elastase may be mediated via PAR-1.
Figure 3.



PAR-1 agonists induce lung epithelial apoptosis. Apoptosis was assessed using histone-associated DNA fragments (Cell Death Detection assay; A, B, D, E, G, H) at 4 and 12 h after the treatment with a control peptide (Control) 100 μM, leukocyte elastase (LE) 0.1 U/ml, PAR-1AP 100 μM, or Thrombin 1 U/ml. BEAS-2B cells were used in A and B, HSAE cells were used in D and E, and human alveolar epithelial (HAEC) cells were used in G and H. Values represent the mean ± SD; *P < 0.05 and **P < 0.01 compared with control. (A) n = 4; (B) n = 7; (D, E, G, H) n = 3. Apoptosis of lung epithelial cells, including BEAS-2B cells (C), HSAE cells (F), and HAEC (I) was determined using TUNEL labeling. Representative data from one of three experiments are shown.
Leukocyte Elastase Induces Epithelial Apoptosis via PAR-1
To supply direct evidence for a role for PAR-1 in leukocyte elastase–induced apoptosis, two approaches were undertaken. First, we used a selective pharmacological inhibitor of PAR-1, SCH79797 (34). Treatment of cells with this inhibitor significantly diminished epithelial apoptosis induced by leukocyte elastase, thrombin, or PAR-1 AP (Figure 4A). Second, we employed gene-silencing with short interfering RNA directed at PAR-1. For these experiments, several sequences corresponding to distinct regions of PAR-1 mRNA were synthesized (Table 1) and introduced into BEAS-2B lung epithelial cells by transfection. Alterations in PAR-1 expression were assessed using Western analysis and flow cytometry with anti–PAR-1 antibodies (Figures 4B and 4C, respectively). These studies revealed that PAR-1 expression was significantly diminished (> 70%) by two of the three siRNAs (PAR-1A and 1C). Additional studies demonstrated that unrelated cellular proteins including α-actin (Figure 4B) and intercellular adhesion molecule-1 (data not shown) were unaffected by this treatment indicating the specificity of gene silencing under these conditions.
Figure 4.

PAR-1 siRNA diminishes PAR-1 expression and inhibits leukocyte elastase (LE)-induced apoptosis in BEAS-2B cells. (A) The selective pharmacologic inhibitor of PAR-1, SCH79797, attenuates epithelial apoptosis by leukocyte elastase and thrombin. The data represent the mean ± SEM of n = 3 experiments. (B) Upper panel represents PAR-1 expression and the lower panel illustrates α-actin protein expression using with Western blot analysis. (C) PAR-1, cell surface receptor as detected by FACS analysis. Cells were incubated with a FITC-conjugated PAR-1 murine monoclonal antibody 72 h after transfection with control siRNA (iv), PAR-1A siRNA (ii), and PAR-1C siRNA (iii). Histogram (i) represents staining with FITC-conjugated isotype control murine IgG1. Representative data each from one of five experiments are shown. (D) PAR-1 siRNA reduces leukocyte elastase–induced lung epithelial apoptosis. BEAS-2B cells were treated with leukocyte elastase 0.1 U/ml or buffer 12 h after transfection with siRNA. Values are mean ± SD; ++P < 0.01 compared with the sample of control siRNA transfection and buffer only incubation, **P < 0.01 compared with the sample of control siRNA transfection and leukocyte elastase treatment (n = 5).
Having established conditions where PAR-1 expression was selectively diminished by siRNA, we then assessed the apoptotic response of these cells to treatment with leukocyte elastase. Figure 4D illustrates that under conditions where PAR-1 expression was attenuated using siRNA, leukocyte elastase–induced apoptosis of lung epithelial cells was diminished. By contrast, leukocyte elastase–induced apoptosis was unaffected in lung epithelial cells transfected with control siRNA. It is noteworthy that treatment with siRNA by itself did not induce apoptosis under these conditions. We conclude that that leukocyte elastase–induced apoptosis is mediated at least in part by PAR-1.
Leukocyte Elastase and PAR-1–Activating Agents Increase Mitochondrial Permeability
To explore the mechanisms of apoptosis mediated by PAR-1, we focused initially on mitochondrial-dependent pathways and assessed alterations in mitochondrial membrane potential (ΔΨ) using the fluorescent probe, JC-1 (Figure 5A). Control lung epithelial cells loaded with JC-1 and viewed under a rhodamine filter exhibited a typical red punctuate distribution of JC-1, indicating accumulation of JC-1 aggregates in mitochondria (35). Valinomycin, used here as a positive control, disrupts the ΔΨ and thus JC-1 translocates to cytoplasm reverting to its monomeric form, indicated by more diffuse fluorescence when viewed under a fluorescein filter (Figure 5A, panel g). Similar effects were observed in cells treated with 0.1 U/ml leukocyte elastase (Figure 5A, panel j), 100 μM of PAR-1AP (Figure 5A, panel h), and 1 U/ml of thrombin treatment (Figure 5A, panel i). These mitochondrial alterations were apparent as early as 30 min after treatment, persisted for at least 3 h, and were dose-dependent (data not shown). Taken together, these data suggest that PAR-1 activation, either by direct receptor activation (PAR-1AP) or by proteolytic cleavage (thrombin or leukocyte elastase), induces apoptosis via alterations in the mitochondrial membrane permeability of lung epithelial cells.
Figure 5.

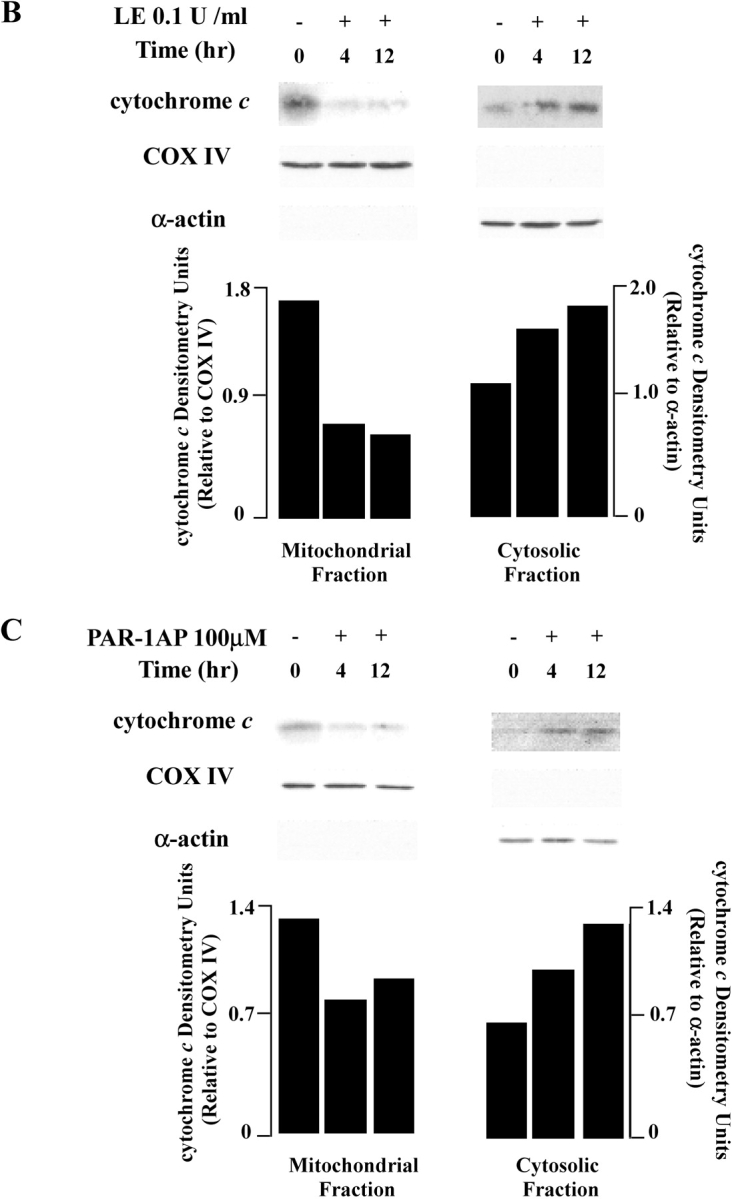
Leukocyte elastase (LE) and PAR-1 agonists reduce mitochondrial membrane potential (ΔΨ) in BEAS-2B cells. (A) Upper panels represent images acquired with a rhodamine filter, middle panels with an FITC filter, and lower panels show DIC images. Cells were treated with a control peptide 100 μM, valinomicin 1 μM, leukocyte elastase 0.1 U/ml, PAR-1AP 100 μM, and thrombin 1 U/ml for 1 h. Representative data each from one of five experiments are shown. (B and C) Cytochrome c release from mitochondria to cytosol in epithelial cells exposed to leukocyte elastase and PAR-1AP. Cells fractions were collected after 0, 4, 12 h of incubation. COX IV is used as a marker for the mitochondrial fraction and α-actin as a marker for the cytosolic fraction. The graph shows densitometry units normalized for COX IV and α-actin levels. Representative data each from one of four experiments are shown.
Given these changes in ΔΨ, we next examined the release of cytochrome c from the mitochondria to the cytosol as an independent assay of alterations mitochondrial membrane permeability. Subcellular fractions were prepared by cell lysis followed by sucrose density gradient ultracentrifugation with analysis of fractions by Western blotting. As illustrated in Figure 5B, after treatment with leukocyte elastase, the amount of mitochondrial cytochrome c was reduced, whereas cytosolic cytochrome c levels increased. Similar effects were noted after treatment of lung epithelial cells with PAR-1AP (Figure 5C). We conclude that PAR-1AP, thrombin, and leukocyte elastase induce release of cytochrome c to the cytosol, supporting the fluorescence studies indicating that these agents alter mitochondrial membrane permeability.
Leukocyte Elastase and PAR-1 Agonists Activate Caspase-9 and -3
We next sought to determine if downstream caspases were activated under these conditions. These experiments revealed that treatment with either leukocyte elastase or PAR-1AP induced cleavage of caspase-9 and -3 in a time-dependent manner, reaching a maximum at 12 h (Figures 6A and 6B).
Figure 6.

Caspase activation by leukocyte elastase (LE) and PAR-1AP exposure in lung epithelial cells. A illustrates cleavage of caspase-9, and B illustrates cleavage of caspase-3 in BEAS2-B epithelial cells by Western blot analysis. The graph represents quantification by densitometry normalized for levels of α-actin. Representative data each from one of three experiments are shown.
Together, these data indicate that activation of PAR-1, either by specific activating peptide or by proteolytic cleavage, increases mitochondrial permeability with associated release of cytochrome c from the mitochondria to the cytosol, and activates caspase-9 and -3 leading to the biochemical and morphologic alterations of apoptosis. As there are multiple pathways that potentially regulate apoptosis, we explored the role of two pathways involving Akt and JNK in mediating PAR-1–induced apoptosis.
Leukocyte Elastase Modulates Akt Phosphorylation through PAR-1
The serine/threonine kinase Akt is known to suppress apoptosis through phosphorylation and inhibition of caspase-9, and phosphorylation of BAD, one of the proapoptotic Bcl-2 members. To determine if PAR-1 activation by leukocyte elastase modulates apoptosis in lung epithelia via this pathway, we assessed the phosphorylation status of Akt in response to leukocyte elastase and to PAR-1AP. These experiments revealed that the phosphorylation of Akt was reduced by leukocyte elastase in a time-dependent manner, whereas total Akt was unchanged (Figure 7A). As noted above (Figure 6A), cleavage of caspase-9 in response to leukocyte elastase transpired with similar kinetics. By comparison, a similar reduction of phospho-Akt was observed after treatment of cells with PAR-1AP at early time points (1–4 h). However, the alteration in response to PAR-1AP was more evanescent, tending to return to baseline levels by 12 h (Figure 7B). These differences may be related to the kinetics of receptor activation by the peptide as compared with proteolytic cleavage by leukocyte elastase. Importantly, gene silencing with PAR-1 siRNA largely prevented the reduction of Akt phosphorylation in response to leukocyte elastase exposure for 12 h (Figure 7C). These data suggest leukocyte elastase and PAR-1AP may modulate epithelial apoptosis via effects on the phosphorylation status of Akt.
Figure 7.
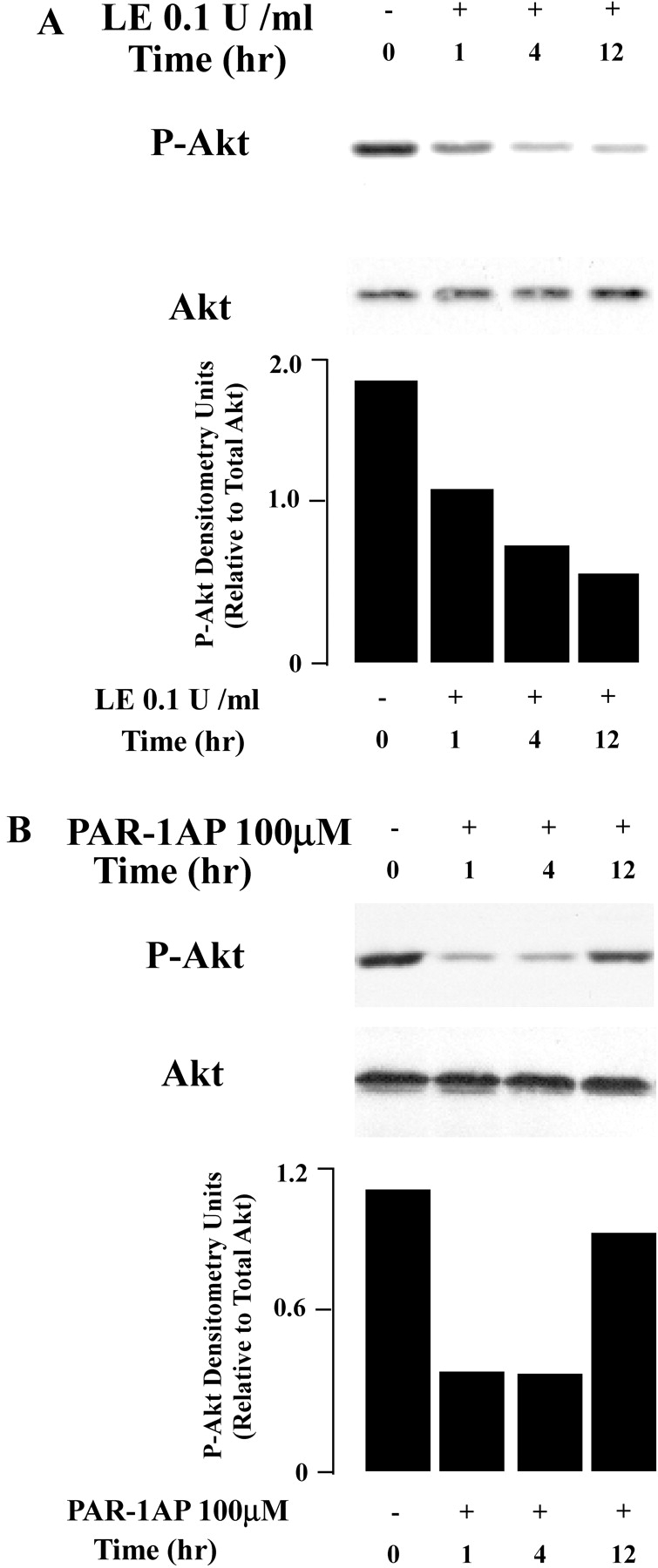

Akt phosphorylation is reduced by exposure to leukocyte elastase (LE) and PAR-1AP. Western analysis of total protein from BEAS-2B cells collected at 0, 1, 4, and 12 h after exposure to leukocyte elastase (A) or PAR-1AP (B). The graph represents quantification by densitometry normalized for levels of total Akt. Representative data from one of five independent experiments are shown. (C) Western analysis of total protein from BEAS-2B cells collected after PAR-1A siRNA or control siRNA transfection followed by incubation with leukocyte elastase or buffer control. Phospho-Akt and total Akt were assessed at 0 and 12 h. The accompanying graph represents quantification by densitometry normalized for levels of total Akt. Representative data from one of three independent experiments are shown.
Leukocyte Elastase Modulates JNK through PAR-1
JNK is a serine/threonine kinase that is activated in response to diverse environmental stresses (36). The role of JNK in regulation of apoptosis is complex and depends on the cell type. JNK has been reported to promote or suppress apoptosis through diverse pathways, including mitochondrial, nuclear factor (NF)-κB–, or AP-1–dependent pathways (37). To determine if leukocyte elastase–induced epithelial apoptosis involves JNK, we examined JNK phosphorylation using Western blot analysis. These studies revealed that leukocyte elastase treatment of lung epithelia induces rapid phosphorylation of JNK peaking at 30 min and returning to baseline by 12 h (Figure 8A). Similar responses were recorded after PAR-1AP treatment of epithelial cells (Figure 8B). To determine the relationship between JNK activation and apoptosis, we treated cells with a specific pharmacologic inhibitor of JNK, SP600125, and monitored apoptosis in response to leukocyte elastase and PAR-1AP. As illustrated in Figure 8D, leukocyte elastase–induced epithelial apoptosis was enhanced when JNK phosphorylation was blocked by SP600125 at a concentration of 10 μM (Figure 8C). We conclude that activation of JNK under these conditions provides an anti-apoptotic signal.
Figure 8.
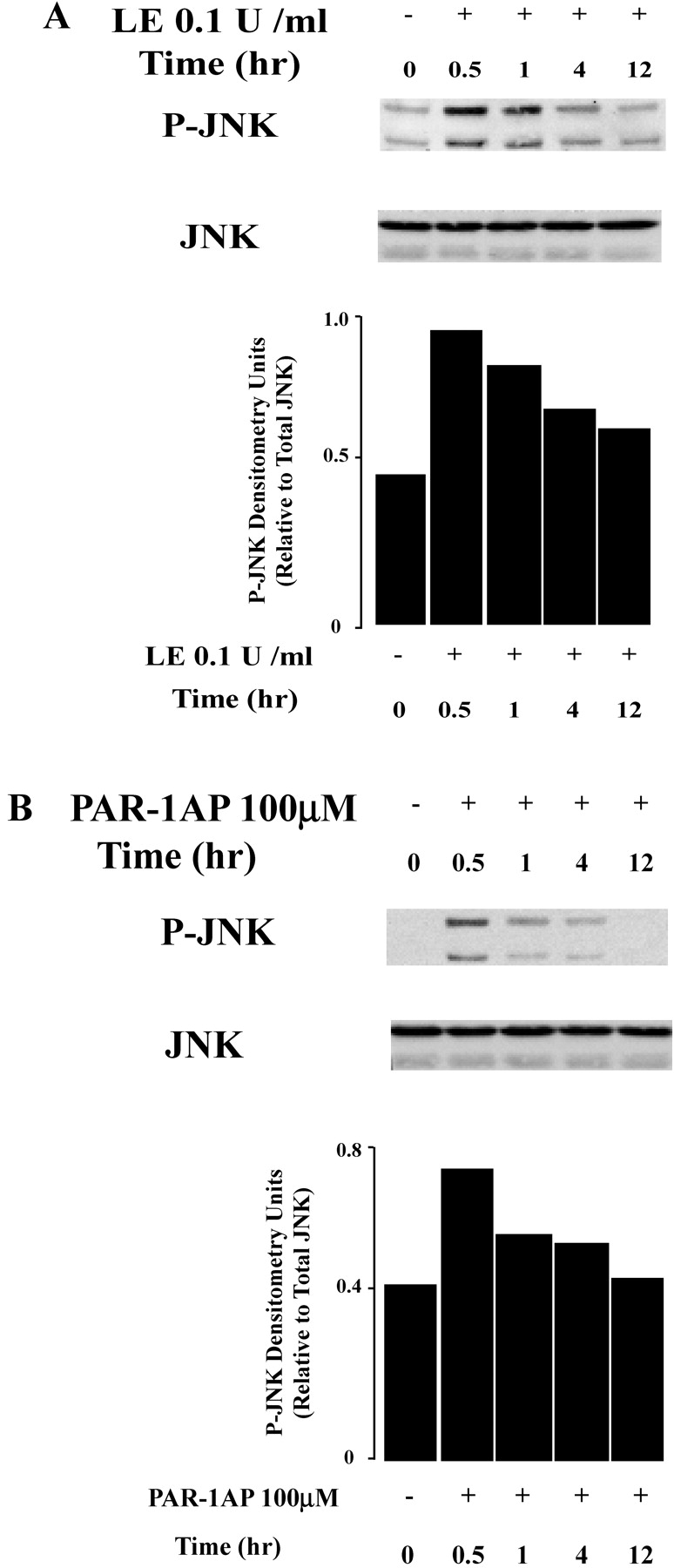
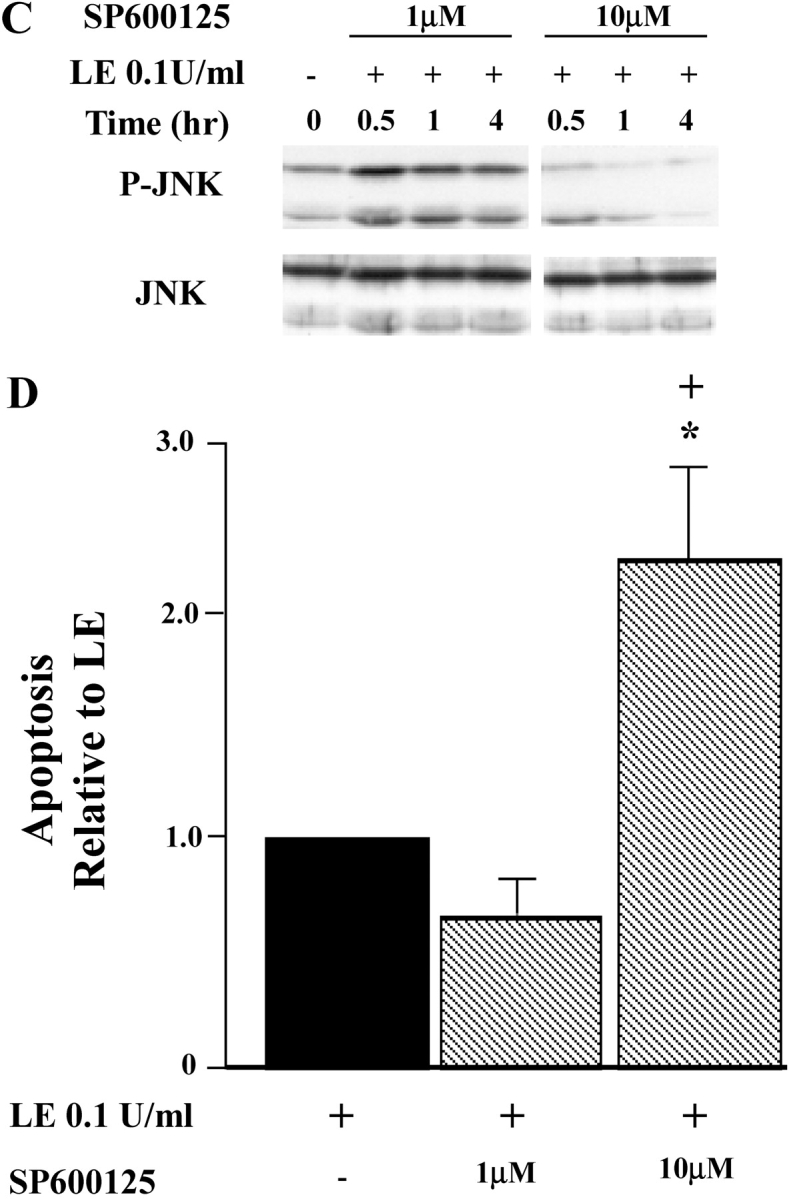
JNK phosphorylation is induced by exposure to leukocyte elastase (LE) and PAR-1AP. BEAS-2B cells were treated with leukocyte elastase (A) and PAR-1AP (B), and total protein was collected at 0, 0.5, 1, 4, and 12 h. The graph represents quantification by densitometry normalized for levels of total JNK. Representative data from one of four independent experiments are shown. (C) Cells were treated with SP600125, a specific JNK inhibitor 1 h before exposure to leukocyte elastase and PAR-1AP. Western blot analysis was used to assess levels of phospho-JNK and total JNK. (D) Treatment of cells with a JNK inhibitor enhances leukocyte elastase–induced epithelial apoptosis. Cells were pretreated with SP600125 1 μM and 10 μM and exposed to leukocyte elastase for an additional 12 h. The graph represents apoptosis relative to leukocyte elastase treatment samples. Values are mean ± SD; *P < 0.05 compared with the samples without SP600125; + P < 0.05 compared with the samples with SP600125 1 μM; n = 3.
DISCUSSION
In this study, we provide evidence that leukocyte elastase induces apoptosis of human lung airway and alveolar epithelial cells through PAR-1. Our data indicate that the pro-apoptotic effects of leukocyte elastase are mediated via the intrinsic apoptotic pathway involving alterations in mitochondrial permeability (as manifest by decrease in ΔΨ), release of cytochrome c into the cytosol, and activation of caspase-9 and -3. In addition, exposure of epithelial cells to leukocyte elastase, in a PAR-1–dependent manner, results in inhibition of Akt and activation of JNK, a member of the MAP kinase family. Both of these effects modulate apoptosis. These observations have important implications for our understanding of the pathogenesis of acute and chronic lung injury including emphysema where inappropriate apoptosis induced by serum or leukocyte-derived proteinases, may contribute to lung epithelial injury and dysfunction.
In the current study, leukocyte elastase, thrombin, and PAR-1–activating peptide all induced apoptosis of human lung epithelial cells with a similar (but not identical) pattern. The similarity in responses to leukocyte elastase and to “authentic” PAR-1 activating agents supports the notion that the effects of elastase are mediated via PAR-1. Consistent with this notion, epithelial apoptosis induced by leukocyte elastase was largely prevented by the selective PAR-1 antagonist, SCH79797 (Figure 4A). Additional and very strong evidence for a role of PAR-1 in elastase-induced epithelial apoptosis is provided by the studies using RNA interference (Figure 4), where PAR-1 siRNA diminished elastase-induced apoptosis by more than 50%. Although both PAR-1–activating peptide and thrombin activate PAR-1 directly (26, 27), the ability of leukocyte elastase to activate PAR-1 has not been previously reported. It is known that leukocyte serine proteinases including elastase, cathepsin G, and proteinase 3 can cleave and inactivate (“disarm”) PAR-1 on human endothelial cells (38). However, in the case of PAR-2, proteolytic cleavage of the receptor by leukocyte elastase can either activate or inactivate the receptor, depending on the site of cleavage (39, 40). By analogy, it is possible that proteolytic cleavage of PAR-1 might also activate or inactivate the receptor depending on the site of cleavage. Our data obtained with the use of the nonpeptide PAR-1 antagonist, SCH 79797, demonstrate clearly that the apoptotic action of leukocyte elastase is due to PAR-1 activation (41). Factors that could influence the site and extent of cleavage of the receptor include the extent of glycosylation of the receptor, the ratio of enzyme to receptor, and the presence of endogenous antiproteinases such as SLPI. These factors might differ between endothelial and epithelial cells and provide an explanation for differential responses between endothelial and epithelial cells. It is also possible that the effects of leukocyte elastase on PAR-1 are indirect and mediated for example, by release of surface bound proteinases such as matrix metalloproteinases (MMPs) that could then cleave and activate PAR-1. Further, the effects of leukocyte elastase might be mediated in part through other epithelial plasma membrane receptors or by release of surface-bound molecules such as growth factors that could in turn bind to their cognate receptors and initiate pro-apoptotic signaling pathways. Thus, although our data clearly indicate a role for PAR-1 in transducing the pro-apoptotic effects of leukocyte elastase, whether these are direct or indirect effects of the enzyme on the receptor remains to be clarified.
The involvement of PAR-1 in regulation of apoptosis has been recently recognized in other cell types including neurons, endothelium, intestinal epithelium, lung fibroblasts, and melanoma cells, suggesting a more generalized mechanism (28, 42–45). Under these circumstances, PAR-1 has been linked to both pro- and anti-apoptotic effects depending on the cell type and the experimental conditions (46). For example, activation of PAR-1 by thrombin can either induce or inhibit apoptosis in cultured neurons and astrocytes (47, 48). In gastrointestinal epithelium, activation of PAR-1 increases apoptosis and intestinal permeability in a caspase-3–dependent fashion (28). In the current study, we demonstrate that leukocyte elastase induces apoptosis in human lung epithelial cells of either airway (BEAS-2B and SAEC) or alveolar (primary alveolar type II cells) origin. The generalizability of these responses across a broad range of lung epithelial cell types supports their physiologic importance.
In the current studies, we noted some differences in the effects of PAR-1–activating peptide and leukocyte elastase on the kinetics of apoptosis (Figure 3). This might be explained by both rapid activation and termination of signals from PAR-1 by the activating peptide compared with more gradual but protracted activation of the receptor by leukocyte elastase. Further, leukocyte elastase may have additional effects including the ability to activate multiple receptors, the ability to generate cleaved peptides with biological activity, and the ability to alter cellular processes independently of cell surface receptor modifications as discussed above.
In the present study, the concentration of leukocyte elastase was chosen on the basis of estimates of the amount of elastase released by neutrophils in vitro (7). In this regard, it is important to appreciate that when leukocytes such as neutrophils are in close proximity to lung parenchymal cells, proteinases can be released into a “protected space” that forms between the epithelial cell and the neutrophil (2, 49). Large molecular weight anti-proteinases do not have access to this space, promoting the unopposed action of proteinases. Moreover, activated neutrophils release reactive oxygen species, which can inactivate anti-proteinases. Thus, it is likely that adjacent epithelial or endothelial cells are exposed to even higher local concentrations of proteinases.
Although it has been previously described that leukocyte-derived proteinases can lead to epithelial apoptosis (8, 50), the mechanisms remain incompletely understood. Research has focused on mechanisms involving modulation of plasma membrane receptors of the death domain family (i.e., the extrinsic pathway). Expression of Fas, a prototypical member of the death-receptor family, is increased in the alveolar epithelium of patients with ARDS and importantly, soluble Fas ligand (FasL) levels are elevated in the BAL fluid of patients with ARDS (51, 52). More recently, Nakamura and coworkers reported that proinflammatory cytokines sensitize the lung epithelium to Fas-induced cell death (53). As leukocyte elastase enhances production of proinflammatory cytokines such as IL-8 and IL-1β, it is possible that this effect may also lead to apoptosis. Elastase exposure has also been shown to increase expression of FasL in liver cells directly (54, 55). By analogy, a similar mechanism may be applicable to ALI/ARDS. Although our results indicate a role for the intrinsic apoptotic pathway, this does not exclude a potential role for the Fas–FasL pathway.
Diverse and interconnected signaling molecules are known to be involved in apoptosis including NF-κB, mitogen-activated protein (MAP) kinases, phosphatidylinositide 3-kinase (PI3K), and tyrosine kinases (44, 46, 56, 57). In the current study, we focused on two additional signaling molecules modulated by leukocyte elastase that lie downstream of PAR-1. The first of these is Akt, a serine/threonine kinase that is known to suppress apoptosis. Akt inactivates the pro-apoptotic protein BAD and caspase-9 by phosphorylation (58). We recently reported that treatment of GI epithelial cells with a pharmacologic inhibitor of Akt, amplifies leukocyte elastase–induced apoptosis (7) and we have observed similar effects in lung epithelial cells (data not shown). Thus under the conditions of our model system, Akt exerts primarily an anti-apoptotic influence and this is prevented by leukocyte elastase in a PAR-1–dependent manner.
In the current study, we provide evidence that elastase-induced activation of JNK, a member of the MAP kinase family, modulates apoptosis. The role of JNK in apoptosis is complex and both pro- and anti-apoptotic effects have been reported (59–63). Evidence for pro-apoptotic effects of JNK includes the observation that mouse embryonic fibroblasts deficient in both JNK1 and JNK2 are resistant to apoptosis (59). In addition, JNK stimulates BAX translocation to the mitochondria in ischemia-induced neuronal apoptosis (64) and phosphorylates Bcl-2 interacting mediators of cell death (BimEL) in apoptosis induced via the p75 neurotrophin receptor (p75NTR) (65). Conversely, JNK activation may promote cell survival. For example, JNK1−/− and JNK2−/− embryos exhibit markedly increased apoptosis in the forebrain (66, 67). In addition, downregulation of JNK using antisense oligonucleotides results in increased apoptosis in human breast and colorectal carcinoma cells (68). In our current study, we observed that JNK phosphorylation rapidly (30–60 min) increased after exposure of epithelial cells to leukocyte elastase and PAR-1–activating peptide. Importantly, inhibition of JNK amplified leukocyte elastase–induced apoptosis indicating that under these circumstances, JNK exerts a net anti-apoptotic effect. These data do not preclude the possibility that JNK exerts both pro- and anti-apoptotic effects with the anti-apoptotic effect predominating under the conditions of the experiments. There is evidence that MAP kinases including JNK lie downstream of PAR-1 (69), but to our knowledge, this is the first report linking PAR-1 modulation of JNK to apoptosis.
A substantial body of experimental and clinical data support the role of apoptosis in the pathogenesis of inflammatory tissue injury in critical illness including acute lung injury and ARDS, myocardial infarction, and renal and gastrointestinal tract failure dysfunction in sepsis (52, 70–72). Although apoptosis is vital in many physiologically important processes, either inappropriate activation or inhibition of apoptosis can lead to disease, either because cells have prolonged survival (ongoing inflammation) or because they die prematurely and produce structural and functional changes in normal tissue. Similarly, in the context of ALI and ARDS, apoptosis may be either beneficial or harmful. For example, apoptosis of neutrophils may reduce inflammatory injury by induction of an anti-inflammatory phenotype in activated alveolar macrophages (72) and epithelial apoptosis is necessary for remodeling of tissue during repair. Conversely, during the acute phase, excessive epithelial apoptosis may aggravate lung injury by compromise of the alveolo–capillary membrane. Indeed, in both animal and human studies, increasing evidence suggests that apoptosis of distal lung epithelial cells appears to be pivotal in the pathogenesis of ALI (8, 10, 50). With lung epithelial loss, the permeability of alveolar capillary barrier is increased, and activated neutrophils as well as plasma proteins (thrombin) move from the bloodstream into the alveolar space. In this milieu, there is potential for alveolar epithelial cells to be exposed to PAR-1–activating proteases that may induce apoptosis and thus exacerbate lung injury. Therapeutic intervention aimed at modulating pro-apoptotic signals from PAR-1 might be considered under these circumstances. Clearly the balance between pro-apoptotic and anti-apoptotic factors in ALI is crucial and any therapeutic strategies directed at these processes must bear this in mind.
In summary, we have provided strong evidence that leukocyte elastase, acting via PAR-1, induces apoptosis of human lung epithelial cells by a mechanism involving increased mitochondrial permeability, release of cytocrome c to the cytosol, and activation of caspase-9 and -3 leading to apoptosis. In addition, PAR-1 activation by leukocyte elastase leads to inhibition of Akt and activation of JNK, responses that are pro-apoptotic. Together, these observations provide a potential mechanistic explanation for apoptosis of lung epithelial induced by exposure to leukocyte-derived proteinases, circumstances that are relevant to inflammatory lung injury such as occurs in ALI and ARDS (8, 11) as well as in chronic obstructive lung disease (11, 73).
This work was supported by operating grants from the Canadian Institutes of Health Research (to G.D. and J.M.), the block term grant from the Ontario Thoracic Society (to G.D.), and the National Institutes of Health, National Heart, Lung, and Blood Institute (HL51856 and P50HL74005 to M.M.) T. Suzuki is a recipient of a Research Fellowship from the Canadian Institutes of Health Research. T.M. is supported by a Duncan Gordon Fellowship from the Hospital for Sick Children. G.D. holds the R. Fraser Elliott Chair in Transplantation Research from the Toronto General Hospital of the University Health Network, is the recipient of a Tier 1 Canada Research Chair in Respiration, and is a scholar of the McLaughlin Center for Molecular Medicine, University of Toronto.
Conflict of Interest Statement: None of the authors have a financial relationship with a commercial entity that has an interest in the subject of this manuscript.
References
- 1.Havemann K, Gramse M. Physiology and pathophysiology of neutral proteinases of human granulocytes. Adv Exp Med Biol 1984;167:1–20. [DOI] [PubMed] [Google Scholar]
- 2.Lee WL, Downey GP. Leukocyte elastase: physiological functions and role in acute lung injury. Am J Respir Crit Care Med 2001;164:896–904. [DOI] [PubMed] [Google Scholar]
- 3.Cochrane CG, Spragg RG, Revak SD, Cohen AB, McGuire WW. The presence of neutrophil elastase and evidence of oxidation activity in bronchoalveolar lavage fluid of patients with adult respiratory distress syndrome. Am Rev Respir Dis 1983;127:S25–S27. [DOI] [PubMed] [Google Scholar]
- 4.Collins JF, Anzueto AA, Peters JI, de los Santos R, Gonzalez DC, Johanson WG Jr, Seidenfeld JJ, Coalson JJ, Jenkinson SG. Elastase activity in bronchoalveolar lavage fluid from oxygen-exposed, Pseudomonas-infected baboons. Lung 1991;169:165–179. [DOI] [PubMed] [Google Scholar]
- 5.Suter PM, Suter S, Girardin E, Roux-Lombard P, Grau GE, Dayer JM. High bronchoalveolar levels of tumor necrosis factor and its inhibitors, interleukin-1, interferon, and elastase, in patients with adult respiratory distress syndrome after trauma, shock, or sepsis. Am Rev Respir Dis 1992;145:1016–1022. [DOI] [PubMed] [Google Scholar]
- 6.Ginzberg HH, Cherapanov V, Dong Q, Cantin A, McCulloch CA, Shannon PT, Downey GP. Neutrophil-mediated epithelial injury during transmigration: role of elastase. Am J Physiol Gastrointest Liver Physiol 2001;281:G705–G717. [DOI] [PubMed] [Google Scholar]
- 7.Ginzberg HH, Shannon PT, Suzuki T, Hong O, Vachon E, Moraes T, Abreu MT, Cherepanov V, Wang X, Chow CW, et al. Leukocyte elastase induces epithelial apoptosis: role of mitochondial permeability changes and Akt. Am J Physiol Gastrointest Liver Physiol 2004;287:G286–G298. [DOI] [PubMed] [Google Scholar]
- 8.Bardales RH, Xie SS, Schaefer RF, Hsu SM. Apoptosis is a major pathway responsible for the resolution of type II pneumocytes in acute lung injury. Am J Pathol 1996;149:845–852. [PMC free article] [PubMed] [Google Scholar]
- 9.Martin TR, Nakamura M, Matute-Bello G. The role of apoptosis in acute lung injury. Crit Care Med 2003;31:S184–S188. [DOI] [PubMed] [Google Scholar]
- 10.Kawasaki M, Kuwano K, Hagimoto N, Matsuba T, Kunitake R, Tanaka T, Maeyama T, Hara N. Protection from lethal apoptosis in lipopolysaccharide-induced acute lung injury in mice by a caspase inhibitor. Am J Pathol 2000;157:597–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li X, Shu R, Filippatos G, Uhal BD. Apoptosis in lung injury and remodeling. J Appl Physiol 2004;97:1535–1542. [DOI] [PubMed] [Google Scholar]
- 12.Calabrese F, Giacometti C, Beghe B, Rea F, Loy M, Zuin R, Marulli G, Baraldo S, Saetta M, Valente M. Marked alveolar apoptosis/proliferation imbalance in end-stage emphysema. Respir Res 2005;6:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aoshiba K, Yokohori N, Nagai A. Alveolar wall apoptosis causes lung destruction and emphysematous changes. Am J Respir Cell Mol Biol 2003;28:555–562. [DOI] [PubMed] [Google Scholar]
- 14.Hajra KM, Liu JR. Apoptosome dysfunction in human cancer. Apoptosis 2004;9:691–704. [DOI] [PubMed] [Google Scholar]
- 15.Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science 1998;281:1305–1308. [DOI] [PubMed] [Google Scholar]
- 16.Budihardjo I, Oliver H, Lutter M, Luo X, Wang X. Biochemical pathways of caspase activation during apoptosis. Annu Rev Cell Dev Biol 1999;15:269–290. [DOI] [PubMed] [Google Scholar]
- 17.Erlich JH, Boyle EM, Labriola J, Kovacich JC, Santucci RA, Fearns C, Morgan EN, Yun W, Luther T, Kojikawa O, et al. Inhibition of the tissue factor-thrombin pathway limits infarct size after myocardial ischemia-reperfusion injury by reducing inflammation. Am J Pathol 2000;157:1849–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Quera R, Shanahan F. Thromboembolism–an important manifestation of inflammatory bowel disease. Am J Gastroenterol 2004;99:1971–1973. [DOI] [PubMed] [Google Scholar]
- 19.Schultz MJ, Millo J, Levi M, Hack CE, Weverling GJ, Garrard CS, van der Poll T. Local activation of coagulation and inhibition of fibrinolysis in the lung during ventilator associated pneumonia. Thorax 2004;59:130–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bone RC, Francis PB, Pierce AK. Intravascular coagulation associated with the adult respiratory distress syndrome. Am J Med 1976;61:585–589. [DOI] [PubMed] [Google Scholar]
- 21.Idell S, Koenig KB, Fair DS, Martin TR, McLarty J, Maunder RJ. Serial abnormalities of fibrin turnover in evolving adult respiratory distress syndrome. Am J Physiol 1991;261:L240–L248. [DOI] [PubMed] [Google Scholar]
- 22.Macfarlane SR, Seatter MJ, Kanke T, Hunter GD, Plevin R. Proteinase-activated receptors. Pharmacol Rev 2001;53:245–282. [PubMed] [Google Scholar]
- 23.Hollenberg MD, Compton SJ. International Union of Pharmacology. XXVIII. Proteinase-activated receptors. Pharmacol Rev 2002;54:203–217. [DOI] [PubMed] [Google Scholar]
- 24.Asokananthan N, Graham PT, Fink J, Knight DA, Bakker AJ, McWilliam AS, Thompson PJ, Stewart GA. Activation of protease-activated receptor (PAR)-1, PAR-2, and PAR-4 stimulates IL-6, IL-8, and prostaglandin E2 release from human respiratory epithelial cells. J Immunol 2002;168:3577–3585. [DOI] [PubMed] [Google Scholar]
- 25.Coughlin SR. Thrombin signalling and protease-activated receptors. Nature 2000;407:258–264. [DOI] [PubMed] [Google Scholar]
- 26.Dery O, Corvera CU, Steinhoff M, Bunnett NW. Proteinase-activated receptors: novel mechanisms of signaling by serine proteases. Am J Physiol 1998;274:C1429–C1452. [DOI] [PubMed] [Google Scholar]
- 27.Buresi MC, Schleihauf E, Vergnolle N, Buret A, Wallace JL, Hollenberg MD, MacNaughton WK. Protease-activated receptor-1 stimulates Ca(2+)-dependent Cl(-) secretion in human intestinal epithelial cells. Am J Physiol Gastrointest Liver Physiol 2001;281:G323–G332. [DOI] [PubMed] [Google Scholar]
- 28.Chin AC, Vergnolle N, MacNaughton WK, Wallace JL, Hollenberg MD, Buret AG. Proteinase-activated receptor 1 activation induces epithelial apoptosis and increases intestinal permeability. Proc Natl Acad Sci USA 2003;100:11104–11109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jesmin S, Gando S, Matsuda N, Sakuma I, Kobayashi S, Sakuraya F, Hattori Y. Temporal changes in pulmonary expression of key procoagulant molecules in rabbits with endotoxin-induced acute lung injury: elevated expression levels of protease-activated receptors. Thromb Haemost 2004;92:966–979. [DOI] [PubMed] [Google Scholar]
- 30.Moffatt JD, Jeffrey KL, Cocks TM. Protease-activated receptor-2 activating peptide SLIGRL inhibits bacterial lipopolysaccharide-induced recruitment of polymorphonuclear leukocytes into the airways of mice. Am J Respir Cell Mol Biol 2002;26:680–684. [DOI] [PubMed] [Google Scholar]
- 31.Frank J, Roux J, Kawakatsu H, Su G, Dagenais A, Berthiaume Y, Howard M, Canessa CM, Fang X, Sheppard D, et al. Transforming growth factor-beta1 decreases expression of the epithelial sodium channel alphaENaC and alveolar epithelial vectorial sodium and fluid transport via an ERK1/2-dependent mechanism. J Biol Chem 2003;278:43939–43950. [DOI] [PubMed] [Google Scholar]
- 32.Wang Q, Downey GP, Herrera-Abreu MT, Kapus A, McCulloch CA. SHP-2 modulates IL-1-induced Ca2+ flux and ERK activation via phosphorylation of PLCgamma 1. J Biol Chem 2005;280:8397–8406. [DOI] [PubMed] [Google Scholar]
- 33.Scacheri PC, Rozenblatt-Rosen O, Caplen NJ, Wolfsberg TG, Umayam L, Lee JC, Hughes CM, Shanmugam KS, Bhattacharjee A, Meyerson M, et al. Short interfering RNAs can induce unexpected and divergent changes in the levels of untargeted proteins in mammalian cells. Proc Natl Acad Sci USA 2004;101:1892–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ma L, Perini R, McKnight W, Dicay M, Klein A, Hollenberg MD, Wallace JL. Proteinase-activated receptors 1 and 4 counter-regulate endostatin and VEGF release from human platelets. Proc Natl Acad Sci USA 2005;102:216–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xu M, Wang Y, Ayub A, Ashraf M. Mitochondrial K(ATP) channel activation reduces anoxic injury by restoring mitochondrial membrane potential. Am J Physiol Heart Circ Physiol 2001;281:H1295–H1303. [DOI] [PubMed] [Google Scholar]
- 36.Yang DD, Kuan CY, Whitmarsh AJ, Rincon M, Zheng TS, Davis RJ, Rakic P, Flavell RA. Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the Jnk3 gene. Nature 1997;389:865–870. [DOI] [PubMed] [Google Scholar]
- 37.Lin A. Activation of the JNK signaling pathway: breaking the brake on apoptosis. Bioessays 2003;25:17–24. [DOI] [PubMed] [Google Scholar]
- 38.Renesto P, Si-Tahar M, Moniatte M, Balloy V, Van Dorsselaer A, Pidard D, Chignard M. Specific inhibition of thrombin-induced cell activation by the neutrophil proteinases elastase, cathepsin G, and proteinase 3: evidence for distinct cleavage sites within the aminoterminal domain of the thrombin receptor. Blood 1997;89:1944–1953. [PubMed] [Google Scholar]
- 39.Uehara A, Muramoto K, Takada H, Sugawara S. Neutrophil serine proteinases activate human nonepithelial cells to produce inflammatory cytokines through protease-activated receptor 2. J Immunol 2003;170:5690–5696. [DOI] [PubMed] [Google Scholar]
- 40.Dulon S, Leduc D, Cottrell GS, D'Alayer J, Hansen KK, Bunnett NW, Hollenberg MD, Pidard D, Chignard M. Pseudomonas aeruginosa elastase disables PAR2 in respiratory epithelial cells. Am J Respir Cell Mol Biol 2005;32:411–419. [DOI] [PubMed] [Google Scholar]
- 41.Ahn HS, Foster C, Boykow G, Stamford A, Manna M, Graziano M. Inhibition of cellular action of thrombin by N3-cyclopropyl-7-[[4-(1-methylethyl)phenyl]methyl]-7H-pyrrolo[3, 2-f]quinazoline-1,3-diamine (SCH 79797), a nonpeptide thrombin receptor antagonist. Biochem Pharmacol 2000;60:1425–1434. [DOI] [PubMed] [Google Scholar]
- 42.Turgeon VL, Milligan CE, Houenou LJ. Activation of the protease-activated thrombin receptor (PAR)-1 induces motoneuron degeneration in the developing avian embryo. J Neuropathol Exp Neurol 1999;58:499–504. [DOI] [PubMed] [Google Scholar]
- 43.Wang H, Ubl JJ, Stricker R, Reiser G. Thrombin (PAR-1)-induced proliferation in astrocytes via MAPK involves multiple signaling pathways. Am J Physiol Cell Physiol 2002;283:C1351–C1364. [DOI] [PubMed] [Google Scholar]
- 44.Chalmers CJ, Balmanno K, Hadfield K, Ley R, Cook SJ. Thrombin inhibits Bim (Bcl-2-interacting mediator of cell death) expression and prevents serum-withdrawal-induced apoptosis via protease-activated receptor 1. Biochem J 2003;375:99–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cheng T, Liu D, Griffin JH, Fernandez JA, Castellino F, Rosen ED, Fukudome K, Zlokovic BV. Activated protein C blocks p53-mediated apoptosis in ischemic human brain endothelium and is neuroprotective. Nat Med 2003;9:338–342. [DOI] [PubMed] [Google Scholar]
- 46.Flynn AN, Buret AG. Proteinase-activated receptor 1 (PAR-1) and cell apoptosis. Apoptosis 2004;9:729–737. [DOI] [PubMed] [Google Scholar]
- 47.Donovan FM, Pike CJ, Cotman CW, Cunningham DD. Thrombin induces apoptosis in cultured neurons and astrocytes via a pathway requiring tyrosine kinase and RhoA activities. J Neurosci 1997;17:5316–5326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huang YQ, Li JJ, Karpatkin S. Thrombin inhibits tumor cell growth in association with up-regulation of p21(waf/cip1) and caspases via a p53-independent, STAT-1-dependent pathway. J Biol Chem 2000;275:6462–6468. [DOI] [PubMed] [Google Scholar]
- 49.Rice WG, Weiss SJ. Regulation of proteolysis at the neutrophil-substrate interface by secretory leukoprotease inhibitor. Science 1990;249:178–181. [DOI] [PubMed] [Google Scholar]
- 50.Bachofen M, Weibel ER. Structural alterations of lung parenchyma in the adult respiratory distress syndrome. Clin Chest Med 1982;3:35–56. [PubMed] [Google Scholar]
- 51.Albertine KH, Soulier MF, Wang Z, Ishizaka A, Hashimoto S, Zimmerman GA, Matthay MA, Ware LB. Fas and fas ligand are up-regulated in pulmonary edema fluid and lung tissue of patients with acute lung injury and the acute respiratory distress syndrome. Am J Pathol 2002;161:1783–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Matute-Bello G, Liles WC, Steinberg KP, Kiener PA, Mongovin S, Chi EY, Jonas M, Martin TR. Soluble Fas ligand induces epithelial cell apoptosis in humans with acute lung injury (ARDS). J Immunol 1999;163:2217–2225. [PubMed] [Google Scholar]
- 53.Nakamura M, Matute-Bello G, Liles WC, Hayashi S, Kajikawa O, Lin SM, Frevert CW, Martin TR. Differential response of human lung epithelial cells to fas-induced apoptosis. Am J Pathol 2004;164:1949–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yang J, Gallagher SF, Haines K, Epling-Burnette PK, Bai F, Gower WR Jr, Mastorides S, Norman JG, Murr MM. Kupffer cell-derived Fas ligand plays a role in liver injury and hepatocyte death. J Gastrointest Surg 2004;8:166–174. [DOI] [PubMed] [Google Scholar]
- 55.Gallagher SF, Yang J, Baksh K, Haines K, Carpenter H, Epling-Burnette PK, Peng Y, Norman J, Murr MM. Acute pancreatitis induces FasL gene expression and apoptosis in the liver. J Surg Res 2004;122:201–209. [DOI] [PubMed] [Google Scholar]
- 56.Lidington EA, Haskard DO, Mason JC. Induction of decay-accelerating factor by thrombin through a protease-activated receptor 1 and protein kinase C-dependent pathway protects vascular endothelial cells from complement-mediated injury. Blood 2000;96:2784–2792. [PubMed] [Google Scholar]
- 57.Rahman A, True AL, Anwar KN, Ye RD, Voyno-Yasenetskaya TA, Malik AB. Galpha(q) and Gbetagamma regulate PAR-1 signaling of thrombin-induced NF-kappaB activation and ICAM-1 transcription in endothelial cells. Circ Res 2002;91:398–405. [DOI] [PubMed] [Google Scholar]
- 58.Kennedy SG, Kandel ES, Cross TK, Hay N. Akt/Protein kinase B inhibits cell death by preventing the release of cytochrome c from mitochondria. Mol Cell Biol 1999;19:5800–5810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tournier C, Hess P, Yang DD, Xu J, Turner TK, Nimnual A, Bar-Sagi D, Jones SN, Flavell RA, Davis RJ. Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science 2000;288:870–874. [DOI] [PubMed] [Google Scholar]
- 60.Sanna MG, da Silva Correia J, Luo Y, Chuang B, Paulson LM, Nguyen B, Deveraux QL, Ulevitch RJ. ILPIP, a novel anti-apoptotic protein that enhances XIAP-mediated activation of JNK1 and protection against apoptosis. J Biol Chem 2002;277:30454–30462. [DOI] [PubMed] [Google Scholar]
- 61.Deng Y, Ren X, Yang L, Lin Y, Wu X. A JNK-dependent pathway is required for TNFalpha-induced apoptosis. Cell 2003;115:61–70. [DOI] [PubMed] [Google Scholar]
- 62.Lamb JA, Ventura JJ, Hess P, Flavell RA, Davis RJ. JunD mediates survival signaling by the JNK signal transduction pathway. Mol Cell 2003;11:1479–1489. [DOI] [PubMed] [Google Scholar]
- 63.Stewart CE, Newcomb PV, Holly JM. Multifaceted roles of TNF-alpha in myoblast destruction: a multitude of signal transduction pathways. J Cell Physiol 2004;198:237–247. [DOI] [PubMed] [Google Scholar]
- 64.Okuno S, Saito A, Hayashi T, Chan PH. The c-Jun N-terminal protein kinase signaling pathway mediates bax activation and subsequent neuronal apoptosis through interaction with bim after transient focal cerebral ischemia. J Neurosci 2004;24:7879–7887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Becker EB, Howell J, Kodama Y, Barker PA, Bonni A. Characterization of the c-Jun N-terminal kinase-BimEL signaling pathway in neuronal apoptosis. J Neurosci 2004;24:8762–8770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kuan CY, Yang DD, Samanta Roy DR, Davis RJ, Rakic P, Flavell RA. The Jnk1 and Jnk2 protein kinases are required for regional specific apoptosis during early brain development. Neuron 1999;22:667–676. [DOI] [PubMed] [Google Scholar]
- 67.Sabapathy K, Jochum W, Hochedlinger K, Chang L, Karin M, Wagner EF. Defective neural tube morphogenesis and altered apoptosis in the absence of both JNK1 and JNK2. Mech Dev 1999;89:115–124. [DOI] [PubMed] [Google Scholar]
- 68.Potapova O, Gorospe M, Dougherty RH, Dean NM, Gaarde WA, Holbrook NJ. Inhibition of c-Jun N-terminal kinase 2 expression suppresses growth and induces apoptosis of human tumor cells in a p53-dependent manner. Mol Cell Biol 2000;20:1713–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Marinissen MJ, Servitja JM, Offermanns S, Simon MI, Gutkind JS. Thrombin protease-activated receptor-1 signals through Gq- and G13-initiated MAPK cascades regulating c-Jun expression to induce cell transformation. J Biol Chem 2003;278:46814–46825. [DOI] [PubMed] [Google Scholar]
- 70.Abbate A, Biondi-Zoccai GG, Bussani R, Dobrina A, Camilot D, Feroce F, Rossiello R, Baldi F, Silvestri F, Biasucci LM, et al. Increased myocardial apoptosis in patients with unfavorable left ventricular remodeling and early symptomatic post-infarction heart failure. J Am Coll Cardiol 2003;41:753–760. [DOI] [PubMed] [Google Scholar]
- 71.Chan LY, Leung JC, Lai KN. Novel mechanisms of tubulointerstitial injury in IgA nephropathy: a new therapeutic paradigm in the prevention of progressive renal failure. Clin Exp Nephrol 2004;8:297–303. [DOI] [PubMed] [Google Scholar]
- 72.Matute-Bello G, Martin TR. Science review: apoptosis in acute lung injury. Crit Care 2003;7:355–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Majo J, Ghezzo H, Cosio MG. Lymphocyte population and apoptosis in the lungs of smokers and their relation to emphysema. Eur Respir J 2001;17:946–953. [DOI] [PubMed] [Google Scholar]