

Abstract
The Coxsackievirus B and Adenovirus Receptor (CAR) plays a dual role as a homotypic junctional adhesion protein and as a viral receptor. CAR is a transmembrane protein and a member of the Immunoglobulin (Ig) superfamily with two extracellular Ig-like domains. The most distal Ig-like domain (D1) mediates the homophilic interaction and is also responsible for the high-affinity binding of the adenovirus (Ad) fiber protein. Currently, no activity has been ascribed to the proximal Ig-like domain (D2). To further understand the function of the extracellular domain in the biological activities of CAR, we created extracellular deletion mutants and evaluated cellular localization, adhesion, and viral infection. Deletion of any segment of the extracellular domain results in loss of adhesion and mislocalization as explained by a model, termed “diffusion trapping,” that suggests adhesion is the driving force in junctional localization. Loss of junctional localization and adhesion was particularly apparent in polarized human airway epithelia, where mutant CAR expression was basolateral but not limited to the lateral junctions between cells. Surprisingly, the D2 domain was required for adenovirus fiber-knob binding and infection. In summary, the entire extracellular domain of CAR is of vital importance to the biology of this highly conserved and important protein.
Keywords: adenovirus, CAR, Coxsackievirus, extracellular domain, receptor
The Coxsackievirus B and Adenovirus Receptor (CAR) is a member of the immunoglobulin (Ig) superfamily with one variable and one constant Ig domain (termed D1 and D2, respectively), a single-pass transmembrane domain, and a cytoplasmic domain. Although our understanding of the function of this molecule is incomplete, CAR has been implicated in several distinct biological roles. These include acting as a viral receptor, adhesion protein, and growth modulator, and recently it was found to traffic certain PDZ domain containing proteins to cell:cell junctions (1, 2).
The homotypic CAR:CAR and viral:CAR interactions have been delimited to the D1 domain in several studies (3–9). The fiber knob of adenovirus (Ad) uses the same surface on the D1 domain for binding CAR as is used for CAR:CAR homodimer formation. Fiber knob binding is predicted to have ∼ 1,000-fold greater affinity allowing viral disruption of CAR D1:D1 dimers (7). This disruption not only has implications for infection but also for escape from polarized epithelial cells, where CAR behaves as an adhesion protein (10). Although there is partial overlap, Coxsackievirus B3 (CB3) uses additional surfaces of CAR D1 for binding (6).
One prominent feature of CAR is its localization at the junctions between cells (11, 12). It is known that the intracellular carboxy terminus (c-terminus) of CAR is important for junctional localization (11, 13). This is particularly apparent in polarized epithelial cells, where CAR is normally trafficked to the basolateral surface and is a component of the junctional adhesion complex (10). Deletion of and certain mutations within the c-terminus allow the receptor to reach the apical membrane. The contribution of the extracellular domain to localization has never been examined.
Based on CAR homodimer formation between adjacent cells via its most distal (D1) domain, we hypothesized that this interaction would be essential for trapping CAR between cells that express CAR, resulting in the junctional localization. No activity has been ascribed to the proximal Ig-like domain (D2), and its importance in dimerization or viral interactions is unknown. To better understand this protein, we have further defined the functional contribution of the extracellular domain in localization and in the physiologic activities of CAR.
MATERIALS AND METHODS
Materials
FLAG M2 antibody (Ab) was purchased from Sigma (F3165; St. Louis, MO). Alexa-488– and -568–conjugated goat anti-mouse or anti-rabbit Abs were from Molecular Probes (Eugene, OR). The CAR Ab RmcB (CRL-2379; ATCC, Manassas, VA) was produced by the University of Iowa Hybridoma Core. Rabbit anti–CAR-1605 was produced in rabbits immunized with a GST fusion to the intracellular c-terminus (aa 261–365). The fragment was cloned by polymerase chain reaction (PCR) and inserted into the vector pGEX-KT (77331; ATCC). The plasmid was transformed into BL21-A cells, induced with IPTG, and isolated by standard methodologies. CAR-positive monkey fibroblast cells (COS-7) and CAR-negative Chinese Hamster Ovary cells (CHO) cells were from ATCC and maintained under standard culture conditions (D-MEM or HAM F12 with 10% fetal calf serum, penicillin, and streptomycin). Ad serotype 5 containing the β-galactosidase (Ad-β-Gal), eGFP, RFP (peGFP-N1, pDSRed1; Clontech, Palo Alto, CA), or CAR gene have previously been described (12, 14). Ad serotype 5 CMV driven CARD0, CARΔD1, and CARΔD2 were created as previously described (12). All Ads were generated by the University of Iowa Gene Vector Core. Ad5GL (wild-type fiber length), Ad5longIGL (27 β repeats, elongated fiber), and Ad5longIIGL (32 β repeats, elongated fiber) were a generous gift from Dr. David Curiel (University of Alabama, Birmingham, AL). Ad-GFP labeled with 3H-thymidine was a generous gift from Dr. Beverly Davidson (University of Iowa, Iowa City, IA).
Cloning of CARD0, CARΔD1, and CARΔD2
The extracellular domains of CAR were deleted via three PCR reactions. The first reaction created the leader and FLAG tag fragment (and the D1 domain for CARΔD2). The second reaction cloned the transmembrane domain and c-terminus for CARD0 and CARΔD2, or the D2 domain plus the transmembrane domain and c-terminus for CARΔD1. These fragments were gel-purified and used as the template for the third PCR that fused the fragments together through homologous regions made in the primers. The resulting product was digested with EcoRI and BamHI and cloned into pcDNA3.1(−) or an Ad5–CMV shuttle plasmid. DNA sequencing and Western blot confirmed the expected protein. The primers included a primer to the 5′ end of CAR (CAR5′: 5′TGGAATTCCCAGGAGCGAGAG), 3′ end of CAR (CAR3′: 5′CGGATCCCTATACTATAGACCCATC), CARΔD1F(5′TATCTGCCG-AAAGCTCCTGGTGTTGCA), CARΔD1R (5′AGGAGCTTT-CGGCAGATAGGCAGTTTCC), CARΔD2F (5′AAGATAAAA-ACAGTCAGAAACAGAGTGG), CARΔD2R (5′TCTGACTGT-TTTTATCTTAAAGTCACTTC), CARD0F (5′TATCTGCCG-ACAGTCAGAAACAGAGTGG), CARD0R (5′TCTGACTGT-CGGCAGATAGGCAGTTTCC). The PCR reactions were performed as follows: 95°C, 5 min; 30 rounds of 94°C, 30 s, 56°C, 45 s, 72°C, 45 s; 72°C, 5 min.
Electroporation of COS Cells
COS-7 cells were electroporated by standard methodologies. Briefly, 10 million cells were mixed with 20 μg of plasmid DNA for single transfection or 15 μg of each DNA for double transfections, 400 μl of cytomix (120 mM KCl, 0.15 mM CaCl2, 10 mM K2HPO4, 10 mM KH2PO4, 25 mM HEPES, 2 mM EGTA, 5 mM MgCl2, 2 mM ATP, and glutathione) and put in an electroporation cuvette (Bio-Rad Laboratories, Hercules, CA) for 30 min on ice. After electroporation, cells were seeded onto 10-cm dishes for immunoprecipitation and collagen-coated glass chamber slides for immunofluorescence studies 2 d later.
Biotinylation and Western Blot
Cells from 100 mm plates were placed on ice, washed once with ice-cold phosphate-buffered saline (PBS), incubated at 4°C with 3 ml of 1 mg/ml EZ-Link Sulfo-NHS-SS-Biotin (Cat. 21331; Pierce, Rockford, IL) in PBS for 45 min, quenched with 100 mM glycine, washed three times with PBS, and lysed with lysis buffer (50 mM Tris-HCl, pH 7.5, 137 mM NaCl, 1% Triton X-100, 5 mM EDTA, 1 mM EGTA, protease inhibitors [10 μg/ml] leupeptin, aprotinin, pepstatin, and 1 mM phenylmethylsulfonyl fluoride) by rocking at 4°C. Cells were scraped, sonicated five times, and spun in a microcentrifuge at full speed for 10 min. Supernatant was incubated with Ultralink Immobilized NeutrAvidin Biotin Binding Protein (Cat. 53150; Pierce) for 2 h followed by a wash with lysis buffer, 10% lysis buffer in TBS (50 mM Tris-HCl, pH 7.5, 137 mM NaCl), and TBS. Beads were suspended in loading buffer (4% sodium dodecyl sulfate, 100 mM dithiothreitol, 20% glycerol, 65 mM Tris [pH 6.8], 0.005% bromophenol blue) and proteins were separated by SDS-poly acrylamide gel electrophoresis. Gels were transferred to a polyvinylidene difluoride membrane (Millipore, Bedford, MA), blocked with 5% bovine serum albumin, washed, probed with primary Ab as indicated, followed by washing and incubation with protein A– or G–conjugated horseradish peroxidase (Pierce). Bands were detected with enhanced chemiluminescence reagents (Pierce).
Transfection and Infection of CHO
CHO cells were plated in 100-mm plates, 6-well dishes, 24-well dishes, or collagen-coated 4-well chamber slides and transfected the next day via calcium phosphate precipitation of Ad-CAR, -CARD0, -CARΔD1, -CARΔD2, or peGFP-N1 as previously described (15). After 48 h cells were analyzed by Western blot and immunofluorescence microscopy. Ad infection was investigated by seeding cells into 24-well dishes (n = 6) and infecting with Ad-β-Gal (MOI = 100) for 1 h at 37°C. After 48 h cells were lysed and β-galactosidase expression per mg protein was determined as previously described (12).
To determine the ability of CARΔD2 to mediate infection of an Ad with an artificially extended fiber, CHO cells expressing CAR, CARΔD2, or CARD0 were infected with 1,000 particles of Ad5GL (control fiber shaft length 22 β repeats), Ad5longIGL (longer shaft 27 β repeats), or 2,000 particles Ad5longIIGL (longer shaft 32 β repeats) for 1 h at 37°C, washed once with PBS, and returned to fresh media. Cells were incubated for 48 h, lysed and luciferase expression per mg protein was determined (16).
Binding of adenovirus was determined by 3H-labeled adenovirus. Briefly, cells were transfected as above, placed on ice, washed twice with ice-cold Eagle's Minimal Essential Medium (EMEM), incubated in triplicate with 3H-Ad for 1 h on ice, washed three times with ice-cold PBS, and lysed with 1% Triton X-100. Lysate was put into scintillation vials and then into a scintillation counter. All experiments were done in triplicate.
Immunocytochemistry
CHO cells grown on collagen-coated chamber slides were washed once with PBS, fixed with −20°C methanol plus 1% paraformaldehyde, and blocked with 2% bovine serum albumin in SuperBlock (Pierce). Cells were incubated with primary Ab, washed extensively, and then incubated with goat anti-mouse Alexa-568 Ab. Nuclei were stained with TO-PRO3 (Molecular Probes), washed again, then coverslipped with Vectashield mounting media (Vector Laboratories, Inc., Burlingame, CA). Images were acquired with a Bio-Rad MRC-1024 Laser Scanning Confocal Microscope mounted on a Nikon E600 microscope (Melville, NY) using a ×60 oil immersion lens.
Adhesion
Transfected cells were washed once with Ca2+-free Hanks' Balanced Salt Solution (HBSS, Cat. 14170–112; Invitrogen, Grand Island, NY) and incubated with 1 mM EDTA in HBSS until released from the plate. Cells were pelleted and washed twice with HBSS. Cells were resuspended at 2 × 106 cells/ml in HBSS supplemented with HBSS dialyzed fetal calf serum (2%) and placed on a rotator for 1 h at 37°C. Equal volumes of cells were then gently plated out and supplemented with a small volume of growth medium. Clumps of more than three cells were counted after 1 h at 37°C in at least 10 separate fields of view under ×10 power on a light microscope (Nikon Eclipse TS100).
CAR Expression in Primary Human Airway Epithelia
Primary human airway epithelia (HAE) were isolated from trachea and bronchi of donor lungs and seeded onto collagen-coated, semipermeable membranes (Millipore) and grown at the air–liquid interface as previously described (17, 18). Approximately 2 wk after seeding, cultures were well differentiated and attained a measurable transepithelial resistance. Primary HAE were treated apically with 5 mM EGTA in PBS for 5 min when the transepithelial resistance was reduced to background levels. EGTA was then removed and virus (MOI = 200) in 50 μl of EMEM was added apically and left on the cells for 1 h. Virus was removed and the basolateral media was changed. Immunocytochemistry was performed 24 h after infection.
RESULTS
The Extracellular Domain of CAR Is Not Required for Cell Surface Expression
Based on the boundaries predicted for D1 and D2 (19), we created three deletion mutants for the extracellular domain of CAR (Figure 1A). We first asked whether the mutants would be expressed in cells and make it to the cells' surface. To test this, each mutant was transfected into COS cells and expression was evaluated by cell surface biotinylation and Western blot (Figure 1B). Precipitation using streptavidin-coated beads resulted in the isolation of CAR, CARΔD1, CARΔD2, and CARD0, but not GFP or a control cytoplasmic protein (RafI, data not shown), revealing that biotin labeling was limited to the cell surface. The band for CARD0 showed comparatively lower intensity consistent with either less biotinylation sites, less protein at the cell surface, or both. Similar cell surface expression was observed in CHO cells (data not shown). These data show that the extracellular domain of CAR is not required for targeting CAR to the cell surface.
Figure 1.
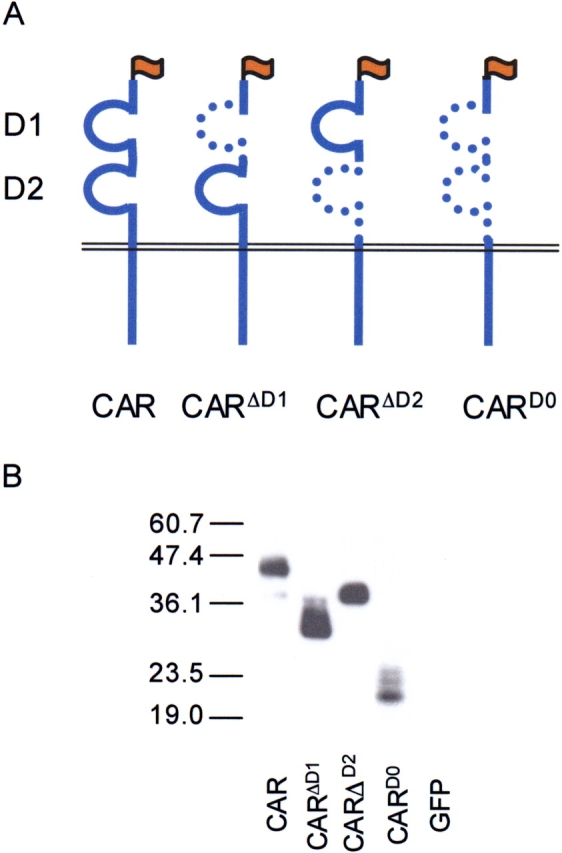
Schematic representation and cell surface expression of the extracellular domain deletion mutants of CAR. (A) CARΔD1 contains aa 1–161+213–365, CARΔD2 contains 1–40+213–365, and CARD0 was a fusion of aa 1–40+213–365. (B) Western immunoblot for expression of FLAG-tagged (CAR) cell surface biotinylated proteins in electroporated COS cells. Bands were present for CAR, CARΔD1, CARΔD2, and CARD0 at the appropriate molecular weight, whereas no bands were detectable from eGFP-expressing cells.
The Extracellular Domain of CAR Is Required for Cell:Cell Junction Localization and Adhesion
We hypothesized that although the extracellular domain is not critical for cell surface expression, it may be essential for junctional localization. We further hypothesized that the D1 domain would be essential for cell:cell junction localization if it occurs by trapping CAR molecules on adjacent cells. D2 would only be essential if the distance was too great for a short CAR molecule to engage another on an adjacent cell. To determine the cellular localization of these mutants, immunocytochemistry was performed on CHO cells transfected with CAR, CARD0, CARΔD1, CARΔD2, or GFP (Figure 2A). CHO cells transfected with CAR show some diffuse staining with significant accumulation at the junctions between cells expressing CAR (see arrow). In contrast, cells expressing CARD0 or CARΔD1 showed diffuse staining throughout the cells and did not accumulate specifically between cells. Surprisingly, CARΔD2 did not localize at the junctions between cells as well. This indicates that, in addition to the intracellular domain, the extracellular domain of CAR is essential for junctional localization.
Figure 2.
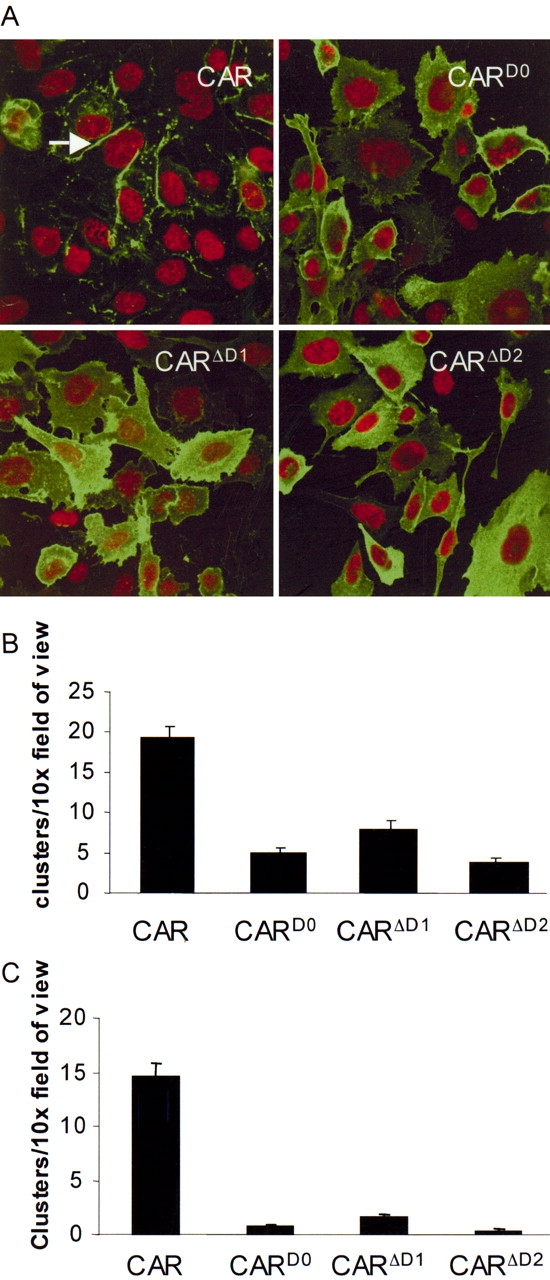
The extracellular domain of CAR is required for cell:cell localization and adhesion. (A) Immunofluorescence in CHO cells expressing CAR, CARD0, CARΔD1, and CARΔD2. Although each of the mutant forms is found at the cell surface, CAR is more often found limited to the junctions between cells expressing CAR (arrow). (B and C) Cell clustering of CHO cells expressing CAR, CARD0, CARΔD1, or CARΔD2. B represents clusters of > 3 cells and C represents clusters > 7. CAR showed significantly more and larger clusters than mutant forms of CAR (n = 3).
We also investigated the role of the extracellular domain of CAR in cell:cell adhesion. CHO cells were transfected as above. Incubating the cells with EDTA generated a single cell suspension, and clumping was assessed after 1 h in a Ca2+-free medium. CHO cells expressing CAR had significantly more clumping (Figure 2B) and significantly larger clumps of cells (Figure 2C) than CARΔD1, CARΔD2, or CARD0. Additionally, there was no difference in clumping between CARΔD2 and the other mutants, suggesting that the D2 domain plays a role in cell:cell adhesion.
The Extracellular Domain of CAR Is Required for Appropriate Localization in Primary Airway Epithelia
In airway epithelia, we and others have shown that CAR localizes to the junctional adhesion complex (10, 11, 20, 21). Considering that the entire c-terminus of CAR is present in the mutants in this study and that trafficking signals have been found in the c-terminus, we hypothesized that CARD0, CARΔD1, and CARΔD2 would traffic to the basolateral side of the epithelia, but without the homotypic interaction they would be found diffusely throughout the basolateral side. Differentiated primary HAE were infected with Ad vectors expressing each construct and immunostained for the FLAG tag (Figure 3, green) on each resulting protein as well as the tight junction marker ZO-1 (red) and the nucleus (blue). Although expression of all proteins appeared basolateral, there was an obvious difference between their localization. Wt-CAR results in mostly lateral expression with a decreasing gradient of expression from ZO-1 toward the basal side of the cell. In contrast, CARΔD1 and CARΔD2 show no preference for basal or lateral expression. CARD0 shows primarily diffuse expression throughout the cell. These data suggest that CAR reaches the lateral wall and that full-length extracellular CAR finds CAR molecules on adjacent cells, resulting in lateral retention, while the deletion mutants continue to diffuse to the basal side (22).
Figure 3.
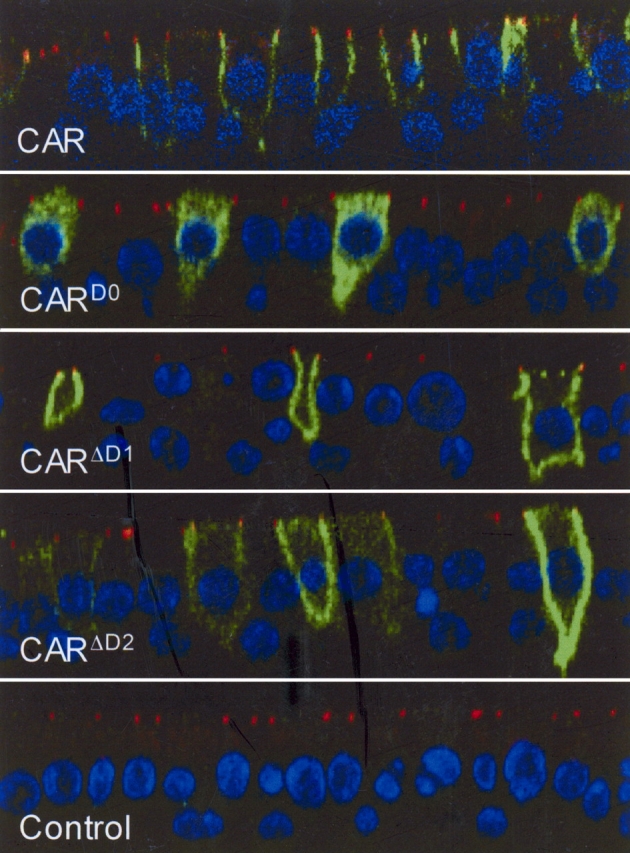
The extracellular domain of CAR is required for lateral localization in polarized airway epithelial cells. Primary HAE were infected with Ad-CAR, CARD0, CARΔD1, or CARΔD2 and immunostained for the FLAG epitope (green), the tight junction protein ZO-1 (red), and the nucleus (blue). Although all forms remained basolateral, CAR was found mainly on the lateral surface, whereas CARΔD1 and CARΔD2 show no preference between the basal and lateral surface. CARD0 staining was diffuse, indicating that a significant amount of this construct remains in the cell.
The Extracellular Domain of CAR Is Required for Ad Binding and Infection
Several studies have delimited the D1 domain of CAR as essential for Ad binding (4–9); thus, we hypothesized that CARD0 and CARΔD1 would not support Ad binding or infection, whereas CARΔD2 may bind and allow infection. As expected, compared with wt-CAR, the deletion of D1 in CARD0 and CARΔD1 rendered these molecules unable to bind or support Ad-mediated gene transfer above control (GFP) levels (Figures 4A and 4B, respectively). Binding and infection through CARΔD2 was significantly attenuated but not eliminated with ∼ 15% residual activity compared with wt-CAR. Interestingly, both the binding and infectivity were equivalent, suggesting that binding was affected; but, once bound, CARΔD2 was functional for infection. These data suggest that the D2 domain of CAR plays an important role in facilitating D1:fiber knob interactions.
Figure 4.
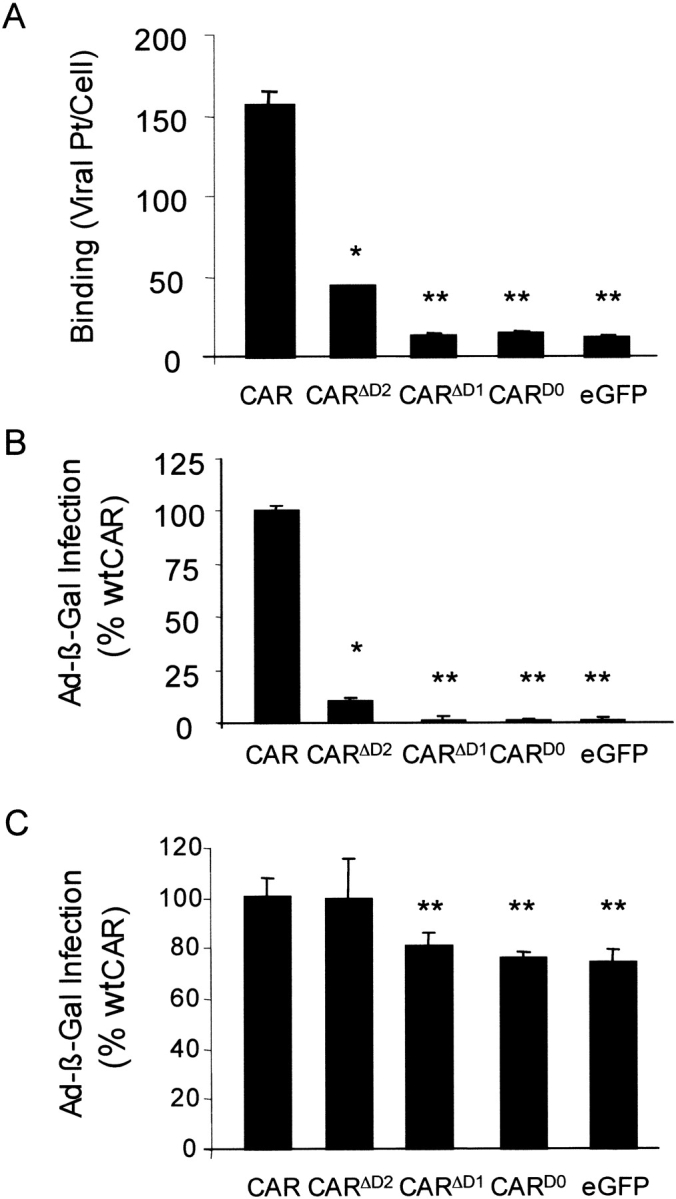
CAR extracellular deletion mutants do not bind or mediate efficient adenovirus infection in CHO cells and do not have a dominant-negative effect on Ad infection in COS cells. (A) The binding of 3H-Ad-GFP on CHO cells expressing CAR, CARD0, CARΔD1, CARΔD2, or eGFP (n = 3, one representative experiment shown). (B) Compared with Ad-mediated β-galactosidase gene expression in CHO cells expressing CAR, cells expressing either CARD0 or CARΔD1 fail to mediate infection and CARΔD2-mediated infection is significantly attenuated (n = 3). (C) Infection of Ad-β-Gal in COS cells expressing CAR, CARD0, CARΔD1, or CARΔD2. Additional expression of CAR and CARΔD2 in COS cells, which express endogenous amounts of CAR, augments Ad infection, whereas expression of CARD0 or CARΔD1 does not affect Ad-mediated gene expression (n = 3, one representative experiment shown).
Deletion of the Extracellular Domain of CAR Does Not Have a Dominant-Negative Effect
Although the D1 domain of CAR is essential for Ad binding and infection, we hypothesized that CARΔD1 or CARD0 may interact with intracellular proteins or the D2 domain of wt-CAR and affect the function or activity of wt-CAR. We expressed CAR, CARD0, CARΔD1, CARΔD2, or GFP in COS cells that express endogenous CAR. We then tested for Ad infection by incubating the cell with Ad-β-Gal and tested for β-galactosidase activity 48 h later (Figure 4C). We found that overexpression of wt-CAR resulted in significantly increased Ad infection over control cells that expressed GFP, suggesting that baseline CAR expression is rate limiting. Expression of CARD0 or CARΔD1 had no effect on Ad-mediated gene transfer, suggesting that they do not act in a dominant-negative fashion. Finally, overexpression of CARΔD2 significantly augmented Ad infection to a degree similar to that of wt-CAR (CAR or CARΔD2 versus CARD0, CARΔD1, or GFP P < 0.001), suggesting that in the presence of wt-CAR, CARΔD2 can synergistically augment Ad binding and infection.
CARΔD2 Does Not Rescue Infection of Ad with Elongated Fibers
We hypothesized that an Ad with an elongated fiber may be able to interact and infect through the short CARΔD2. CHO cells expressing CAR, CARD0, or CARΔD2 were infected with equal particle numbers of AdGL (wild-type fiber, 22 β repeats), AdLongIGL (27 β repeats), or AdLongIIGL (32 β repeats) (16) and analyzed for luciferase activity 48 h later (Figure 5, n = 3). As previously shown by Seki and coworkers, CAR-mediated infection of AdGL was greater than AdLongIGL, which in turn was significantly greater than AdLongIIGL (16). As expected, CARΔD2-mediated infection with AdGL (wild-type fiber length) was attenuated (P < 0.001 versus CAR) but not ablated as compared with CARD0 (P < 0.01). However, AdLongIGL and AdLongIIGL failed to infect via CARΔD2. CARD0 did not support infection of any of the three viruses. These data suggest that the failure of infection via CARΔD2 cannot be corrected simply by increasing the length of fiber.
Figure 5.

CARΔD2 does not rescue infection of cells with adenoviruses with elongated fibers. CHO cells expressing CAR, CARΔD2, or CARD0 were infected with Ad-GL, Ad-IGL, or Ad-IIGL at a dose of 1,000 pt/cell. At 48 h, cells were analyzed for luciferase activity per milligram protein (n = 3, one representative experiment shown). Elongating the Ad fiber does not rescue infection via CARΔD2.
DISCUSSION
Our study demonstrates that the intact extracellular domain of CAR is important for the localization of CAR at cell:cell junctions and on the lateral wall of the basolateral membrane of polarized cells. It is also required for efficient Ad binding and infection.
Previous studies have shown that the intracellular domain of CAR plays a role in apical versus basolateral sorting of CAR in polarized cells (10, 11, 20, 21). This study reveals that although the extracellular domain does not play a role in basolateral cell surface targeting, it is important for localizing CAR on the lateral membrane at and below the tight junction. Junctional localization plays an implicit role in the adhesion activity of CAR. However, adhesion may be the driving force for junctional localization. One potential model for defining the localization of CAR has been termed “diffusion trapping.” This model predicts relatively free movement in the membrane until it is stabilized by engaging its ligand (i.e., another CAR molecule) on an adjacent cell. Such a model has been proposed for other adhesion proteins such as platelet-endothelial cell adhesion molecule-1 (PECAM-1/CD31) (23). Junctional localization of PECAM-1/CD31 requires the transmembrane domain, the juxta-membrane portion of the c-terminus, and only occurred with an extracellular domain capable of adhesion. Point mutations that rendered PECAM-1 unable to interact with adjacent PECAM-1 molecules resulted in distribution across the membrane with no obvious preference for junctions. This appears to be true for CAR and may be a general phenomenon for adhesion proteins. Interestingly, the D2 domain of CAR is also required, suggesting that the steric positioning of D1, when extended by D2, is required to bind to the other D1 on adjacent cells.
Ad infection also requires the D2 domain. There are at least three potential reasons for this requirement. First, the distance between the cell membrane and the capsid is determined by the length of fiber and CAR and is precisely important. Second, D2 may play a role in infection such as interacting with other CAR molecules, co-receptors, proteins involved in internalization or endosomal escape. Third, D2 is required for the proper folding of the CAR extracellular domain. Our data indicate that the D2 segment requirement during Ad infection is limited to Ad binding. A precise fiber:extracellular domain interaction requires the extension of the D1 domain. The fiber shaft contains a variable number of ∼ 14 amino acid repeats that form a triple β-spiral fold (24, 25). A wild-type Ad serotype 2 or 5 virus has 22 β repeats, but the repeat number can range from 6 (Ad3, Ad11, and Ad35) to 23 (Ad12). Substantially increasing or decreasing the length from 22 significantly alters the efficiency of Ad infection (16, 24–27). We hypothesized that viruses modified to contain elongated fibers may compensate for the shortening of CAR. However, a simple elongation of fiber, which would hypothetically reduce the charge repulsion between the cell surface and Ad particle, did not improve the ability of CARΔD2 to mediate infection, further revealing the complexity of viral infection. Interestingly, when CARΔD2 was expressed in conjunction with endogenous CAR, it increased the ability of Ad to infect COS cells. Ad interaction with cells has been shown to exhibit positive cooperative binding (4, 28). This is explained by both the multivalency of the virus attachment (multiple fibers on the virion) and the capacity of CAR to migrate and cluster around the virus. Thus, we speculate that once one fiber has bound to the D1 domain of wt-CAR, CARΔD2 can participate in cooperative binding.
The hypothesis that CARΔD2 would support Ad infection stemmed from a study investigating reovirus interactions with its receptor Junctional Adhesion Molecule A (JAM-A) (29). CAR and JAM-A are part of the same Ig subfamily, and although they are both adhesion proteins and viral receptors, other known biological activities are distinct (30). Reovirus binds to JAM-A through a fiber-like protein known as σ1, which has several characteristics in common with the Ad fiber, such as a triple β-spiral motif in the shaft, similar globular structures in the knob equivalent domain, and the overall length (31). Interestingly, deletion of the D2 domain of JAM-A or swapping it with the CAR D2 domain did not affect reovirus infection. These results can be explained by the inherent flexibility of the σ1 protein, which has a portion of the shaft composed of α-helical coiled coils. In contrast, the Ad shaft is made up almost entirely of β-spirals and known to be rigid. In addition, a JAM-A-CAR chimeric molecule, in which the D1 domain of CAR was cloned in place of the JAM-A D1 domain, became specific for Ad and allowed infection with an efficiency similar to that of wt-CAR. This experiment suggests that the converse would be true and that replacing the CAR D2 domain with JAM-A D2 would allow CAR to maintain its activity in Ad binding. This indicates that the D2 domain of CAR only plays a role as a spacer in Ad infection and may have an effect on the predicted ability of a trimeric fiber to bind to multiple CAR molecules in close proximity.
In conclusion, the extracellular domain plays an important role in the biology of CAR from epithelial subcellular localization and adhesion to viral infection. CAR is an extremely well-conserved protein that likely plays key roles in the biology of the cell with effects on protein trafficking, localization, cell signaling, growth, and adhesion. We would predict that mutations in this extracellular domain that disrupt the adhesion characteristic would have a significant effect on CAR-mediated activities within the cell and potentially affect communication between cells.
Acknowledgments
The authors thank Jamie Kesselring for assistance with manuscript preparation, Michael Welsh for discussions, David Curiel and Beverly Davidson for their valuable contributions, the Central Microscopy Research Facility, the Gene Transfer Vector Core, the Gene Transfer Morphology Core (supported by the NIDDK [DK54759] and the Cystic Fibrosis Foundation, ENGELH98S0), the Hybridoma/Tissue Culture Facility, the In Vitro Cell Models Core (supported by the National Heart, Lung and Blood Institute, the Cystic Fibrosis Foundation, and the National Institutes of Diabetes and Digestive and Kidney Diseases [DK54759]), and the Iowa Statewide Organ Procurement.
This work was supported by a PPG grant from the NIH (HL51670-11). KE is supported by a fellowship from the Parker B. Francis Foundation (PBF).
Conflict of Interest Statement: K.J.D.A.E. has no declared conflicts of interest; G.L.T. has no declared conflicts of interest; and J.Z. has no declared conflicts of interest.
References
- 1.Excoffon KJ, Hruska-Hageman A, Klotz M, Traver GL, Zabner J. A role for the PDZ-binding domain of the coxsackie B virus and adenovirus receptor (CAR) in cell adhesion and growth. J Cell Sci 2004;117:4401–4409. [DOI] [PubMed] [Google Scholar]
- 2.Coyne CB, Voelker T, Pichla SL, Bergelson JM. The coxsackievirus and adenovirus receptor interacts with the multi-PDZ domain protein-1 (MUPP-1) within the tight junction. J Biol Chem 2004;279:48079–48084. [DOI] [PubMed] [Google Scholar]
- 3.Jiang S, Jacobs A, Laue TM, Caffrey M. Solution structure of the coxsackievirus and adenovirus receptor domain 1. Biochemistry 2004;43:1847–1853. [DOI] [PubMed] [Google Scholar]
- 4.Lortat-Jacob H, Chouin E, Cusack S, van Raaij MJ. Kinetic analysis of adenovirus fiber binding to its receptor reveals an avidity mechanism for trimeric receptor-ligand interactions. J Biol Chem 2001;276:9009–9015. [DOI] [PubMed] [Google Scholar]
- 5.Kirby I, Davison E, Beavil AJ, Soh CP, Wickham TJ, Roelvink PW, Kovesdi I, Sutton BJ, Santis G. Identification of contact residues and definition of the CAR-binding site of adenovirus type 5 fiber protein. J Virol 2000;74:2804–2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.He Y, Chipman PR, Howitt J, Bator CM, Whitt MA, Baker TS, Kuhn RJ, Anderson CW, Freimuth P, Rossmann MG. Interaction of coxsackievirus B3 with the full length coxsackievirus-adenovirus receptor. Nat Struct Biol 2001;8:874–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van Raaij MJ, Chouin E, van der Zandt H, Bergelson JM, Cusack S. Dimeric structure of the coxsackievirus and adenovirus receptor D1 domain at 1.7 A resolution. Struct Fold Des 2000;8:1147–1155. [DOI] [PubMed] [Google Scholar]
- 8.Roelvink PW, Mi Lee G, Einfeld DA, Kovesdi I, Wickham TJ. Identification of a conserved receptor-binding site on the fiber proteins of CAR-recognizing adenoviridae. Science 1999;286:1568–1571. [DOI] [PubMed] [Google Scholar]
- 9.Bewley MC, Springer K, Zhang YB, Freimuth P, Flanagan JM. Structural analysis of the mechanism of adenovirus binding to its human cellular receptor, CAR. Science 1999;286:1579–1583. [DOI] [PubMed] [Google Scholar]
- 10.Walters R, Freimuth P, Moninger T, Ganske I, Zabner J, Welsh M. Adenovirus fiber disrupts CAR-mediated intercellular adhesion allowing virus escape. Cell 2002;110:789–799. [DOI] [PubMed] [Google Scholar]
- 11.Cohen CJ, Gaetz J, Ohman T, Bergelson JM. Multiple regions within the coxsackievirus and adenovirus receptor cytoplasmic domain are required for basolateral sorting. J Biol Chem 2001;276:25392–25398. [DOI] [PubMed] [Google Scholar]
- 12.Ashbourne Excoffon KJ, Moninger T, Zabner J. The coxsackie B virus and adenovirus receptor resides in a distinct membrane microdomain. J Virol 2003;77:2559–2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Walters RW, van't Hof W, Yi SM, Schroth MK, Zabner J, Crystal RG, Welsh MJ. Apical localization of the coxsackie-adenovirus receptor by glycosyl- phosphatidylinositol modification is sufficient for adenovirus-mediated gene transfer through the apical surface of human airway epithelia. J Virol 2001;75:7703–7711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Walters RW, Grunst T, Bergelson JM, Finberg RW, Welsh MJ, Zabner J. Basolateral localization of fiber receptors limits adenovirus infection from the apical surface of airway epithelia. J Biol Chem 1999;274:10219–10226. [DOI] [PubMed] [Google Scholar]
- 15.Fasbender A, Lee JH, Walters RW, Moninger TO, Zabner J, Welsh MJ. Incorporation of adenovirus in calcium phosphate precipitates enhances gene transfer to airway epithelia in vitro and in vivo. J Clin Invest 1998;102:184–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Seki T, Dmitriev I, Kashentseva E, Takayama K, Rots M, Suzuki K, Curiel DT. Artificial extension of the adenovirus fiber shaft inhibits infectivity in coxsackievirus and adenovirus receptor-positive cell lines. J Virol 2002;76:1100–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zabner J, Wadsworth SC, Smith AE, Welsh MJ. Adenovirus-mediated generation of cAMP-stimulated Cl- transport in cystic fibrosis airway epithelia in vitro: effect of promoter and administration method. Gene Ther 1996;3:458–465. [PubMed] [Google Scholar]
- 18.Karp PH, Moninger T, Weber SP, Nesselhauf TS, Launspach J, Zabner J, Welsh MJ. An in vitro model of differentiated human airway epithelia: methods and evaluation of primary cultures. In: Wise C, editor. Epithelial cell culture protocols. Totowa, NJ: Humana Press, Inc.; 2002. pp. 115–137. [DOI] [PubMed]
- 19.Bergelson JM, Cunningham JA, Droguett G, Kurt-Jones EA, Krithivas A, Hong JS, Horwitz MS, Crowell RL, Finberg RW. Isolation of a common receptor for coxsackie B viruses and adenoviruses 2 and 5. Science 1997;275:1320–1323. [DOI] [PubMed] [Google Scholar]
- 20.Zabner J, Winter M, Excoffon KJ, Stoltz D, Ries D, Shasby S, Shasby M. Histamine alters E-cadherin cell adhesion to increase human airway epithelial permeability. J Appl Physiol 2003;95:394–401. [DOI] [PubMed] [Google Scholar]
- 21.Cohen CJ, Shieh JT, Pickles RJ, Okegawa T, Hsieh JT, Bergelson JM. The coxsackievirus and adenovirus receptor is a transmembrane component of the tight junction. Proc Natl Acad Sci USA 2001;98:15191–15196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mostov K, Su T, ter Beest M. Polarized epithelial membrane traffic: conservation and plasticity. Nat Cell Biol 2003;5:287–293. [DOI] [PubMed] [Google Scholar]
- 23.Sun J, Paddock C, Shubert J, Zhang HB, Amin K, Newman PJ, Albelda SM. Contributions of the extracellular and cytoplasmic domains of platelet-endothelial cell adhesion molecule-1 (PECAM-1/CD31) in regulating cell-cell localization. J Cell Sci 2000;113:1459–1469. [DOI] [PubMed] [Google Scholar]
- 24.Wu E, Pache L, Von Seggern DJ, Mullen TM, Mikyas Y, Stewart PL, Nemerow GR. Flexibility of the adenovirus fiber is required for efficient receptor interaction. J Virol 2003;77:7225–7235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ambriovic-Ristov A, Mercier S, Eloit M. Shortening adenovirus type 5 fiber shaft decreases the efficiency of postbinding steps in CAR-expressing and nonexpressing cells. Virology 2003;312:425–433. [DOI] [PubMed] [Google Scholar]
- 26.Shayakhmetov DM, Lieber A. Dependence of adenovirus infectivity on length of the fiber shaft domain. J Virol 2000;74:10274–10286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roelvink PW, Lizonova A, Lee JG, Li Y, Bergelson JM, Finberg RW, Brough DE, Kovesdi I, Wickham TJ. The coxsackievirus-adenovirus receptor protein can function as a cellular attachment protein for adenovirus serotypes from subgroups A, C, D, E, and F. J Virol 1998;72:7909–7915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Persson R, Wohlfart C, Svensson U, Everitt E. Virus-receptor interaction in the adenovirus system: characterization of the positive cooperative binding of virions on HeLa cells. J Virol 1985;54:92–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Forrest JC, Campbell JA, Schelling P, Stehle T, Dermody TS. Structure-function analysis of reovirus binding to junctional adhesion molecule 1. Implications for the mechanism of reovirus attachment. J Biol Chem 2003;278:48434–48444. [DOI] [PubMed] [Google Scholar]
- 30.Raschperger E, Engstrom U, Pettersson RF, Fuxe J. CLMP, a novel member of the CTX family and a new component of epithelial tight junctions. J Biol Chem 2004;279:796–804. [DOI] [PubMed] [Google Scholar]
- 31.Stehle T, Dermody TS. Structural similarities in the cellular receptors used by adenovirus and reovirus. Viral Immunol 2004;17:129–143. [DOI] [PubMed] [Google Scholar]